
A Literature-Derived Knowledge Graph Augments the Interpretation of Single Cell RNA-seq Datasets
 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Generation of Cell Type Vocabulary
2.2. Generation of Gene Vocabulary
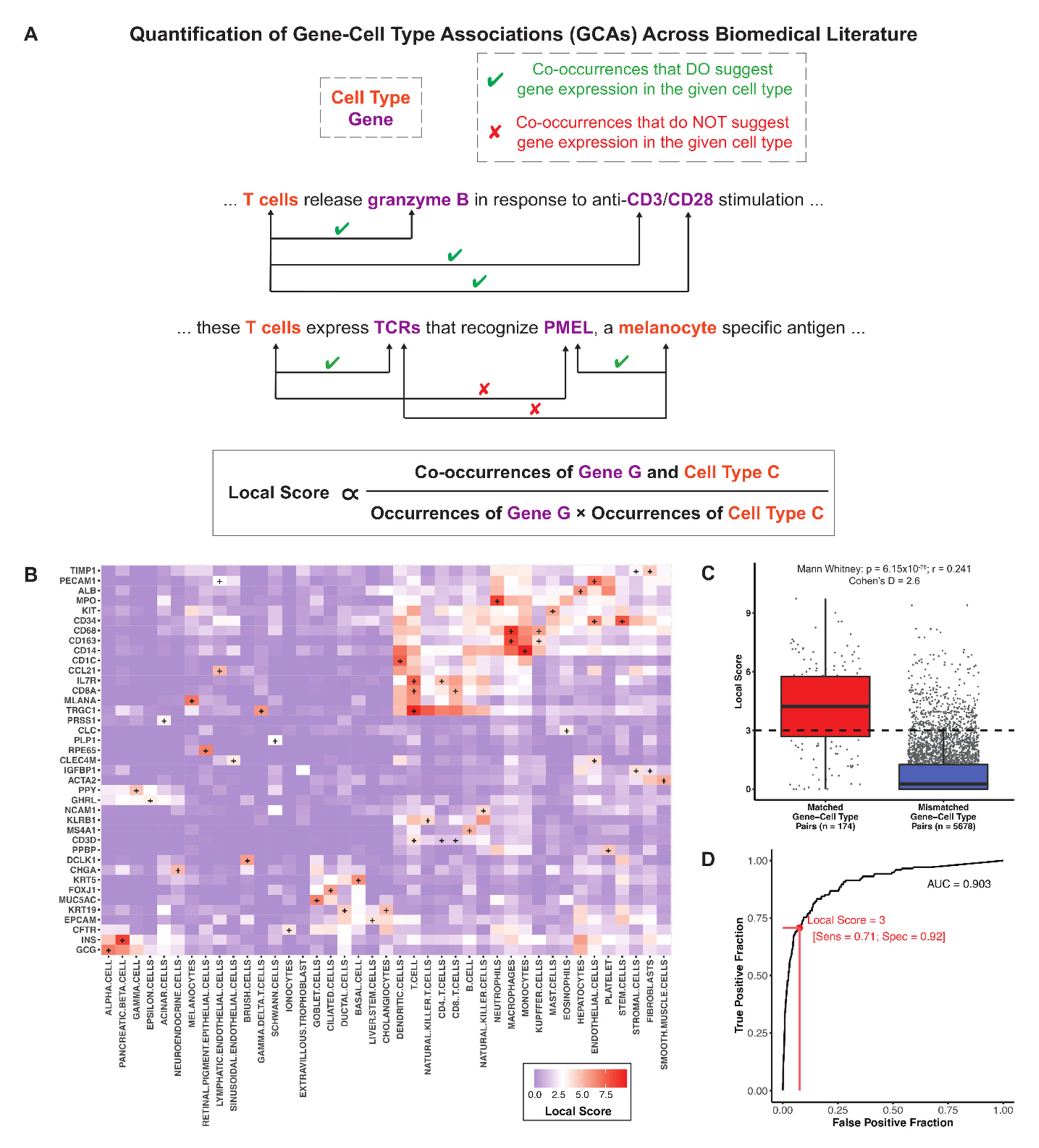
2.3. Quantification of Literature Associations between Genes and Cell Types
2.4. Curation of Canonical Cell Type Defining Genes
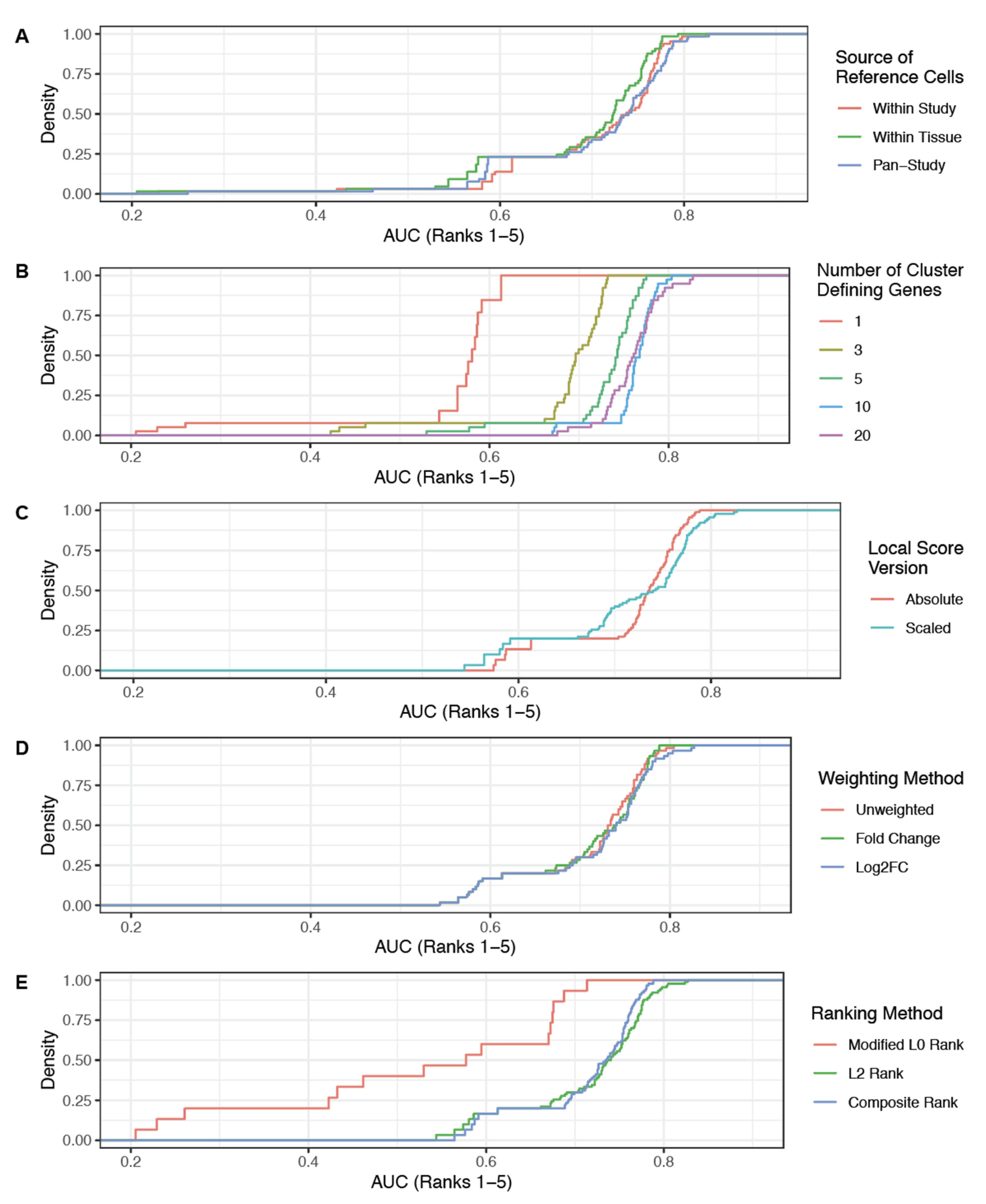
2.5. ROC Analysis of Local Scores to Classify Matched GCAs versus Mismatched GCAs
2.6. Processing of scRNA-seq Studies
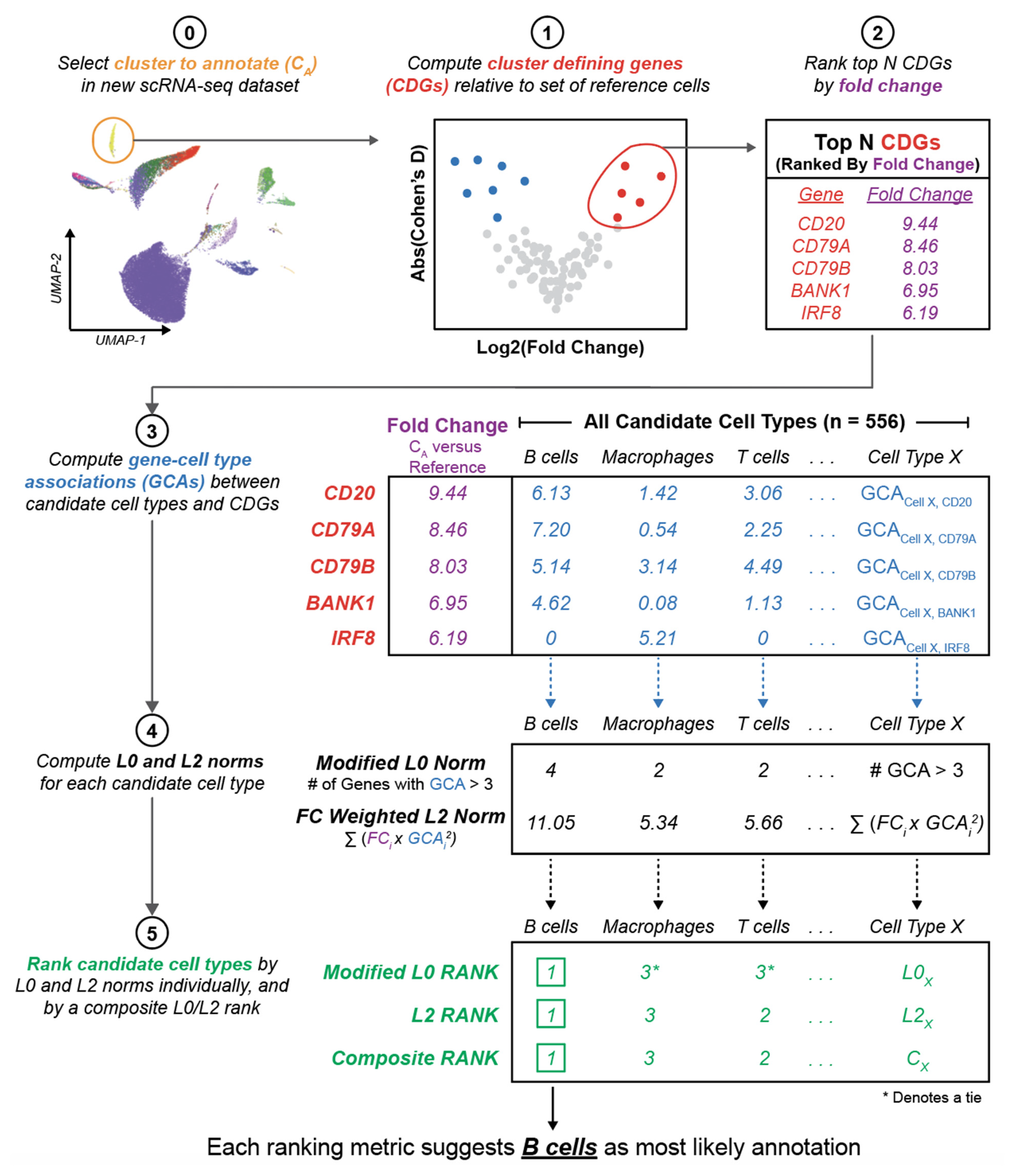
2.7. Cell Type Annotation Algorithm
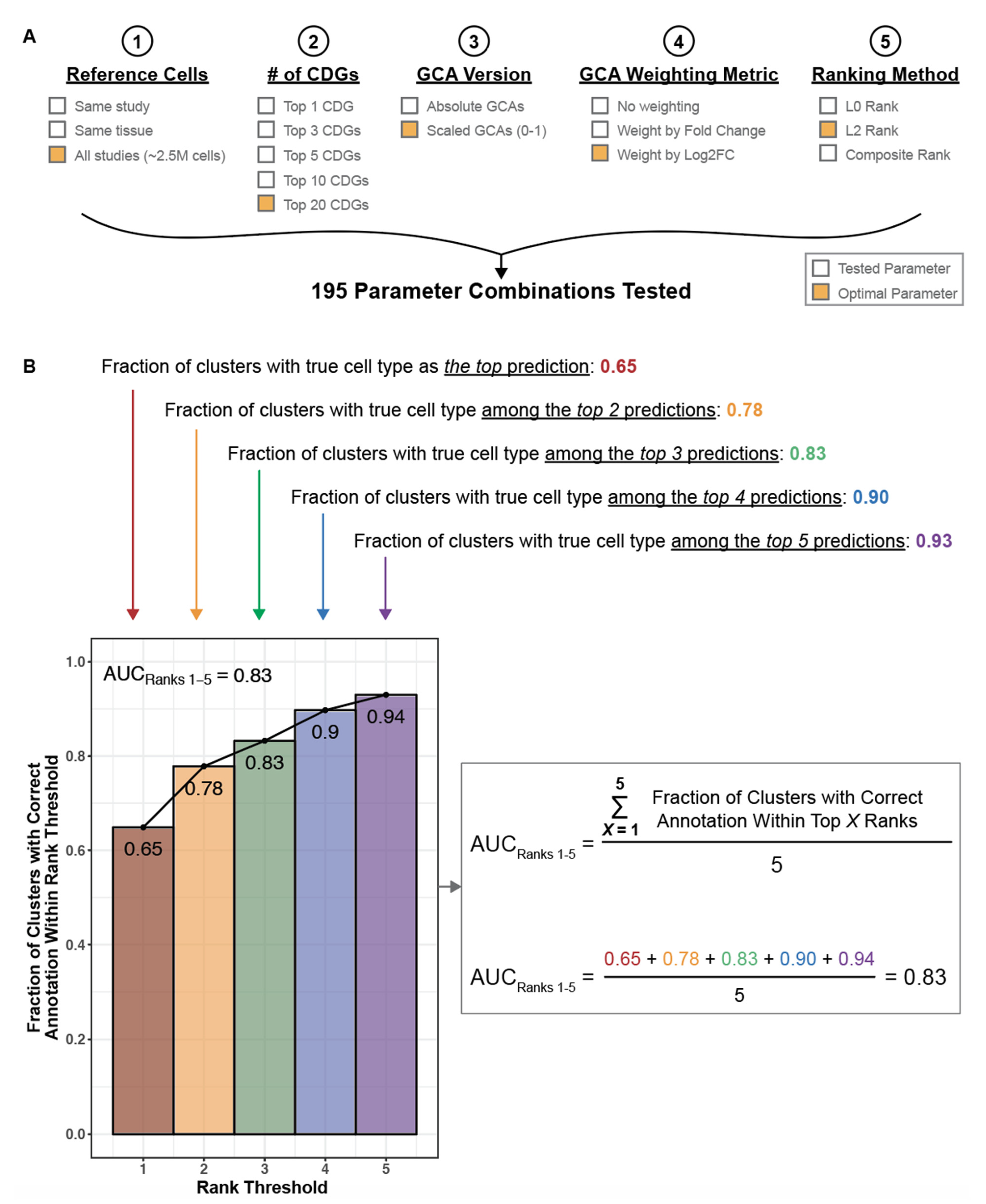
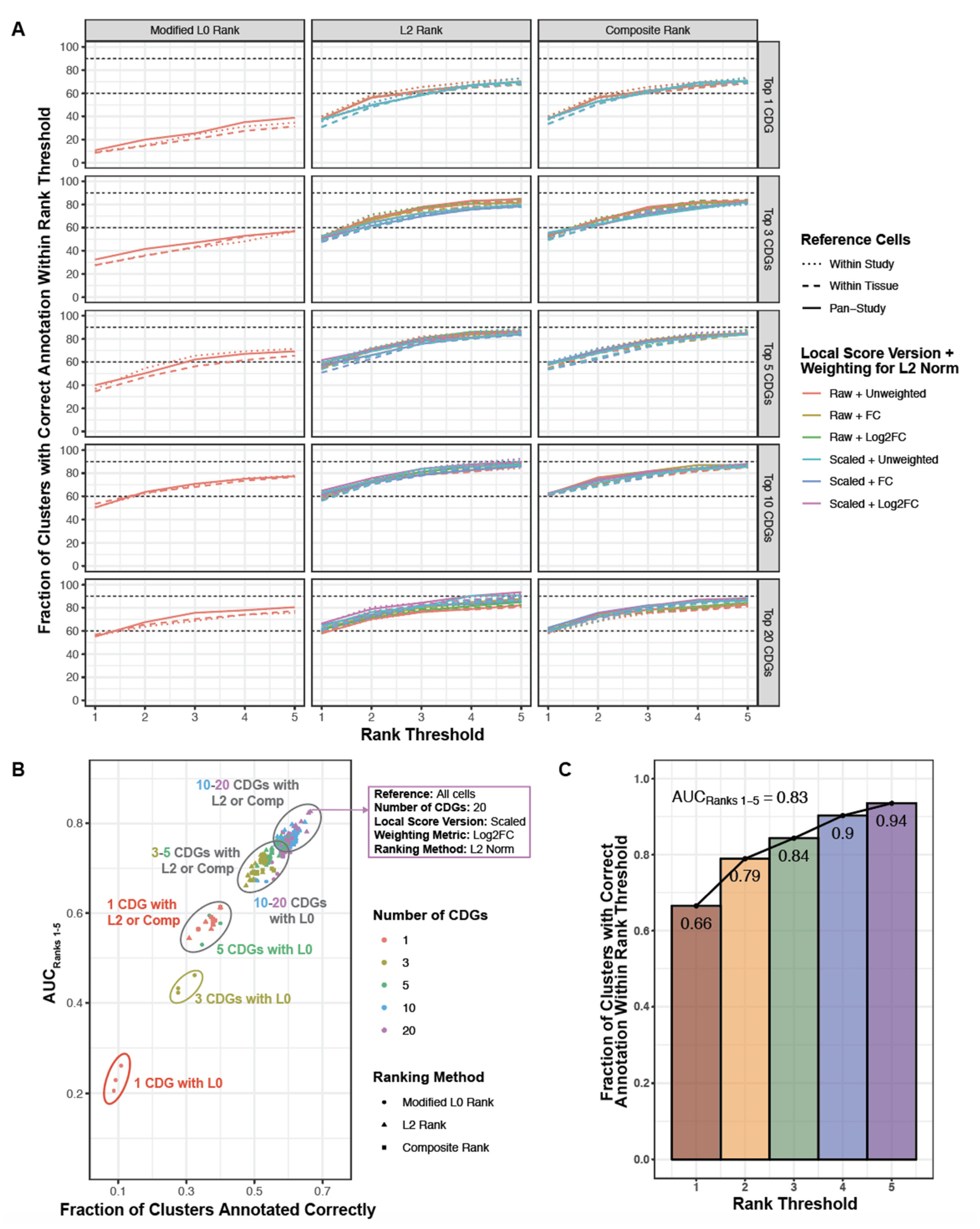
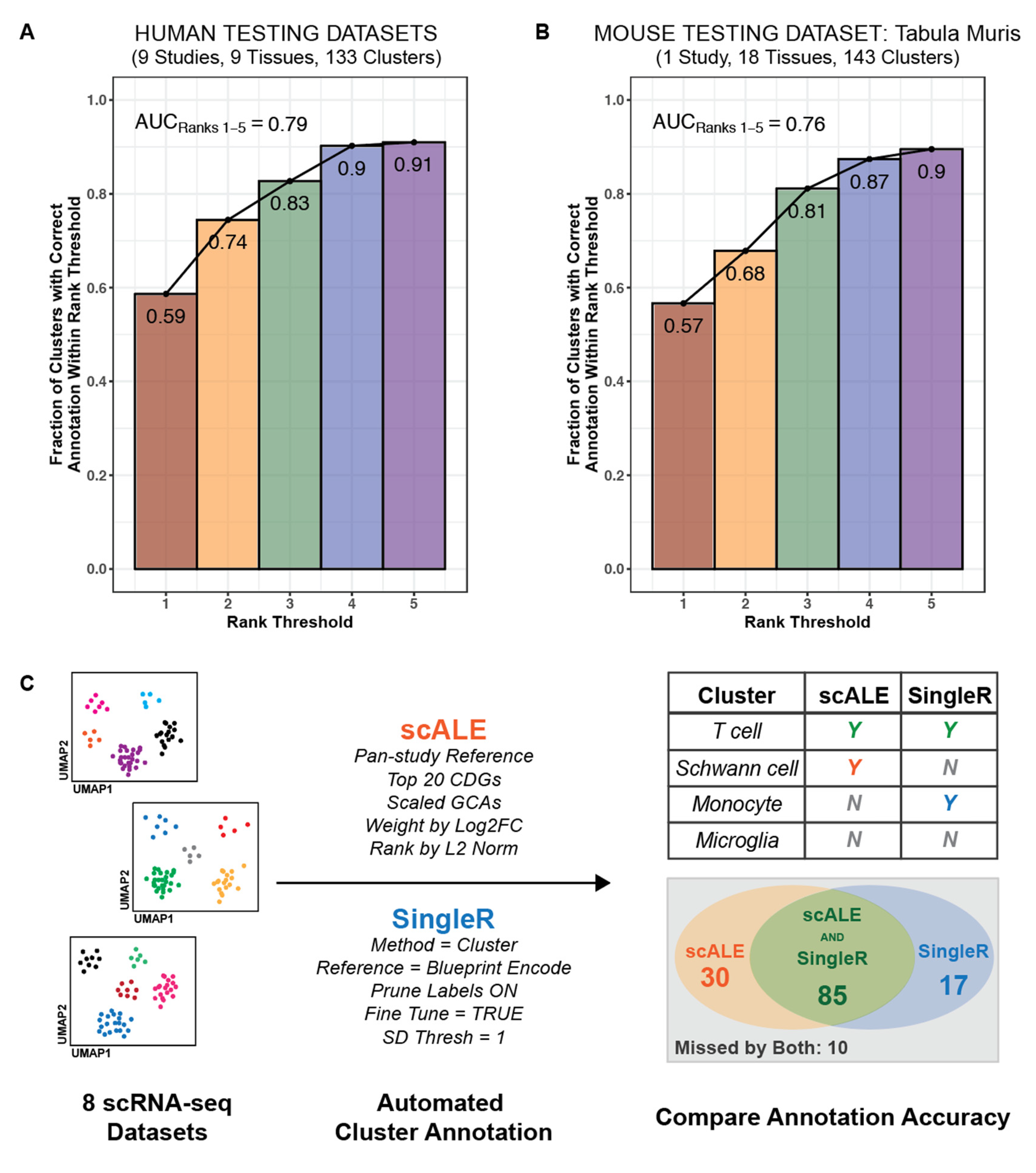
2.8. Optimization and Evaluation of Cell Type Annotation Algorithm
2.9. Application of scALE to Annotate to Murine scRNA-seq Data
2.10. Comparison of Cell Type Annotations from scALE versus SingleR
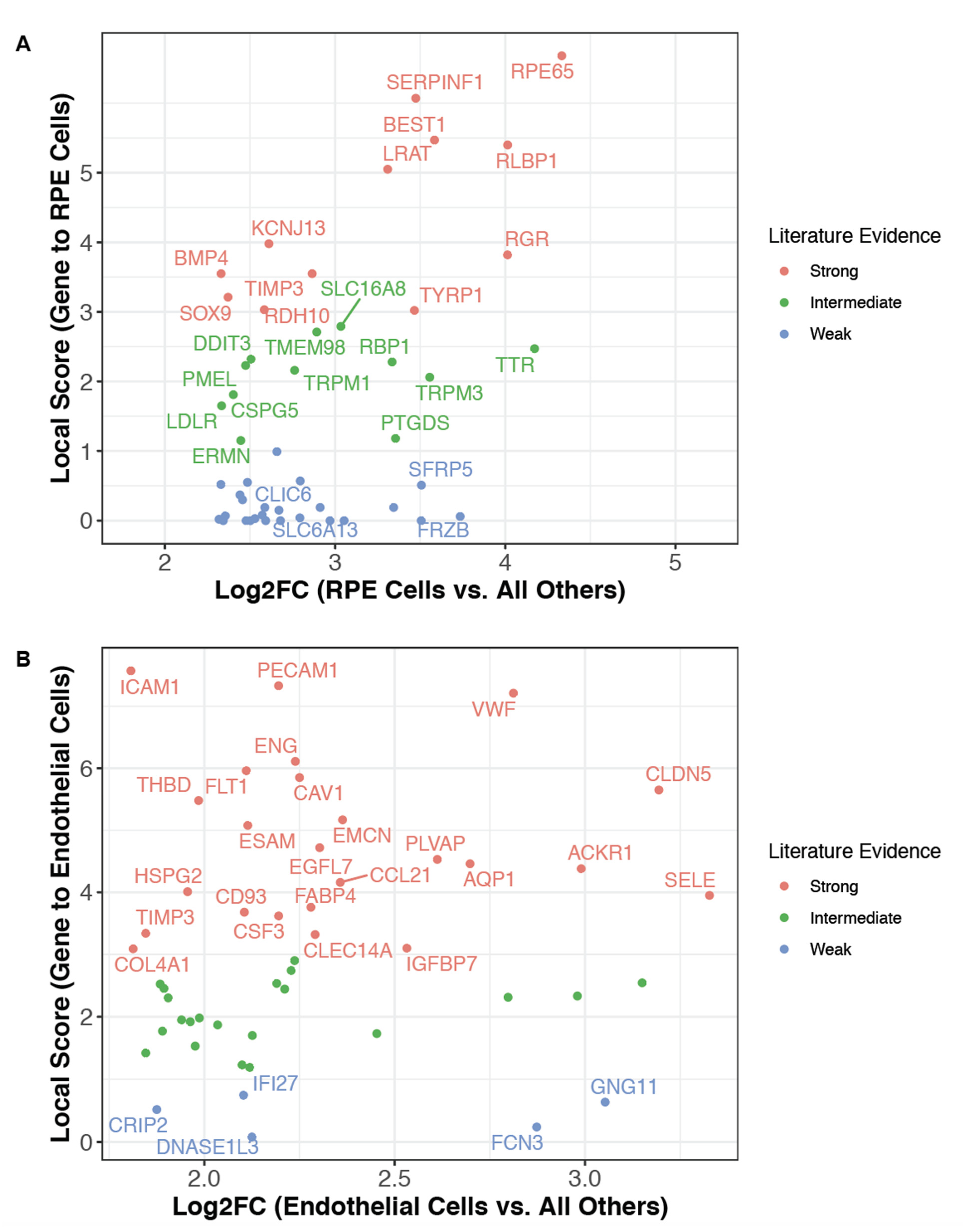
2.11. Identification of Poorly Characterized Cell Type Markers
2.12. Computation of Endothelial Gene Signatures
2.13. Statistical Analysis
3. Results
3.1. A Literature Derived Knowledge Graph Recapitulates Canonical Gene-Cell Type Associations
3.2. Literature Associations Facilitate Augmented Annotation of Single Cell RNA-seq Datasets
3.3. The Literature Knowledge Graph Highlights Uncharacterized Markers of Established Cell Types
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- de Kanter, J.K.; Lijnzaad, P.; Candelli, T.; Margaritis, T.; Holstege, F.C.P. CHETAH: A selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019, 47, e95. [Google Scholar] [CrossRef] [Green Version]
- Hou, R.; Denisenko, E.; Forrest, A.R.R. scMatch: A single-cell gene expression profile annotation tool using reference datasets. Bioinformatics 2019, 35, 4688–4695. [Google Scholar] [CrossRef] [Green Version]
- Pliner, H.A.; Shendure, J.; Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 2019, 16, 983–986. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Peng, G. SCSA: A Cell Type Annotation Tool for Single-Cell RNA-seq Data. Front. Genet. 2020, 11, 490. [Google Scholar] [CrossRef]
- Shao, X.; Liao, J.; Lu, X.; Xue, R.; Ai, N.; Fan, X. scCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 2020, 23, 100882. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.-J.; Wei, L.; Lu, S.; Yang, D.-C.; Gao, G. Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. Nat. Commun. 2020, 11, 3458. [Google Scholar] [CrossRef]
