
Ocular Involvement in Hereditary Transthyretin Amyloidosis: A Case Series Describing Novel Potential Biomarkers

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Data Acquisition
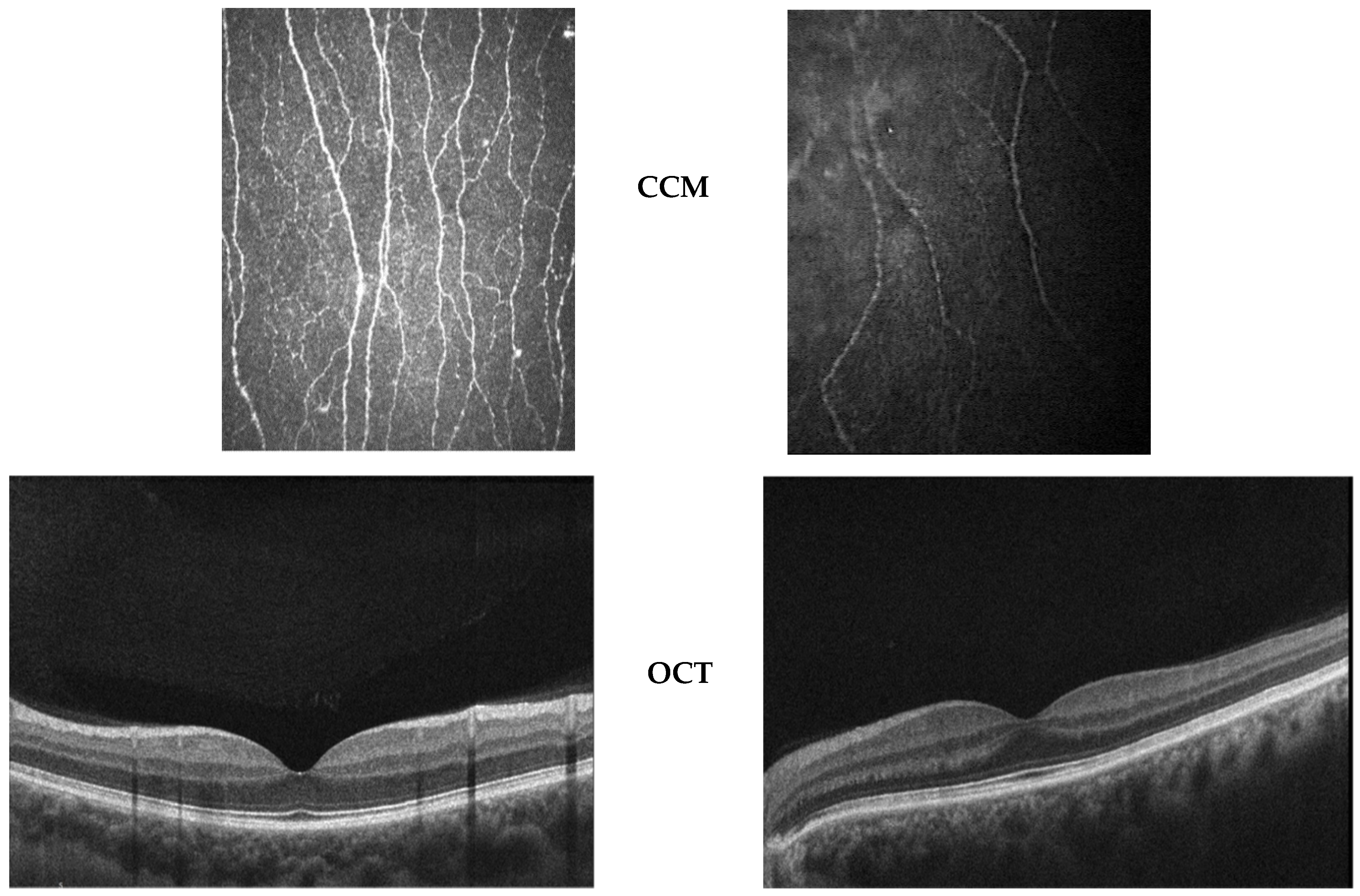
2.3. OCT Assessment
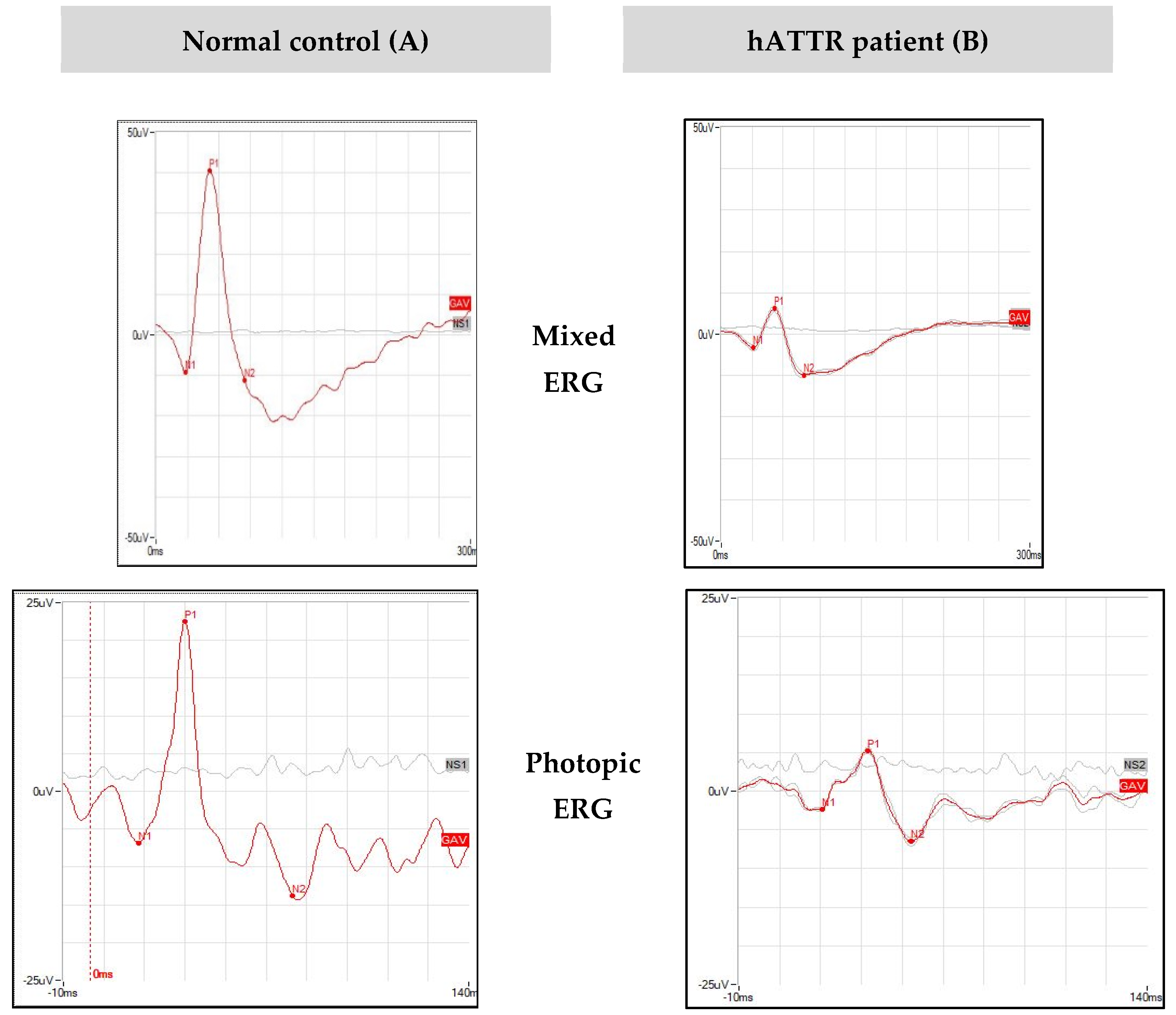
2.4. Electroretinogram Assessment
2.5. Statistical Analyses
3. Results
3.1. Demographic, Genetic and Systemic Findings
3.2. Ocular Findings
- (a)
- Best Corrected Visual Acuity
- (b)
- Anterior Segment
- (c)
- Posterior Segment
- (d)
- Functional Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Planté-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef]
- Saraiva, M.J.; Birken, S.; Costa, P.P.; Goodman, D.S. Family studies of the genetic abnormality in transthyretin (prealbumin) in Portuguese patients with familial amyloidotic polyneuropathy. Ann. N. Y. Acad. Sci. 1984, 435, 86–100. [Google Scholar] [CrossRef]
- Richardson, S.J. Cell and molecular biology of transthyretin and thyroid hormones. Int. Rev. Cytol. 2007, 258, 137–193. [Google Scholar]
- Martins, A.C.; Rosa, A.M.; Costa, E.; Tavares, C.; Quadrado, M.J.; Murta, J.N. Ocular Manifestations and Therapeutic Options in Patients with Familial Amyloid Polyneuropathy: A Systematic Review. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y. Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef]
- Adams, D.; Lozeron, P.; Lacroix, C. Amyloid neuropathies. Curr. Opin. Neurol. 2012, 25, 564–572. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Ferreira, A.; Lalu, T.; Zaros, C.; Lacroix, C.; Adams, D.; Said, G. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology 2007, 69, 693–698. [Google Scholar] [CrossRef]
- Luigetti, M.; Conte, A.; Del Grande, A.; Bisogni, G.; Madia, F.; Lo Monaco, M.; Laurenti, L.; Obici, L.; Merlini, G.; Sabatelli, M. TTR-related amyloid neuropathy: Clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol. Sci. 2013, 34, 1057–1063. [Google Scholar] [CrossRef]
