
Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome


1
Department of Neurology, Washington University in St Louis, St Louis, MO 63130, USA
2
Department of Developmental Biology, Washington University in St Louis, St Louis, MO 63130, USA
3
Paley Orthopaedic and Spine Institute, West Palm Beach, FL 33407, USA
*
Author to whom correspondence should be addressed.
Genes 2021, 12(6), 943; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060943
Submission received: 17 May 2021
/
Revised: 10 June 2021
/
Accepted: 16 June 2021
/
Published: 20 June 2021
(This article belongs to the Special Issue Genetic Conditions Affecting the Skeleton: Congenital, Idiopathic Scoliosis and Arthrogryposis)
Abstract
:Distal arthrogryposis and lethal congenital contracture syndromes describe a broad group of disorders that share congenital limb contractures in common. While skeletal muscle sarcomeric genes comprise many of the first genes identified for Distal Arthrogyposis, other mechanisms of disease have been demonstrated, including key effects on peripheral nerve function. While Distal Arthrogryposis and Lethal Congenital Contracture Syndromes display superficial similarities in phenotype, the underlying mechanisms for these conditions are diverse but overlapping. In this review, we discuss the important insights gained into these human genetic diseases resulting from in vitro molecular studies and in vivo models in fruit fly, zebrafish, and mice.
1. Introduction
Arthrogryposis (arth = joint; grp = curved; osis = pathological state) describes a broad range of phenotypes consisting of multiple congenital joint contractures presenting at birth [1]. About 1 in 3000 live births presents with some form of arthrogryposis, many of which are nonprogressive and improve with physiotherapy. The core root of arthrogryposis is fetal akinesia, or lack of fetal movement, that results in contractures forming in the joints [1,2,3]. Movement is required for normal joint development; it influences the structure of the joints, as well as promoting cellular signaling that guides normal tissue development. Mechanical forces also influence bone morphology, affecting organization of chondrocytes, bone elongation, and differential growth, all affecting the shape of bones as they develop. Fetal akinesia impairs joint formation, which may lead to joint fusions. Furthermore, tension is required for normal tendon development, forming a connection between bone and muscle [4]. Arrested movement during development has significant impact on the formation of the skeleton, joints, muscle, and connective tissues.
The full range of joint movement in utero can be perturbed both intrinsically and extrinsically. Intrinsically, mutations affecting the muscle, bone, connective tissue, and neural system can affect the range of movement of joints. Currently, there are over 400 genes associated with arthrogryposis broadly, encapsulating a wide diversity of genes affecting different pathways, including genes associated with axon structure, circulatory development, or synaptic transmission [5]. Extrinsically, maternal disease or exposures, uterine space limitation, and decreased blood supply are also root causes for contraction defects [1,2]. Because joint motion is affected by many different systems, a wide range of issues during development can arrest joint motion.
A subset of arthrogryposis is described as Distal Arthrogryposis (DA), a group of genetically induced contractures that predominantly affect the joints of the distal limbs, including the hands, wrists, ankles, and feet. Clinically, the lower extremity manifestations commonly include clubfoot and vertical talus. There are currently 10 classifications of DA, including Sheldon-Hall syndrome (DA2B) and Freeman-Sheldon syndrome (DA2A) [6,7,8,9]. Freeman-Sheldon syndrome is considered the most severe form of DA, and also presents with facial contractures [9].
Currently, DA patients are offered supportive care to improve quality of life, including occupational therapy, physical therapy, and surgery [10]. While these treatments improve outcome for patients, they often fall short of complete restoration of range of motion in the joints and functionality. This strategy also fails to address underlying causes for DA, such as muscle weakness and impaired neurotransmission. Therefore, further investigation is necessary to understand the impact of disease variants which will allow us to determine the most effective treatment options for patients.
Lethal Congenital Contracture Syndromes (LCCS) are included in this review, as some of the same genes and disease mechanisms apply to this serious condition, which is typically fetal or neonatal lethal. LCCS presents with severe generalized contractures, along with many other typical features including incomplete lung development, and polyhydramnios [11]. In contrast to DA, which is most often inherited as an autosomal dominant condition, LCCS has only been described in the autosomal recessive state. Eleven subtypes have been described to date [10].
Various disease models have been developed to examine the mechanisms behind DA and to test therapeutic interventions (Table 1). Molecular and single-cell studies are useful for precisely examining the effects of DA-causing variants on the affected proteins and tissues. Access to human tissues, particularly from muscle biopsies, has facilitated molecular analysis for research, yet is clinically useful only in select cases [12,13]. In addition, protein modeling can help predict the impact of various amino acid substitutions on molecular interactions [14,15]. On the other hand, animal models are necessary to analyze the effect of single gene variants on organisms on scales larger than single cells. The effect of zygosity and gene dosage may also be better studied in animal models to assess interactions between normal and abnormal gene products. Animal models are also useful for studying experimental interventions that may improve patient quality of life and outcome, acting as stand-ins for potential human patients.
Models of human disease are rapidly becoming more sophisticated, with the ability to knock-in single nucleotide variants and create conditional (tissue specific or time-dependent) knockouts [16,17]. Loss-of-function alleles, which are often easier to generate, provide critical information about gene function, but may not fully explain autosomal dominant phenotypes in which gain-of-function or dominant negative effects can cause markedly different phenotypes. Conditional knockouts, while very helpful in defining gene function, rarely replicate the human phenotype in its entirety, but may be required when early lethality limits further study. These methods allow researchers to design models that more accurately represent these human conditions, and replicate pathogenic effects broadly or in specific tissues.
This review will examine genetic models of DA and LCCS, and the impact they have had in understanding the underlying pathophysiology. While we will describe both in vitro and in vivo approaches, we will focus primarily on vertebrate models, as these have the potential to provide insight into the multifaceted effects of disease variants on the multiple tissue types that contribute to these complex human phenotypes. We will also examine the current trajectory of DA research, and how these research strategies can help those afflicted by DA.