- Xu, C.; Lopez, R.; Mehlman, E.; Regier, J.; Jordan, M.I.; Yosef, N. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol. Syst. Biol. 2021, 17, e9620. [Google Scholar] [CrossRef]
- Pique-Regi, R.; Romero, R.; Tarca, A.L.; Sendler, E.D.; Xu, Y.; Garcia-Flores, V.; Leng, Y.; Luca, F.; Hassan, S.S.; Gomez-Lopez, N. Single cell transcriptional signatures of the human placenta in term and preterm parturition. eLife 2019, 8. [Google Scholar] [CrossRef]
- Grive, K.J.; Hu, Y.; Shu, E.; Grimson, A.; Elemento, O.; Grenier, J.K.; Cohen, P.E. Dynamic transcriptome profiles within spermatogonial and spermatocyte populations during postnatal testis maturation revealed by single-cell sequencing. PLoS Genet. 2019, 15, e1007810. [Google Scholar] [CrossRef] [Green Version]
- Sfakianos, J.P.; Daza, J.; Hu, Y.; Anastos, H.; Bryant, G.; Bareja, R.; Badani, K.K.; Galsky, M.D.; Elemento, O.; Faltas, B.M.; et al. Epithelial plasticity can generate multi-lineage phenotypes in human and murine bladder cancers. Nat. Commun. 2020, 11, 2540. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.-C.S.; Rancan, C.; et al. Intratumoral CD4+ T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Bard, J.; Rhee, S.Y.; Ashburner, M. An ontology for cell types. Genome Biol. 2005, 6, R21. [Google Scholar] [CrossRef] [Green Version]
- Meehan, T.F.; Masci, A.M.; Abdulla, A.; Cowell, L.G.; Blake, J.A.; Mungall, C.J.; Diehl, A.D. Logical development of the cell ontology. BMC Bioinform. 2011, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Diehl, A.D.; Augustine, A.D.; Blake, J.A.; Cowell, L.G.; Gold, E.S.; Gondré-Lewis, T.A.; Masci, A.M.; Meehan, T.F.; Morel, P.A.; Nijnik, A.; et al. Hematopoietic cell types: Prototype for a revised cell ontology. J. Biomed. Inform. 2011, 44, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Bodenreider, O. The Unified Medical Language System (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, D267–D270. [Google Scholar] [CrossRef] [Green Version]
- Braschi, B.; Denny, P.; Gray, K.; Jones, T.; Seal, R.; Tweedie, S.; Yates, B.; Bruford, E. Genenames.org: The HGNC and VGNC resources in 2019. Nucleic Acids Res. 2019, 47, D786–D792. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Puranik, A.; Anand, A.; Zemmour, D.; Yao, X.; Wu, X.; Chilaka, R.; Murakowski, D.K.; Standish, K.; Raghunathan, B.; et al. Knowledge synthesis of 100 million biomedical documents augments the deep expression profiling of coronavirus receptors. eLife 2020, 9. [Google Scholar] [CrossRef]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [Green Version]