- Ando, E.; Ando, Y.; Okamura, R.; Uchino, M.; Ando, M.; Negi, A. Ocular manifestations of familial amyloidotic polyneuropathy type I: Long-term follow up. Br. J. Ophthalmol. 1997, 81, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Haraoka, K.; Ando, Y.; Ando, E.; Sandgren, O.; Hirata, A.; Nakamura, M.; Terazaki, H.; Tajiri, T.; Tanoue, Y.; Sun, X.; et al. Amyloid deposition in ocular tissues of patients with familial amyloidotic polyneuropathy (FAP). Amyloid 2002, 9, 183–189. [Google Scholar] [CrossRef]
- Beirão, J.M.; Malheiro, J.; Lemos, C.; Matos, E.; Beirão, I.; Pinho-Costa, P.; Torres, P. Impact of liver transplantation on the natural history of oculopathy in Portuguese patients with transthyretin (V30M) amyloidosis. Amyloid 2015, 22, 31–35. [Google Scholar] [CrossRef]
- Hara, R.; Kawaji, T.; Ando, E.; Ohya, Y.; Ando, Y.; Tanihara, H. Impact of liver transplantation on transthyretin-related ocular amyloidosis in Japanese patients. Arch. Ophthalmol. 2010, 128, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Galli-Resta, L.; Falsini, B.; Rossi, G.; Piccardi, M.; Ziccardi, L.; Fadda, A.; Minnella, A.M.; Marangoni, D.; Placidi, G.; Campagna, F.; et al. Bilateral Symmetry of Visual Function Loss in Cone–Rod Dystrophies. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3759–3768. [Google Scholar] [CrossRef] [Green Version]
- Abed, E.; Piccardi, M.; Rizzo, D.; Chiaretti, A.; Ambrosio, L.; Petroni, S.; Parrilla, R.; Dickmann, A.; Riccardi, R.; Falsini, B. Functional Loss of the Inner Retina in Childhood Optic Gliomas Detected by Photopic Negative Response. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2469–2474. [Google Scholar] [CrossRef]
- Abed, E.; Placidi, G.; Campagna, F.; Federici, M.; Minnella, A.; Guerri, G.; Bertelli, M.; Piccardi, M.; Galli-Resta, L.; Falsini, B. Early impairment of the full-field photopic negative response in patients with Stargardt disease and pathogenic variants of the ABCA4 gene. Clin. Exp. Ophthalmol. 2018, 46, 519–530. [Google Scholar] [CrossRef]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Russo, M.; Obici, L.; Bartolomei, I.; Cappelli, F.; Luigetti, M.; Fenu, S.; Cavallaro, T.; Chiappini, M.G.; Gemelli, C.; Pradotto, L.G.; et al. ATTRv amyloidosis Italian Registry: Clinical and epidemiological data. Amyloid 2020, 27, 259–265. [Google Scholar] [CrossRef]