2. Muscle-Related Distal Arthrogryposis
2.1. MYH3
Missense mutations in MYH3, the earliest expressed embryonic myosin heavy chain gene that is predominantly expressed in myotubes destined to become fast-twitch myofibers [9], are strongly associated with DA clinically, and contribute to multiple subtypes including DA1, DA2A, and DA2B with varying degrees of severity [7,8,9,59,60]. MYH3-associated DA is almost always caused by single missense variants, as frameshift knockout or premature stop mutations are frequently observed in healthy population controls [9]. However, nonsense MYH3 variants may contribute to autosomal recessive spondylocarpotarsal syndrome in the compound heterozygous state when presenting along with a missense allele [21], and have also been described with autosomal dominant spondylocarpotarsal syndrome [61]. DA-associated pathogenic variants cluster in the motor domain, but have also been found in the tail region of the protein [9]. Many of these missense variants are de novo, but some segregate in families with complete, or nearly complete penetrance [9]. MYH3 appears to be one of the most common genes associated with DA, therefore various in vitro and in vivo studies, including protein modeling, cell models, and vertebrate studies, have been performed to elucidate the effects of MYH3 variants on muscle function and the subsequent effects on the joints and skeleton.
2.1.1. Biochemical and Cell Models for MYH3-Associated Distal Arthrogryposis
Single molecule and single cell studies are useful to examine the precise impact of a variant on protein function. The effects of amino acid substitutions are difficult to predict without mechanistic examination. Single-molecule studies facilitate understanding the effect of a missense variant on protein function and can later be translated into an understanding of how small mechanical differences affect tissues and whole body systems. Missense variants can be studied in human skeletal muscle biopsies. However, these are not routinely performed for DA diagnosis, which makes these studies challenging. To study this mechanistic link between DA phenotype and gene variant, Racca et al. performed contractility studies on isolated muscle cells and myofibrils derived from biopsied muscle tissue from DA2A patients [12]. They found that a DA2A-associated MYH3 variant inhibited cross-bridge detachment, thereby slowing muscle relaxation and lowering force production. A later study replicated these results while examining multiple MYH3 variants associated with DA2 [54]. In addition to slower actin-myosin detachment, ATP binding and ATPase activity were lower in variant MYH3 molecules.
Development of single cell models, in which MYH3 variants are exogenously expressed or overexpressed from a plasmid or virus, have been limited by the difficulties of expressing large genes, like MYH3, in vitro. Other key challenges of in vitro modeling include the paucity of skeletal muscle cell lines other than C2C12 cells, and the propensity of muscle cells to form a syncytium. In addition, difficulties in differentiating human induced pluripotent stem cells (iPSCs) into skeletal muscle have also limited their use for arthrogryposis disease modeling. Likewise, because many features of DA are due to complex relationships between different cell types, co-cultures of muscle cells with tenocytes and bone may be required to recapitulate the human condition. Thus, many investigators have preferred to study DA genes in whole organisms.
2.1.2. Invertebrate Models for MYH3-Associated Distal Arthrogryposis
Drosophila melanogaster (fruit flies) are useful tools for studying muscle function and myofibril assembly, particularly as introduction of single variants are traditionally simpler in this system compared to other models. MYH3 and the Drosophila myosin heavy chain gene, Mhc, are highly conserved. Drosophila have the advantage of having only a single myosin heavy chain, which eliminates the possible obscuration of effects due to compensation by other myosin heavy chain analogs. Therefore, the effect of variants on protein function can be examined in a setting without other myosin heavy chain isoforms.
Drosophila transgenic models have been generated by overexpressing Mhc constructs containing DA variants [14,25]. Guo et al. predicted that a DA1 variant would perturb a hydrophobic interaction, while a DA2B mutation would introduce a hydrogen bond that was not present in the wild type. The effect of these predicted interactions was tested mechanistically in Drosophila models. Muscle fibers containing the DA alleles were extracted and found to have lower actin affinity, reduced power output, and increased stiffness, which may explain the motor deficits [14].
Morphological studies of Drosophila skeletal muscle expressing three DA2A Mhc transgenes (R672H, R672C, and T178I) showed branching and splitting defects, which were most severe in the R672C variant, which caused Z-discs to be split and malformed. In addition, Z-disc distance was shorter in the transgenic flies indicating an overall shortening of the sarcomeres, perhaps due to an enhanced contractile state of the myofibers. Presumably, the shortened sarcomeres observed in Drosophila contribute to the formation of contractures in human patients. Indeed, ATPase activity was reduced in these transgenic flies, leading to functional defects in muscle activity [25].
In studying the intact adult Drosophila, Guo et al. showed that the MhcF437I mutants had a much longer lifespan than MhcA234T mutants, consistent with the less severe phenotype of DA1 patients compared to DA2B patients [14]. In addition, the researchers found that both mutants displayed aberrant myofibril assembly, as well as misaligned sarcomere structure including distorted M and Z lines [14]. Again, this phenotype was more severe in A234T mutants than in the F437I mutants. MhcF437I mutants displayed essentially normal myofibers and sarcomeres, while MhcA234T mutants had small myofibers with disrupted morphology, as well as abnormal sarcomeres [14]. Das et al. also found that decreased climbing capability of adult flies also correlated with the phenotypic severity in humans [25].
Like Drosophila, Caenorhabditis elegans (C. elegans) has also been used to study myosin heavy chain genes [62]. There are many advantages of C. elegans and Drosophila for disease modeling, including large numbers of progeny, knowledge of ontogeny of individual cells, and ease of functional studies for drug screening. However, as described in Gil-Galvez et al., the evolutionary distance, and differences in number of myosin heavy chain genes between invertebrates and humans makes it difficult to determine whether the gene being studied has the same function, particularly in terms of spatial and temporal expression, as its human counterpart. Furthermore, Gil-Galvarez et al. caution against overexpression studies in C. elegans broadly, citing interference with muscle cell function overall [62]. The major drawback of invertebrate models is, quite obviously, the lack of skeletal structures which limits their use in understanding the complex relationship between muscle, nerve, and bone.