- Seurat—Guided Clustering Tutorial. Available online: https://satijalab.org/seurat/articles/pbmc3k_tutorial.html (accessed on 13 March 2021).
- 3k PBMCs from a Healthy Donor (v1), Single Cell Gene Expression Dataset by Cell Ranger 1.1.0, 10× Genomics. Available online: https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc3k? (accessed on 13 March 2021).
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Franzén, O.; Gan, L.-M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Grün, D.; Muraro, M.J.; Boisset, J.-C.; Wiebrands, K.; Lyubimova, A.; Dharmadhikari, G.; van den Born, M.; van Es, J.; Jansen, E.; Clevers, H.; et al. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell 2016, 19, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.P.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.; Mohammadi, S.; Davila-Velderrain, J.; Goods, B.A.; Cadwell, T.D.; Xing, Y.; Stemmer-Rachamimov, A.; Shalek, A.K.; Love, J.C.; Kellis, M.; et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 2019, 10, 4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deprez, M.; Zaragosi, L.-E.; Truchi, M.; Becavin, C.; Ruiz García, S.; Arguel, M.-J.; Plaisant, M.; Magnone, V.; Lebrigand, K.; Abelanet, S.; et al. A Single-Cell Atlas of the Human Healthy Airways. Am. J. Respir. Crit. Care Med. 2020, 202, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Stewart, B.J.; Ferdinand, J.R.; Young, M.D.; Mitchell, T.J.; Loudon, K.W.; Riding, A.M.; Richoz, N.; Frazer, G.L.; Staniforth, J.U.L.; Vieira Braga, F.A.; et al. Spatiotemporal immune zonation of the human kidney. Science 2019, 365, 1461–1466. [Google Scholar] [CrossRef]
- Immune Cell Atlas: Blood Mononuclear Cells (2 Donors, 2 Sites). Available online: https://singlecell.broadinstitute.org/single_cell/study/SCP345/ica-blood-mononuclear-cells-2-donors-2-sites (accessed on 1 September 2019).
- Madissoon, E.; Wilbrey-Clark, A.; Miragaia, R.J.; Saeb-Parsy, K.; Mahbubani, K.T.; Georgakopoulos, N.; Harding, P.; Polanski, K.; Huang, N.; Nowicki-Osuch, K.; et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019, 21, 1. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell RNA-seq. PLoS ONE 2018, 13, e0206785. [Google Scholar]
- Zhang, Y.; Yan, Z.; Qin, Q.; Nisenblat, V.; Chang, H.-M.; Yu, Y.; Wang, T.; Lu, C.; Yang, M.; Yang, S.; et al. Transcriptome landscape of human folliculogenesis reveals oocyte and granulosa cell interactions. Mol. Cell 2018, 72, 1021–1034.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Song, W.; Wang, J.; Wang, T.; Xiong, X.; Qi, Z.; Fu, W.; Yang, X.; Chen, Y.-G. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 2020, 217, jem.20191130. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The adult human testis transcriptional cell atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Kurtenbach, S.; Sargi, Z.B.; Harbour, J.W.; Choi, R.; Kurtenbach, S.; Goss, G.M.; Matsunami, H.; Goldstein, B.J. Single-cell analysis of olfactory neurogenesis and differentiation in adult humans. Nat. Neurosci. 2020, 23, 323–326. [Google Scholar] [CrossRef]
- Yu, Z.; Liao, J.; Chen, Y.; Zou, C.; Zhang, H.; Cheng, J.; Liu, D.; Li, T.; Zhang, Q.; Li, J.; et al. Single-cell transcriptomic map of the human and mouse bladders. J. Am. Soc. Nephrol. 2019, 30, 2159–2176. [Google Scholar] [CrossRef]
- Vijay, J.; Gauthier, M.-F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.; et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2020, 2, 97–109. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar]
- James, K.R.; Gomes, T.; Elmentaite, R.; Kumar, N.; Gulliver, E.L.; King, H.W.; Stares, M.D.; Bareham, B.R.; Ferdinand, J.R.; Petrova, V.N.; et al. Distinct microbial and immune niches of the human colon. Nat. Immunol. 2020, 21, 343–353. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [Green Version]
- Popescu, D.-M.; Botting, R.A.; Stephenson, E.; Green, K.; Webb, S.; Jardine, L.; Calderbank, E.F.; Polanski, K.; Goh, I.; Efremova, M.; et al. Decoding human fetal liver haematopoiesis. Nature 2019, 574, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Robinson, M.L.; Croote, D.; Sahoo, M.K.; Sanz, A.M.; Ortiz-Lasso, E.; Albornoz, L.L.; Rosso, F.; Montoya, J.G.; Goo, L.; et al. Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Natl. Acad. Sci. USA 2018, 115, E12363–E12369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 2020, 181, 1475–1488.e12. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [Green Version]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yang, M.; Zhang, Y.; Xiao, S.; Lai, X.; Tan, A.; Du, S.; Li, S. Dissecting the Single-Cell Transcriptome Network Underlying Gastric Premalignant Lesions and Early Gastric Cancer. Cell Rep. 2019, 27, 1934–1947.e5. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Artibani, M.; Alsaadi, A.; Wietek, N.; Morotti, M.; Shi, T.; Zhong, Z.; Santana Gonzalez, L.; El-Sahhar, S.; KaramiNejadRanjbar, M.; et al. The repertoire of serous ovarian cancer non-genetic heterogeneity revealed by single-cell sequencing of normal Fallopian tube epithelial cells. Cancer Cell 2020, 37, 226–242.e7. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational identification of cell Doublets in Single-cell transcriptomic data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Chou, C.-H.; Jain, V.; Gibson, J.; Attarian, D.E.; Haraden, C.A.; Yohn, C.B.; Laberge, R.-M.; Gregory, S.; Kraus, V.B. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci. Rep. 2020, 10, 10868. [Google Scholar] [CrossRef]