- Luigetti, M.; Guglielmino, V.; Antonini, G.; Casali, C.; Ceccanti, M.; Chiappini, M.; De Giglio, L.; Di Lazzaro, V.; Di Muzio, A.; Goglia, M.; et al. ATTRv in Lazio-Italy: A High-Prevalence Region in a Non-Endemic Country. Genes 2021, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Frishman, L.; Sustar, M.; Kremers, J.; McAnany, J.J.; Sarossy, M.; Tzekov, R.; Viswanathan, S. ISCEV extended protocol for the photopic negative response (PhNR) of the full-field electroretinogram. Doc. Ophthalmol. 2018, 136, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Falsini, B.; Anselmi, G.M.; Marangoni, D.; D’Esposito, F.; Fadda, A.; Di Renzo, A.; Campos, E.C.; Riva, C.E. Subfoveal choroidal blood flow and central retinal function in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1064–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Graham, J.; Dabbah, M.A.; Petropoulos, I.N.; Ponirakis, G.; Asghar, O.; Alam, U.; Marshall, A.; Fadavi, H.; Ferdousi, M.; et al. Small nerve fiber quantification in the diagnosis of diabetic sensorimotor polyneuropathy: Comparing corneal confocal microscopy with intraepidermal nerve fiber density. Diabetes Care 2015, 38, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, M.; Marshall, A.; Thompson, L.; Kenny, M.; Waldek, S.; Efron, N.; Malik, R.A. Corneal confocal microscopy: A novel noninvasive means to diagnose neuropathy in patients with Fabry disease. Muscle Nerve 2009, 40, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.; Cauquil, C.; Dupas, B.; Labbé, A.; Baudouin, C.; Barreau, E.; Théaudin, M.; Lacroix, C.; Guiochon-Mantel, A.; Benmalek, A.; et al. Potential Role of In Vivo Confocal Microscopy for Imaging Corneal Nerves in Transthyretin Familial Amyloid Polyneuropathy. JAMA Ophthalmol. 2016, 134, 983–989. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | Sex | Age | Age of Onset | Age at Diagnosis | Follow-Up (Months) | TTR Pathogenic Variant | FAP Stage | NIS (0–244) | Kumamoto sco–re (0–120) | Inheritance | Systemic invo–lvement | Treatment (Months Since Starting) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PN | CM | GI | ||||||||||||