2.1.3. Vertebrate Models for MYH3-Associated Distal Arthrogryposis
Germline loss of Myh3 in mice results in altered muscle fiber size, fiber number, fiber type, and misregulation of genes, and adult Myh3 null mice develop scoliosis [32]. However, the molecular defect may make these mice a better model for recessive MYH3 spondylocarpotarsal synostosis syndromes than for autosomal dominant DA [21,61]. Notably, many patients with spondylocarpotarsal synostosis syndrome also have congenital contractures, which highlights the phenotypic overlap between DA and spondylocarpotarsal synostosis syndrome. Interestingly, MYH3 was also shown to be expressed in bone, which the authors state may explain the effects of MYH3 variants on both skeletal muscle and bone, particularly for patients with spondylocarpotarsal synostosis syndrome and bony fusions [61].
To more accurately model DA2A, Whittle et al. recently introduced one of the most common Freeman-Sheldon syndrome MYH3 variants, R672H, into an analogous gene in zebrafish (Danio rerio) (smyhc1R673H) [16]. Zebrafish breed profusely and are cost-effective compared to mice. They also mature quickly, develop in vitro, and are transparent in the first few days of life, which facilitates imaging. Zebrafish are also vertebrates, making them more closely related to humans than Drosophila or C. elegans. Because two zebrafish lines were created, including smyhc1 null and smyhc1R673H lines, gene dosage effects were studied by examining the variant in the context of different zygosities. Indeed, smyhc1R673H homozygotes displayed severe, early lethal phenotype compared to smyhc1R673H heterozygotes, indicating that the smyhc1R673H mutation acts as a hypermorph [16]. This result suggests human fetal lethality if a DA missense variant occurs in the homozygous state, which has not yet been described.
Zebrafish larvae harboring the smyhc1R673H variant demonstrated severe notochord kinks [1], and adults had vertebral fusions that were similar to those seen in patients autosomal dominant spondylocarpotarsal synostosis due to MYH3 variants. On histological examination, skeletal muscle showed severely shortened and misshapen muscle fibers. Similar to studies in Drosophila, the somite length was reduced in smyhc1R673H mutants, consistent with shortening of the sarcomere.
A major advantage of zebrafish is the ease with which drugs can be administered for therapeutic investigations and drug screening. Based on knowledge that myosin ATPase inhibitors are now being evaluated to treat human cardiomyopathy due to similar variants in cardiac myosin genes [63], Whittle et al. preemptively treated embryos with para-aminoblebbistatin to prevent contractures from forming in larvae. Para-aminoblebbistatin inhibits myosin heavy chain ATPase activity, which chemically relaxed the skeletal muscle and prevented the curved phenotype of the treated smyhc1 mutant fish (Figure 1) [16]. Previous molecular and single fiber studies predicted this mechanistic effect. Based on this experimental work, myosin ATPase inhibitors may be a viable avenue for MYH3-associated DA treatment, but will most likely require development of skeletal-muscle-specific inhibitors and treatment at an appropriately early developmental window.
2.2. MYBPC1 and MYBPC2
Strong evidence now exists linking variants in the slow skeletal muscle myosin binding protein C1 (MYBPC1) to dominantly inherited DA1 [30], DA2 [58], arthrogryposis multiplex congenita [28], myopathy with tremor [49], and, in the recessive state, to lethal congenital contracture syndrome LCCS4 [11].
Morpholino knockdown of mybpc1 in zebrafish resulted in embryos with severe body curvature, as well as impaired motor excitation with defective myofibril organization and reduced sarcomere numbers [31]. Furthermore, overexpression of human MYBPC1 DA1-associated variants in zebrafish resulted in hypermorphic effects with body curvature, decreased motor activity, and impaired survival. No effect was seen with overexpression of wild-type transcripts, suggesting that overexpression studies in zebrafish could be an efficient model for future functional testing of the human variants of uncertain clinical significance.
In contrast to MYBPC1, which is strongly implicated in human disease, the role of fast skeletal muscle myosin binding protein C2 (MYBPC2) in DA is less clear, as there is only a single report of MYBPC2 variants in DA patients in whom other known arthrogryposis gene variants were also observed, suggesting a possible role as a modifier [18]. Knockdown of MYBPC2 with morpholino oligonucleotides produced a myopathic phenotype [37], but single variants have not yet been studied.
2.3. TPM2
TPM2 variants cause a spectrum of phenotypes, including DA1, DA2, as well as nemaline myopathy and cap myopathy (reviewed in (Tajsharghi et al., 2012)) [64]. All are autosomal dominant with the exception of a pathogenic null variant identified in a consanguineous family with Escobar variant of multiple pterygium syndrome that was observed in the recessive state [65]. Biochemical studies were undertaken to study TPM2 gain-of-function phenotypes, including in vitro motility assays, which showed variable effects on calcium sensitivity and tropomyosin flexibility [20,39]. In addition, TPM2 was recently shown to have noncanonical roles other than its sarcomeric function, where it binds thin filament actin to regulate muscle contraction. In this work, TPM2 directly regulated muscle morphogenesis by directing myotubes toward tendon attachment sites [56]. Muscle morphology was disrupted in both flies and zebrafish expressing DA1-associated TPM2 variants, likely by causing myofiber hypercontraction (Figure 2).
2.4. TNNI2
While the function of the fast skeletal muscle Troponin I (TNNI2) has been described in flightless Drosophila models [53], only a single DA disease-associated missense variant has been modeled in mice, which accurately recapitulated the human disease [17]. However, the small body size of mice carrying the TNNI2 DA variant could not be explained by the direct effect of the variant on skeletal muscle morphology or function. Rather, TNNI2 was shown to be expressed in osteoblasts and chondrocytes of long bone growth plates, through which its effects on growth was predicted to occur. Therefore, like the studies described above for MYH3, this model provides evidence that some DA phenotypes may be directly attributable to expression in non-muscle tissue, such as bone.
2.5. TNNT3
DA-associated variants in fast skeletal muscle Troponin T (TNNT3) are also dominantly inherited missense variants, and therefore, like those in many genes described previously, cause disease through a gain-of-function manner and therefore cannot be adequately modeled using a simple knockout approach. Therefore, while knockout approaches have shown a critical function of TNNT3 during vertebral development [34], no models with disease-specific missense variants have been generated, and future studies are needed.