- Fan, X.; Bialecka, M.; Moustakas, I.; Lam, E.; Torrens-Juaneda, V.; Borggreven, N.V.; Trouw, L.; Louwe, L.A.; Pilgram, G.S.K.; Mei, H.; et al. Single-cell reconstruction of follicular remodeling in the human adult ovary. Nat. Commun. 2019, 10, 3164. [Google Scholar] [CrossRef]
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehrborn, C.G.; et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep. 2018, 25, 3530–3542.e5. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.B.; Sedgewick, A.J.; Finnegan, A.I.; Harirchian, P.; Lee, J.; Kwon, S.; Fassett, M.S.; Golovato, J.; Gray, M.; Ghadially, R.; et al. Transcriptional programming of normal and inflamed human epidermis at single-cell resolution. Cell Rep. 2018, 25, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.-S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508.e20. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium; Pisco, A.O.; Schaum, N.; McGeever, A.; Karkanias, J.; Neff, N.F.; Darmanis, S.; Wyss-Coray, T.; Quake, S.R. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the mouse cell atlas by Microwell-seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajbhandari, P.; Arneson, D.; Hart, S.K.; Ahn, I.S.; Diamante, G.; Santos, L.C.; Zaghari, N.; Feng, A.-C.; Thomas, B.J.; Vergnes, L.; et al. Single cell analysis reveals immune cell-adipocyte crosstalk regulating the transcription of thermogenic adipocytes. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.-M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R Core Team R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Park, J.; Marquez, A.L.; Puranik, A.; Rajasekharan, A.; Aravamudan, M.; Garcia-Rivera, E. Recapitulation and Retrospective Prediction of Biomedical Associations Using Temporally-enabled Word Embeddings. bioRxiv 2019, 627513. [Google Scholar]
- Morimura, H.; Fishman, G.A.; Grover, S.A.; Fulton, A.B.; Berson, E.L.; Dryja, T.P. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3088–3093. [Google Scholar] [CrossRef] [Green Version]