| #1 | F | 87 | 79 | 81 | 84 | V30M | III | 140.25 | 30 | U | Yes | Yes | Yes | Diflunisal (12) |
| #2 | M | 64 | 58 | 59 | 76 | V30M | II | 73.75 | 30 | U | Yes | Yes | Yes | Patisiran (48) |
| #3 | M | 79 | 77 | 79 | 11 | V122I | I | 21 | 36 | U | Yes | Yes | No | Tafamidis (10) |
| #4 | M | 76 | 65 | 70 | 19 | A109S | II | 58.7 | 28 | U | Yes | No | Yes | Patisiran (16) |
| #5 | F | 52 | - | - | 10 | V30M | 0 | 0 | 0 | Maternal | No | No | No | None |
| #6 | F | 58 | 58 | 58 | 19 | F64L | I | 4 | 3 | Paternal | Yes | No | No | Tafamidis (8) |
| #7 | M | 51 | 51 | 51 | 19 | F64L | I | 12.50 | 0 | Paternal | Yes | Yes | No | Tafamidis (8) |
| #8 | M | 56 | 47 | 48 | 20 | T59K | II | 123 | 35 | Maternal | Yes | Yes | Yes | Tafamidis (77) + Inotersen (7) |
| #9 | M | 78 | 71 | 71 | 8 | F64L | II | 88 | 28 | U | Yes | No | No | Tafamidis (72) + Patisiran (8) |
| Patient | Eye | BCVA (ETDRS Letters) | IOP (mmHg) | Lens Condition | Vitreous Opacities on Ophthalmos Copy | OCT Finding | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vitreous Opacity (0–5) | V-R Interface Alterations (0–5) | OR Alterations (0–5) | CMT and SFCT (µ) | ONL (µ) | ||||||
| #1 | RE | 77 | 18 | Pseudofakia, PCO | No | 3 | 4 | 1 | 245 and 320 | 69.6 |
| LE | 56 | 15 | Pseudofakia, PCO | No | 3 | 3 | 4 | 290 and 330 | 52.8 | |
| #2 | RE | 90 | 14 | PPC | No | 1 | 1 | 0 | 270 and 330 | 70.4 |
| LE | 87 | 12 | PPC | No | 1 | 1 | 1 | 280 and 350 | 61.8 | |
| #3 | RE | 85 | 11 | Normal | No | 0 | 0 | 0 | 244 and 180 | 82.2 |
| LE | 85 | 13 | Normal | No | 0 | 0 | 0 | 231 and 160 | 84.8 | |
| #4 | RE | 88 | 10 | CNC | No | 3 | 1 | 0 | 279 and 300 | 79.4 |
| LE | 87 | 10 | CNC | No | 2 | 1 | 0 | 287 and 270 | 74.2 | |
| #5 | RE | 90 | 13 | Normal | No | 2 | 1 | 0 | 211 and 300 | 70.6 |
| LE | 90 | 16 | Normal | No | 1 | 0 | 0 | 207 and 310 | 71.8 | |
| #6 | RE | 85 | 15 | Normal | No | 2 | 2 | 0 | 281 and 330 | 73.2 |
| LE | 85 | 16 | Normal | No | 2 | 2 | 0 | 276 and 244 | 71.2 | |
| #7 | RE | 88 | 12 | Normal | No | 0 | 1 | 0 | 262 and 250 | 75.4 |
| LE | 87 | 12 | Normal | No | 0 | 1 | 0 | 266 and 250 | 77.8 | |