2.6. MYLPF
Exome sequencing recently identified MYLPF, a phosphorylatable fast skeletal muscle regulatory light chain, as a cause of DA [23]. Some affected individuals were homozygous for rare variants in the gene, while other individuals have autosomal dominant disease, a finding similar to what was described for MYH3-related disorders. However, unlike MYH3-related disorders, the phenotypes for MYLPF autosomal dominant and recessive conditions are apparently indistinguishable. Protein modeling of MYLPF alleles suggested that the autosomal dominant pathogenic variants cause disease through their direct interaction with myosin, while the recessive alleles only indirectly affect the interaction with myosin [23].
Previously described mylpf knockout mice did not develop either fast or slow type skeletal muscle mass, which resulted in early death before or after delivery, presumably due to respiratory failure [66]. Similarly, an individual with recessive MYLPF-associated DA1 was found to have absent skeletal muscle in an amputated foot, which could best be described as amyoplasia. The recessive MYLPF pathogenic variant in this individual was hypothesized to be hypomorphic. Therefore, to further model hypomorphic alleles of MYLPF, a zebrafish mylpfa mutant was characterized. Because zebrafish have 2 MYLPF genes, mylpfa and mylpfb, the more prominently expressed gene (mlfpfa) was chosen to model hypomorphic MYLPF alleles [23]. Of note, zebrafish had an evolutionary genome duplication event that has resulted in many human genes being represented twice in the zebrafish genome. Some of these genes have subsequently evolved to occupy additional temporal or spatial expression patterns and/or adopted new developmental roles. While this duplication event may complicate identification of the relevant human gene, gene duplications can also be an advantage, allowing genes to be studied whose knockout is early embryonic lethal in other species.
Mylpfa zebrafish null mutants were found to have paralyzed pectoral fins, an impaired escape response, and consistently lower trunk muscle force compared to wildtype [23]. In in vitro studies, myosin extracted from mylpfa mutant larvae propelled significantly more slowly than their wild type protein. Skeletal muscle fibers were also found to degenerate in mutant larvae, collapsing and losing structure, developing membrane abnormalities, indicating that mylpf is necessary to maintain cellular integrity in muscle cells [23]. Thus, MYLPF may be unique among the DA genes in also causing amyoplasia, which may have important implications for personalized therapeutic strategies.
3. Neural-Related Distal Arthrogryposis
3.1. PIEZO2
DA is not only associated with muscle proteins. Whole genome sequencing was performed on individuals with DA5, in which arthrogryposis occurs in combination with ptosis, ophthalmoplegia, and facial dysmorphism, and gain of function variants (I802F and E2727del) were discovered in PIEZO2 [24], a mechanosensitive cation channel responsible for mediating cation currents in primary sensory neurons [24,40,55]. Because of their mechanosensitivity, these channels are also termed “stretch-activated ion channels”. PIEZO2 was found to affect the skeleton non-autonomously in mice. Loss of PIEZO2 specifically in skeletal tissue did not affect bone development; however, loss of gene function in proprioceptive neurons caused spine malalignment [67]. This suggests both that the neural system is necessary in maintaining normal skeletal development, and that PIEZO2 is a critical gene in this process.
To test the mechanistic effects of these pathogenic variants, one group recently transfected human embryonic kidney cells with wild type PIEZO2, or PIEZO2 with missense variants encoding I802F, or E2727del [24]. Both of these disease-associated variants caused the channel to recover more quickly from inactivation and resulted in increased channel activity following a mechanical stimulus. This supports the hypothesis that DA5 is caused by gain of function mutations that alter mechanosensory nerves. Although additional studies are needed, overstimulation may directly affect the neuromuscular pathway that controls muscle tone in developing fetuses, perhaps causing near-constitutive contractions that constrain the developing joint.
3.2. ECEL1
Variants in ECEL1, which encodes the endothelin-converting enzyme like-1, were identified in the recessive state in several families with DA5D, a rare form of arthrogryposis in which affected individuals have contractures as well as distinctive facial features and ptosis [41]. ECEL1, which is expressed in brain and nerve, is required for post-natal development in mice. Loss of Ecel1 in mice results in abnormal terminal branching of motor neurons at the skeletal muscle endplate [68]. The mechanism by which ECEL1 directs motor neuron branching is currently unknown; however, the resulting contractures in patients with ECEL1 variants were proposed to be caused by a similar mechanism to those caused by genes such as CHRNG that causes multiple pterygium syndrome that impairs neurotransmission at the neuromuscular junction [69].
4. Lethal Congenital Contracture Syndrome
In contrast with DA, which are more common and often autosomal dominant, Lethal Congenital Contracture Syndromes (LCCSs) are a group of rare autosomal recessive forms of arthrogryposis. LCCS are characterized by lack of fetal movement (akinesia), micrognathia, incomplete lung development, polyhydramnios, characteristic contractures of the limbs (clubfoot, hyperextended knees, elbow and wrist flexion contractures) and motoneuron degeneration. Eleven subtypes of LCCS have been characterized. However, there are likely to be many more genes that result in these conditions as more genetic studies are performed on products of conception due to spontaneous abortion or stillbirth. LCCS is more common in communities with high rates of consanguinity consistent with the recessive inheritance pattern. Variable expression of LCCS phenotypes may be due to residual gene function in patients with missense variants or modifier genes. The recessive phenotypes of LCCS have made them more amenable to study by complete knockdown of gene expression.
4.1. Nuclear mRNA Export (GLE1, ERBB3, and PIP5K1C)
The first three LCCS subtypes may all act through a similar pathway by supporting nuclear mRNA export. LCCS type 1 (LCCS1) is caused by mutations in GLE1 RNA Export Mediator (GLE1), a regulator of post-transcriptional gene expression [70]. GLE1 acts as an mRNA export factor, as well as by mediating translation initiation and termination [15]. In mice, in situ hybridization showed marked expression in the neural tube of 11 dpf embryos, specifically in the ventral portion from which motoneurons generate [70]. In zebrafish, the gene is expressed prominently in the central nervous system during development [33].
A mutation in GLE1, FinMajor, has been linked to LCCS1 by causing a splice-site mutation that results in a 3 amino acid insertion in the coiled-coil domain [70]. The coiled-coil domain is required for the protein to self-associates to form oligomers, and one group examined the effect of the FinMajor mutation on polypeptide self-association in vitro and in vivo [29]. Both in vitro and in living cells, the GLE1 protein self-aggregated, and FinMajor mutant oligomers were malformed. In human cell culture and in the yeast model, these malformed oligomers were found to perturb mRNA export from the nucleus [29].