- Dalvin, L.A.; Abou Chehade, J.E.; Chiang, J.; Fuchs, J.; Iezzi, R.; Marmorstein, A.D. Retinitis pigmentosa associated with a mutation in BEST1. Am. J. Ophthalmol. Case Rep. 2016, 2, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chen, X.; Xia, T.; Ghiassi-Nejad, M.; Tainsh, L.; Adelman, R.A.; Rizzolo, L.J. Partially differentiated neuroretinal cells promote maturation of the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.C.; ten Brink, J.B.; Swagemakers, S.M.A.; Verkerk, A.J.M.H.; Essing, A.H.W.; van der Spek, P.J.; Bergen, A.A.B. A new strategy to identify and annotate human RPE-specific gene expression. PLoS ONE 2010, 5, e9341. [Google Scholar] [CrossRef] [Green Version]
- Skeie, J.M.; Mahajan, V.B. Proteomic landscape of the human choroid-retinal pigment epithelial complex. JAMA Ophthalmol. 2014, 132, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Hongisto, H.; Jylhä, A.; Nättinen, J.; Rieck, J.; Ilmarinen, T.; Veréb, Z.; Aapola, U.; Beuerman, R.; Petrovski, G.; Uusitalo, H.; et al. Comparative proteomic analysis of human embryonic stem cell-derived and primary human retinal pigment epithelium. Sci. Rep. 2017, 7, 6016. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.W.Y.; Serpas, L.; Ni, M.; Volpi, S.; Hiraki, L.T.; Tam, L.-S.; Rashidfarrokhi, A.; Wong, P.C.H.; Tam, L.H.P.; Wang, Y.; et al. Plasma DNA Profile Associated with DNASE1L3 Gene Mutations: Clinical Observations, Relationships to Nuclease Substrate Preference, and In Vivo Correction. Am. J. Hum. Genet. 2020, 107, 882–894. [Google Scholar] [CrossRef]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Özçakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Jamil, M.A.; Singer, H.; Al-Rifai, R.; Nüsgen, N.; Rath, M.; Strauss, S.; Andreou, I.; Oldenburg, J.; El-Maarri, O. Molecular Analysis of Fetal and Adult Primary Human Liver Sinusoidal Endothelial Cells: A Comparison to Other Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 7776. [Google Scholar] [CrossRef]
- De Smedt, J.; van Os, E.A.; Talon, I.; Ghosh, S.; Toprakhisar, B.; Furtado Madeiro Da Costa, R.; Zaunz, S.; Vazquez, M.A.; Boon, R.; Baatsen, P.; et al. PU.1 drives specification of pluripotent stem cell-derived endothelial cells to LSEC-like cells. Cell Death Dis. 2021, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Chen, S.; Hoshi, M.; Plongthongkum, N.; Salamon, D.; Knoten, A.; Vijayan, A.; Venkatesh, R.; Kim, E.H.; Gao, D.; et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat. Commun. 2019, 10, 2832. [Google Scholar] [CrossRef]
- Wilber, A.; O’Connor, T.P.; Lu, M.L.; Karimi, A.; Schneider, M.C. Dnase1l3 deficiency in lupus-prone MRL and NZB/W F1 mice. Clin. Exp. Immunol. 2003, 134, 46–52. [Google Scholar] [CrossRef]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Ozçakar, Z.B.; Foster, J., 2nd; Diaz-Horta, O.; Kasapcopur, O.; Fan, Y.-S.; Yalçınkaya, F.; Tekin, M. DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum. 2013, 65, 2183–2189. [Google Scholar] [CrossRef]