| #8 | RE | 88 | 10 | Pseudofakia | No | 2 | 2 | 0 | 237 and 248 | 74 |
| LE | 88 | 11 | Normal | No | 1 | 1 | 0 | 237 and 240 | 82.8 | |
| #9 | RE | 86 | 16 | Pseudofakia | No | 0 | 0 | 1 | 269 and 238 | 61.2 |
| LE | 85 | 16 | Pseudofakia | No | 1 | 0 | 0 | 260 and 150 | 73.2 | |
| Patients | Eye | CCM | Other Remarks | ||
|---|---|---|---|---|---|
| Absent/Rarefied Subepithelial NP (Extension and Density) | Nerve Segmentation and/or Fragmentation | Thinning of Stromal Nerves | Deposits between Bowman and Stroma | ||
| #1 | RE | yes | no | no | yes |
| LE | yes | no | no | yes | |
| #2 | RE | yes | no | no | yes |
| LE | yes | no | no | yes | |
| #3 | RE | yes | yes | no | no |
| LE | yes | yes | no | no | |
| #4 | RE | yes | yes | no | no |
| LE | no | yes | no | no | |
| #5 | RE | no | yes | no | no |
| LE | no | yes | no | no | |
| #6 | RE | no | no | yes | no |
| LE | no | no | yes | no | |
| #7 | RE | no | yes | yes | yes |
| LE | no | yes | yes | yes | |
| #8 | RE | yes | yes | no | no |
| LE | yes | yes | no | no | |
| #9 | RE | yes | yes | no | no |
| LE | yes | yes | no | no | |
| Patient | Eye | Mixed ERG | Photopic ERG | PhNR | |||
|---|---|---|---|---|---|---|---|
| B Wave Amplitude (µV) | B Wave Peak Time (ms) | B Wave Amplitude (µV) | B Wave Peak Time (ms) | Amplitude (µV) | Peak Time (ms) | ||
| #1 | RE | 53.22 | 53.91 | 29.84 | 37.2 * | 7.19 * | 55.08 * |
| LE | 47.47 | 52.73 | 33.63 | 36.62 * | 4.35 * | 51.86 * | |
| #2 | RE | 25.73 | 50.39 | 18.60 | 33.40 * | 7.67 * | 51.86 * |
| LE | 24.76 * | 50.39 | 19.46 | 33.40 * | 5.83 * | 50.39 * | |
| #3 | RE | 34.97 | 60.35 * | 15.97 | 39.84 * | 8.43 * | 58.59* |
| LE | 42.33 | 59.18 | 17.70 | 41.31 * | 7.04 * | 50.98 * | |
| #4 | RE | 14.06 * | 50.98 | 10.67 | 37.21 * | 4.62 * | 54.79 * |
| LE | 9.39 * | 51.56 | 7.63 * | 37.21 * | 7.03 * | 53.32 * | |
| #5 | RE | 23.38 | 46.29 | 22.12 | 32.81 | 7.66 * | 47.75 |
| LE | 23.44 * | 46.88 | 23.00 | 32.52 | 9.44 | 47.17 | |
| #6 | RE | 34.45 | 49.80 | 29.91 | 33.40 * | 8.12 * | 50.10 * |
| LE | 35.78 | 49.22 | 28.06 | 33.69 * | 9.70 | 50.39* | |
| #7 | RE | 25.46 | 50.98 | 22.06 | 34.57 * | 5.94 * | 50.39 * |