Because the FinMajor mutation reduced function of the GLE1 protein in mRNA transport, gle1 knockdown and knockouts were studied in zebrafish to understand its effects on development. Knockouts developed with small eyes and underdeveloped jaws and pectoral fins [33]. Cell death was also observed in the head and spinal cord, and there were fewer motoneurons than in wild type fish. Motoneurons also exhibit aberrant branching that worsened with age. Maternal gle1 mRNA is loaded into the yolk sac of oocytes, where it contributes to zebrafish embryogenesis; therefore, morpholino oligonucleotides were also used to knock down expression of the mRNA in embryos. This exacerbated the phenotype, with CNS cell death becoming apparent earlier in development, at 1 dpf, which suggests an important role for gle1 for early development. Notably, this phenotype is rescued in morphants injected with human wild type GLE1, but not when injected with the FinMajor allele [33]. Thus, this zebrafish model may be a viable tool for screening and determining the pathogenicity of human alleles.
LCCS2 is due to loss-of-function mutations in Erb-B2 Receptor Tyrosine Kinase 3 (ERBB3), which encodes HER3, a known modulator of the phosphatidylinositol pathway [44]. Interestingly, variants in LCCS3 were found to be due to variants in Phosphatidylinositol-4-Phosphate 5-Kinase Type 1 (PIP5K1C), which encodes the enzyme PIPK-gamma of the phosphatidylinositol pathway [43]. Nouslainen et al. realized that both ERBB3 and PIP5K1C are involved in the synthesis of inositol hexakisphosphate, which binds directly to yeast Gle1, activating Dbp5 for mRNA transport [70]. Because Gle1 is expressed in the neural tube during development, pathogenic variants in this gene can be devastating to development of the nervous system, as Gle1 is integral to mRNA transport [70].
4.2. Peripheral Nerve (CNTNAP1, ADGRG6, GLDN)
The genes responsible for LCCS7, LCCS9, and LCCS11 are all highly expressed in peripheral nerves and required for proper peripheral nerve function. Contactin Associated Protein 1 (CNTNAP1), which causes LCCS7, is a contactin-associated protein that is required for localization of the paranodal junction proteins contactin and neurofascin. CNTNAP1 is also required for the normal spatial expression patterns of neuronal sodium and potassium channels [19]. Likewise, the causative gene for LCCS11, gliomedin (GLDN), is a ligand for neurofascin and Nrcam, which are axonal immunoglobulin cell adhesion molecules critical for association with sodium channels at the nodes of Ranvier [71]. Adhesion G Protein Coupled Receptor G6 (ADGRG6), which is also known as GPR126, is required for normal Schwann cell development. Thus, defects in all three genes likely result in similar peripheral nerve dysfunction at very early stages in development that leads to the LCCS phenotype.
5. Conclusions
Many techniques and organisms have been used for modeling arthrogryposis, each of which provides complementary information that is essential for understanding basic mechanisms and will yield translational benefits to human patients. There is an expanding list of genes that are associated with limb contractures, as one of many clinical features, beyond those discussed in this review article. Other genes are yet to be discovered, and disease models are often needed to provide evidence of causality. Furthermore, as exome sequencing becomes standard care, disease models may be helpful to facilitate variant interpretation. However, it will be essential to develop more efficient methods for introducing and studying large numbers of individual variants.
Although most genes responsible for distal arthrogryposis and LCCS are skeletal muscle sarcomeric genes or genes critical for neuronal function and neuromuscular transmission, crucial aspects remain to be established using disease models. It is important to determine whether common pathways and mechanisms supported by the genetic data will predict a unifying approach to therapy. Furthermore, now that gene therapies are becoming viable treatment mechanisms, where and when the defect needs to be corrected to prevent development of the DA or LCCS phenotype needs to be elucidated. Disease models will be essential to improve treatment for these challenging disorders.
Author Contributions
J.W. and C.A.G. wrote the first draft of the manuscript. J.W., A.J., M.B.D. and C.A.G. wrote or critically reviewed the manuscript, and reviewed and approved the final version. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases under Award Numbers R01AR067715 and R01AR070299, Eunice Kennedy Shriver National Institutes of Child Health and Human Development of the National Institutes of Health under the Award Numbers R03 HD104065 an P01 HD084387, Washington University Institute of Clinical and Translational Sciences grant UL1 TR002345 from the National Center for Advancing Translational Sciences of the National Institutes of Health, Washington University Musculoskeletal Research Center (NIH/NIAMS P30 AR057235) (NIH/NIAMS P30 AR074992), and the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number P50 HD103525 to the Intellectual and Developmental Disabilities Research Center at Washington University. This study was funded with support from Shriners Hospital for Children, and the Children’s Discovery Institute of Washington University and St Louis Children’s Hospital.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hall, J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014, 57, 464–472. [Google Scholar] [CrossRef]
- Hall, J.G. Arthrogryposis multiplex congenita: Etiology, genetics, classification, diagnostic approach, and general aspects. J. Pediatric Orthop. 1997, 6, 159–166. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Felsenthal, N.; Zelzer, E. Mechanical regulation of musculoskeletal system development. Development 2017, 144, 4271–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, J.; Hall, J.G. Gene ontology analysis of arthrogryposis (multiple congenital contractures). In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; Wiley Online Library: Hoboken, NJ, USA, 2019; pp. 310–326. [Google Scholar]