- Svensson, V.; da Veiga Beltrame, E.; Pachter, L. A curated database reveals trends in single-cell transcriptomics. Database 2020, 2020. [Google Scholar] [CrossRef]
- Friedli, M.; Guipponi, M.; Bertrand, S.; Bertrand, D.; Neerman-Arbez, M.; Scott, H.S.; Antonarakis, S.E.; Reymond, A. Identification of a novel member of the CLIC family, CLIC6, mapping to 21q22.12. Gene 2003, 320, 31–40. [Google Scholar] [CrossRef]
- Nishizawa, T.; Nagao, T.; Iwatsubo, T.; Forte, J.G.; Urushidani, T. Molecular cloning and characterization of a novel chloride intracellular channel-related protein, parchorin, expressed in water-secreting cells. J. Biol. Chem. 2000, 275, 11164–11173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffon, N.; Jeanneteau, F.; Prieur, F.; Diaz, J.; Sokoloff, P. CLIC6, a member of the intracellular chloride channel family, interacts with dopamine D(2)-like receptors. Brain Res. Mol. Brain Res. 2003, 117, 47–57. [Google Scholar] [CrossRef]
- Chuang, J.-Z.; Chou, S.-Y.; Sung, C.-H. Chloride intracellular channel 4 is critical for the epithelial morphogenesis of RPE cells and retinal attachment. Mol. Biol. Cell 2010, 21, 3017–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, K.-S.; Otsu, W.; Li, Y.; Wang, H.-C.; Chen, S.; Tsang, S.H.; Chuang, J.-Z.; Sung, C.-H. CLIC4 regulates late endosomal trafficking and matrix degradation activity of MMP14 at focal adhesions in RPE cells. Sci. Rep. 2019, 9, 12247. [Google Scholar] [CrossRef] [Green Version]
- Ferofontov, A.; Strulovich, R.; Marom, M.; Giladi, M.; Haitin, Y. Inherent flexibility of CLIC6 revealed by crystallographic and solution studies. Sci. Rep. 2018, 8, 6882. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Study Title | Reference |
|---|---|---|
| Blood | Immune Cell Atlas: Blood Mononuclear Cells (2 donors, 2 sites) | Immune Cell Atlas |
| Blood | A single-cell atlas of the peripheral immune response in patients with severe COVID-19 | PMID 32514174 |
| Kidney | Spatiotemporal immune zonation of the human kidney | PMID 31604275 |
| Liver | Resolving the fibrotic niche of human liver cirrhosis at single-cell level | PMID 31597160 |
| Liver | A human liver cell atlas reveals heterogeneity and epithelial progenitors. | PMID 31292543 |
| Lung | A cellular census of human lungs identifies novel cell states in health and in asthma | PMID 31209336 |
| Lung | A single-cell atlas of the human healthy airways | PMID 32726565 |