| LE | 30.53 | 52.73 | 21.83 | 34.57 * | 5.55 * | 50.98* | |
| #8 | RE | 30.22 | 48.63 | 28.75 | 34.28 * | 7.28 * | 49.51 * |
| LE | 19.03 * | 49.22 | 17.61 | 34.57 * | 6.40 * | 50.39 * | |
| #9 | RE | 22.34 | 50.98 | 21.83 | 34.86 * | 9.99 | 73.24* |
| LE | 24.75 * | 52.15 | 18.70 | 35.45 * | 5.68 * | 74.12 * | |
| Mixed ERG | Photopic ERG | PhNR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amplitude (μV) | Peak Time (ms) | Amplitude (μV) | Peak Time (ms) | Amplitude (μV) | Peak Time (ms) | |||||||
| RE | LE | RE | LE | RE | LE | RE | LE | RE | LE | RE | LE | |
| hATTR patients | ||||||||||||
| Mean | 29.31 | 28.60 | 51.36 | 51.56 | 22.19 | 20.84 | 35.28 | 35.48 | 7.43 | 6.78 | 54.59 | 53.28 |
| Std. deviation | 10.40 | 11.13 | 3.71 | 3.25 | 6.19 | 6.85 | 2.18 | 2.49 | 1.42 | 1.68 | 7.3 | 7.5 |
| N of patients | 9 | 9 | 9 | |||||||||
| Normal controls | ||||||||||||
| Mean | 50.55 | 49.28 | 52 | 52 | 25.70 | 25.76 | 29 | 29 | 19.17 | 19.17 | 40 | 40 |
| Std. deviation | 16.36 | 10.86 | 4 | 4 | 9.23 | 8.35 | 2 | 2 | 5 | 5 | 4 | 4 |
| N of patients | 40 | 40 | 40 | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minnella, A.M.; Rissotto, R.; Maceroni, M.; Romano, A.; Fasciani, R.; Luigetti, M.; Sabatelli, M.; Rizzo, S.; Falsini, B. Ocular Involvement in Hereditary Transthyretin Amyloidosis: A Case Series Describing Novel Potential Biomarkers. Genes 2021, 12, 927. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060927
Minnella AM, Rissotto R, Maceroni M, Romano A, Fasciani R, Luigetti M, Sabatelli M, Rizzo S, Falsini B. Ocular Involvement in Hereditary Transthyretin Amyloidosis: A Case Series Describing Novel Potential Biomarkers. Genes. 2021; 12(6):927. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060927
Chicago/Turabian StyleMinnella, Angelo Maria, Roberta Rissotto, Martina Maceroni, Angela Romano, Romina Fasciani, Marco Luigetti, Mario Sabatelli, Stanislao Rizzo, and Benedetto Falsini. 2021. "Ocular Involvement in Hereditary Transthyretin Amyloidosis: A Case Series Describing Novel Potential Biomarkers" Genes 12, no. 6: 927. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060927