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A review and update. J. Bone. Jt. Surgery. Am. Vol. 2009, 91, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Shively, K.; Tang, A.; Bamshad, M.J. Genotype-phenotype relationships in Freeman–Sheldon syndrome. Am. J. Med. Genet. Part A 2014, 164, 2808–2813. [Google Scholar] [CrossRef]
- Scala, M.; Accogli, A.; De Grandis, E.; Allegri, A.; Bagowski, C.P.; Shoukier, M.; Maghnie, M.; Capra, V. A novel pathogenic MYH3 mutation in a child with Sheldon–Hall syndrome and vertebral fusions. Am. J. Med. Genet. Part A 2018, 176, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Toydemir, R.M.; Rutherford, A.; Whitby, F.G.; Jorde, L.B.; Carey, J.C.; Bamshad, M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006, 38, 561–566. [Google Scholar] [CrossRef]
- Desai, D.; Stiene, D.; Song, T.; Sadayappan, S. Distal Arthrogryposis and Lethal Congenital Contracture Syndrome–An Overview. Front. Physiol. 2020, 11, 689. [Google Scholar] [CrossRef]
- Markus, B.; Narkis, G.; Landau, D.; Birk, R.Z.; Cohen, I.; Birk, O.S. Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1. Hum. Mutat. 2012, 33, 1435–1438. [Google Scholar] [CrossRef]
- Racca, A.W.; Beck, A.E.; McMillin, M.J.; Korte, F.S.; Bamshad, M.J.; Regnier, M. The embryonic myosin R672C mutation that underlies Freeman-Sheldon syndrome impairs cross-bridge detachment and cycling in adult skeletal muscle. Hum. Mol. Genet. 2015, 24, 3348–3358. [Google Scholar] [CrossRef] [Green Version]
- Dieterich, K.; Le Tanno, P.; Kimber, E.; Jouk, P.S.; Hall, J.; Giampietro, P. The diagnostic workup in a patient with AMC: Overview of the clinical evaluation and paraclinical analyses with review of the literature. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; Wiley Online Library: Hoboken, NJ, USA, 2019; pp. 337–344. [Google Scholar]
- Guo, Y.; Kronert, W.A.; Hsu, K.H.; Huang, A.; Sarsoza, F.; Bell, K.M.; Suggs, J.A.; Swank, D.M.; Bernstein, S.I. Drosophila myosin mutants model the disparate severity of type 1 and type 2B distal arthrogryposis and indicate an enhanced actin affinity mechanism. Skelet. Muscle 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Folkmann, A.W.; Dawson, T.R.; Wente, S.R. Insights into mRNA export-linked molecular mechanisms of human disease through a Gle1 structure–function analysis. Adv. Biol. Regul. 2014, 54, 74–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, J.; Antunes, L.; Harris, M.; Upshaw, Z.; Sepich, D.S.; Johnson, A.N.; Mokalled, M.; Solnica-Krezel, L.; Dobbs, M.B.; Gurnett, C.A. MYH 3-associated distal arthrogryposis zebrafish model is normalized with para-aminoblebbistatin. EMBO Mol. Med. 2020, 12, e12356. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, F.; Zhao, Y.; Yang, P.; Chen, J.; Sun, H.; Liu, L.; Li, W.; Pan, L.; Guo, Y. A gain-of-function mutation in Tnni2 impeded bone development through increasing Hif3a expression in DA2B mice. PLoS Genet. 2014, 10, e1004589. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Karaca, E.; Akdemir, Z.C.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.A.; Rios, J.C.; Lu, Y.; Garcia-Fresco, G.P.; Ching, W.; Martin, M.S.; Li, J.; Einheber, S.; Chesler, M.; Rosenbluth, J. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 2001, 30, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Karpicheva, O.E.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Piers, A.; Redwood, C.S.J.B. The reason for a high Ca2+-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 2017, 494, 681–686. [Google Scholar] [CrossRef]
- Cameron-Christie, S.R.; Wells, C.F.; Simon, M.; Wessels, M.; Tang, C.Z.; Wei, W.; Takei, R.; Aarts-Tesselaar, C.; Sandaradura, S.; Sillence, D.O. Recessive Spondylocarpotarsal synostosis syndrome due to compound heterozygosity for variants in MYH3. Am. J. Hum. Genet. 2018, 102, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; Brennan, K.; Scheidel, N.; McGettigan, P.; Lavin, P.T.; Carter, S.; Ennis, S.; Dorkins, H.; Ghali, N.; Blacque, O.E.; et al. Recessive NEK9 mutation causes a lethal skeletal dysplasia with evidence of cell cycle and ciliary defects. Hum. Mol. Genet. 2016, 25, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.X.; Talbot, J.C.; Teets, E.M.; Previs, S.; Martin, B.L.; Shively, K.M.; Marvin, C.T.; Aylsworth, A.S.; Saadeh-Haddad, R.; Schatz, U.A. Mutations in MYLPF cause a novel segmental amyoplasia that manifests as distal arthrogryposis. Am. J. Hum. Genet. 2020, 107, 293–310. [Google Scholar] [CrossRef]
- Coste, B.; Houge, G.; Murray, M.F.; Stitziel, N.; Bandell, M.; Giovanni, M.A.; Philippakis, A.; Hoischen, A.; Riemer, G.; Steen, U. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc. Natl. Acad. Sci. USA 2013, 110, 4667–4672. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kumar, P.; Verma, A.; Maiti, T.K.; Mathew, S.J. Myosin heavy chain mutations that cause Freeman-Sheldon syndrome lead to muscle structural and functional defects in Drosophila. Dev. Biol. 2019, 449, 90–98. [Google Scholar] [CrossRef]
- Di Paolo, G.; Moskowitz, H.S.; Gipson, K.; Wenk, M.R.; Voronov, S.; Obayashi, M.; Flavell, R.; Fitzsimonds, R.M.; Ryan, T.A.; De Camilli, P.J.N. Impaired PtdIns (4, 5) P 2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 2004, 431, 415–422. [Google Scholar] [CrossRef]
- Durieux, A.-C.; Vignaud, A.; Prudhon, B.; Viou, M.T.; Beuvin, M.; Vassilopoulos, S.; Fraysse, B.; Ferry, A.; Lainé, J.; Romero, N.B.J.H.m.g. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010, 19, 4820–4836. [Google Scholar] [CrossRef] [Green Version]
- Ekhilevitch, N.; Kurolap, A.; Oz-Levi, D.; Mory, A.; Hershkovitz, T.; Ast, G.; Mandel, H.; Baris, H. Expanding the MYBPC1 phenotypic spectrum: A novel homozygous mutation causes arthrogryposis multiplex congenita. Clin. Genet. 2016, 90, 84–89. [Google Scholar] [CrossRef]