| Pancreas | De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data | PMID 27345837 |
| Pancreas | Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes | PMID 27667667 |
| Pancreas | A Single-Cell Transcriptome Atlas of the Human Pancreas | PMID 27693023 |
| Placenta | Single-cell reconstruction of the early maternal-fetal interface in humans | PMID 30429548 |
| Retina | Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration | PMID 31653841 |
| Retina | Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. | PMID 31712411 |
| Tissue | Study Title | Reference |
|---|---|---|
| Breast | Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment | PMID 29961579 |
| Colon | Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis | PMID 31348891 |
| Heart | Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function | PMID 31915373 |
| Joint (Knee) | Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis | PMID 32616761 |
| Ovary | Single-cell reconstruction of follicular remodeling in the human adult ovary | PMID 31320652 |
| Prostate | A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra | PMID 30566875 |
| Skin | Transcriptional Programming of Normal and Inflamed Human Epidermis at Single-Cell Resolution | PMID 30355494 |
| Skin | Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq | PMID 27124452 |
| Small Intestine (Ileum) | Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy | PMID 31474370 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doddahonnaiah, D.; Lenehan, P.J.; Hughes, T.K.; Zemmour, D.; Garcia-Rivera, E.; Venkatakrishnan, A.J.; Chilaka, R.; Khare, A.; Kasaraneni, A.; Garg, A.; et al. A Literature-Derived Knowledge Graph Augments the Interpretation of Single Cell RNA-seq Datasets. Genes 2021, 12, 898. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060898
Doddahonnaiah D, Lenehan PJ, Hughes TK, Zemmour D, Garcia-Rivera E, Venkatakrishnan AJ, Chilaka R, Khare A, Kasaraneni A, Garg A, et al. A Literature-Derived Knowledge Graph Augments the Interpretation of Single Cell RNA-seq Datasets. Genes. 2021; 12(6):898. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060898
Chicago/Turabian StyleDoddahonnaiah, Deeksha, Patrick J. Lenehan, Travis K. Hughes, David Zemmour, Enrique Garcia-Rivera, A. J. Venkatakrishnan, Ramakrishna Chilaka, Apoorv Khare, Akhil Kasaraneni, Abhinav Garg, and et al. 2021. "A Literature-Derived Knowledge Graph Augments the Interpretation of Single Cell RNA-seq Datasets" Genes 12, no. 6: 898. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060898