- Folkmann, A.W.; Collier, S.E.; Zhan, X.; Ohi, M.D.; Wente, S.R. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 2013, 155, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnett, C.A.; Desruisseau, D.M.; McCall, K.; Choi, R.; Meyer, Z.I.; Talerico, M.; Miller, S.E.; Ju, J.-S.; Pestronk, A.; Connolly, A.M. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010, 19, 1165–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Buchan, J.G.; Alvarado, D.M.; Mccall, K.; Vydyanath, A.; Luther, P.K.; Goldsmith, M.I.; Dobbs, M.B.; Gurnett, C.A. MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Hum. Mol. Genet. 2013, 22, 4967–4977. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.; Sharma, A.; Kumar, P.; Kumar, A.; Bharadwaj, A.; Saini, M.; Kardon, G.; Mathew, S.J. Myosin heavy chain-embryonic regulates skeletal muscle differentiation during mammalian development. Development 2020, 147, dev184507. [Google Scholar] [CrossRef] [PubMed]
- Jao, L.-E.; Appel, B.; Wente, S.R. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Development 2012, 139, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.; Li, J.; Xie, C.; Ritchlin, C.T.; Xing, L.; Hilton, M.J.; Schwarz, E.M. Troponin T3 expression in skeletal and smooth muscle is required for growth and postnatal survival: Characterization of Tnnt3tm2a (KOMP) Wtsi mice. Genesis 2013, 51, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsopoulos, O.S.; Kretz, C.; Weller, C.M.; Roux, A.; Mojzisova, H.; Böhm, J.; Koch, C.; Toussaint, A.; Heckel, E.; Stemkens, D.; et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur. J. Hum. Genet. 2013, 21, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laquérriere, A.; Maluenda, J.; Camus, A.; Fontenas, L.; Dieterich, K.; Nolent, F.; Zhou, J.; Monnier, N.; Latour, P.; Gentil, D.; et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum. Mol. Genet. 2014, 23, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Andersson-Lendahl, M.; Sejersen, T.; Arner, A. Knockdown of fast skeletal myosin-binding protein C in zebrafish results in a severe skeletal myopathy. J. Gen. Physiol. 2016, 147, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maluenda, J.; Manso, C.; Quevarec, L.; Vivanti, A.; Marguet, F.; Gonzales, M.; Guimiot, F.; Petit, F.; Toutain, A.; Whalen, S.; et al. Mutations in GLDN, encoding gliomedin, a critical component of the nodes of ranvier, are responsible for lethal arthrogryposis. Am. J. Hum. Genet. 2016, 99, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyushenko, A.; Levitsky, D. Molecular mechanisms of pathologies of skeletal and cardiac muscles caused by point mutations in the tropomyosin genes. Biochemistry 2020, 85, 20–33. [Google Scholar] [CrossRef]
- McMillin, M.J.; Beck, A.E.; Chong, J.X.; Shively, K.M.; Buckingham, K.J.; Gildersleeve, H.I.; Aracena, M.I.; Aylsworth, A.S.; Bitoun, P.; Carey, J.C. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 2014, 94, 734–744. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.J.; Below, J.E.; Shively, K.M.; Beck, A.E.; Gildersleeve, H.I.; Pinner, J.; Gogola, G.R.; Hecht, J.T.; Grange, D.K.; Harris, D.J. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013, 92, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Monk, K.R.; Naylor, S.G.; Glenn, T.D.; Mercurio, S.; Perlin, J.R.; Dominguez, C.; Moens, C.B.; Talbot, W.S. AG protein–coupled receptor is essential for Schwann cells to initiate myelination. Science 2009, 325, 1402–1405. [Google Scholar] [CrossRef] [Green Version]
- Narkis, G.; Ofir, R.; Landau, D.; Manor, E.; Volokita, M.; Hershkowitz, R.; Elbedour, K.; Birk, O.S. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKIγ of the phophatidylinsitol pathway. Am. J. Hum. Genet. 2007, 81, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Narkis, G.; Ofir, R.; Manor, E.; Landau, D.; Elbedour, K.; Birk, O.S. Lethal congenital contractural syndrome type 2 (LCCS2) is caused by a mutation in ERBB3 (Her3), a modulator of the phosphatidylinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2007, 81, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Smith, L.L.; Faqeih, E.; Mohamed, J.; Gupta, V.A.; Alkuraya, F.S. ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum. Mol. Genet. 2014, 23, 6584–6593. [Google Scholar] [CrossRef] [Green Version]
- Ravenscroft, G.; Nolent, F.; Rajagopalan, S.; Meireles, A.M.; Paavola, K.J.; Gaillard, D.; Alanio, E.; Buckland, M.; Arbuckle, S.; Krivanek, M.; et al. Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am. J. Hum. Genet. 2015, 96, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riethmacher, D.; Sonnenberg-Riethmacher, E.; Brinkmann, V.; Yamaai, T.; Lewin, G.R.; Birchmeier, C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 1997, 389, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Sandaradura, S.A.; Bournazos, A.; Mallawaarachchi, A.; Cummings, B.B.; Waddell, L.B.; Jones, K.J.; Troedson, C.; Sudarsanam, A.; Nash, B.M.; Peters, G.B.; et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum. Mutat. 2018, 39, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashi, V.; Geist, J.; Lee, Y.; Yoo, Y.; Shin, U.; Schoch, K.; Sullivan, J.; Stong, N.; Smith, E.; Jasien, J. Heterozygous variants in MYBPC1 are associated with an expanded neuromuscular phenotype beyond arthrogryposis. Hum. Mutat. 2019, 40, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.S.; Brassington, A.-M.E.; Grannatt, K.; Rutherford, A.; Whitby, F.G.; Krakowiak, P.A.; Jorde, L.B.; Carey, J.C.; Bamshad, M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genetics 2003, 72, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toydemir, R.M.; Chen, H.; Proud, V.K.; Martin, R.; van Bokhoven, H.; Hamel, B.C.; Tuerlings, J.H.; Stratakis, C.A.; Jorde, L.B.; Bamshad, M.J. Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am. J. Med Genet. Part A 2006, 140, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Veugelers, M.; Bressan, M.; McDermott, D.A.; Weremowicz, S.; Morton, C.C.; Mabry, C.C.; Lefaivre, J.-F.; Zunamon, A.; Destree, A.; Chaudron, J.-M.; et al. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N. Engl. J. Med. 2004, 351, 460–469. [Google Scholar] [CrossRef]
- Vigoreaux, J.O. Genetics of the Drosophila flight muscle myofibril: A window into the biology of complex systems. Bioessays 2001, 23, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Walklate, J.; Vera, C.; Bloemink, M.J.; Geeves, M.A.; Leinwand, L. The most prevalent Freeman-Sheldon Syndrome mutations in the embryonic myosin motor share functional defects. J. Biol. Chem. 2016, 291, 10318–10331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhou, H.; Zhang, M.; Liu, W.; Deng, T.; Zhao, Q.; Li, Y.; Lei, J.; Li, X.; Xiao, B. Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 2019, 573, 225–229. [Google Scholar] [CrossRef]
- Williams, J.; Boin, N.G.; Valera, J.M.; Johnson, A.N. Noncanonical roles for Tropomyosin during myogenesis. Development 2015, 142, 3440–3452. [Google Scholar] [PubMed] [Green Version]
- Chong, J.X.; Burrage, L.C.; Beck, A.E.; Marvin, C.T.; McMillin, M.J.; Shively, K.M.; Harrell, T.M.; Buckingham, K.J.; Bacino, C.A.; Jain, M.; et al. Autosomal-dominant multiple pterygium syndrome is caused by mutations in MYH3. Am. J. Hum. Genet. 2015, 96, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhong, B.; Han, W.; Zhao, N.; Liu, W.; Sui, Y.; Wang, Y.; Lu, Y.; Wang, H.; Li, J. Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS ONE 2015, 10, e0117158. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, D.M.; Buchan, J.G.; Gurnett, C.A.; Dobbs, M.B. Exome sequencing identifies an MYH3 mutation in a family with distal arthrogryposis type 1. J. Bone Jt. Surg. Am. Vol. 2011, 93, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Kezele, P.R.; Shively, K.M.; Carey, J.C.; Regnier, M.; Bamshad, M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. Part A 2013, 161, 550–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieba, J.; Zhang, W.; Chong, J.X.; Forlenza, K.N.; Martin, J.H.; Heard, K.; Grange, D.K.; Butler, M.G.; Kleefstra, T.; Lachman, R.S. A postnatal role for embryonic myosin revealed by MYH3 mutations that alter TGFβ signaling and cause autosomal dominant spondylocarpotarsal synostosis. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gil-Gálvez, A.; Carbonell-Corvillo, P.; Paradas, C.; Miranda-Vizuete, A. Cautionary note on the use of Caenorhabditis elegans to study muscle phenotypes caused by mutations in the human MYH7 gene. BioTechniques 2020, 68, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Repetti, G.G.; Toepfer, C.N.; Seidman, J.G.; Seidman, C.E. Novel therapies for prevention and early treatment of cardiomyopathies: Now and in the future. Circ. Res. 2019, 124, 1536–1550. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Ohlsson, M.; Palm, L.; Oldfors, A. Myopathies associated with β-tropomyosin mutations. Neuromuscul. Disord. 2012, 22, 923–933. [Google Scholar] [CrossRef]
- Monnier, N.; Lunardi, J.; Marty, I.; Mezin, P.; Labarre-Vila, A.; Dieterich, K.; Jouk, P.S. Absence of β-tropomyosin is a new cause of Escobar syndrome associated with nemaline myopathy. Neuromuscul. Disord. 2009, 19, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Szczesna-Cordary, D.; Craig, R.; Diaz-Perez, Z.; Guzman, G.; Miller, T.; Potter, J.D. Fast skeletal muscle regulatory light chain is required for fast and slow skeletal muscle development. FASEB J. 2007, 21, 2205–2214. [Google Scholar] [CrossRef]
- Assaraf, E.; Blecher, R.; Heinemann-Yerushalmi, L.; Krief, S.; Vinestock, R.C.; Biton, I.E.; Brumfeld, V.; Rotkopf, R.; Avisar, E.; Agar, G.; et al. Piezo2 expressed in proprioceptive neurons is essential for skeletal integrity. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, A.; Valdenaire, O.; Köster, A.; Lang, Y.; Schmitt, G.; Lenz, B.; Bluethmann, H.; Rohrer, J. Neonatal lethality in mice deficient in XCE, a novel member of the endothelin-converting enzyme and neutral endopeptidase family. J. Biol. Chem. 1999, 274, 20450–20456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, N.V.; Brueton, L.A.; Cox, P.; Greally, M.T.; Tolmie, J.; Pasha, S.; Aligianis, I.A.; van Bokhoven, H.; Marton, T.; Al-Gazali, L. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006, 79, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Nousiainen, H.O.; Kestilä, M.; Pakkasjärvi, N.; Honkala, H.; Kuure, S.; Tallila, J.; Vuopala, K.; Ignatius, J.; Herva, R.; Peltonen, L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 2008, 40, 155–157. [Google Scholar] [CrossRef]
- Eshed, Y.; Feinberg, K.; Poliak, S.; Sabanay, H.; Sarig-Nadir, O.; Spiegel, I.; Bermingham, J.R., Jr.; Peles, E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 2005, 47, 215–229. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The curved spinal phenotype associated with both smyhc1+/R673H and smyhc1R673H/R673H genotypes is normalized with the myosin inhibitor para-aminoblebbistatin. Embryos were treated from 24–48 hpf and photographed at 48 hpf. Treated embryos are shown below DMSO treated controls. Unlike the newer myosin inhibitors that are being developed, para-aminoblebbistatin has many toxic effects, including lethal cardiac edema, which limits its use as a human therapeutic. These images are similar to those published in [16].
Figure 1.
The curved spinal phenotype associated with both smyhc1+/R673H and smyhc1R673H/R673H genotypes is normalized with the myosin inhibitor para-aminoblebbistatin. Embryos were treated from 24–48 hpf and photographed at 48 hpf. Treated embryos are shown below DMSO treated controls. Unlike the newer myosin inhibitors that are being developed, para-aminoblebbistatin has many toxic effects, including lethal cardiac edema, which limits its use as a human therapeutic. These images are similar to those published in [16].

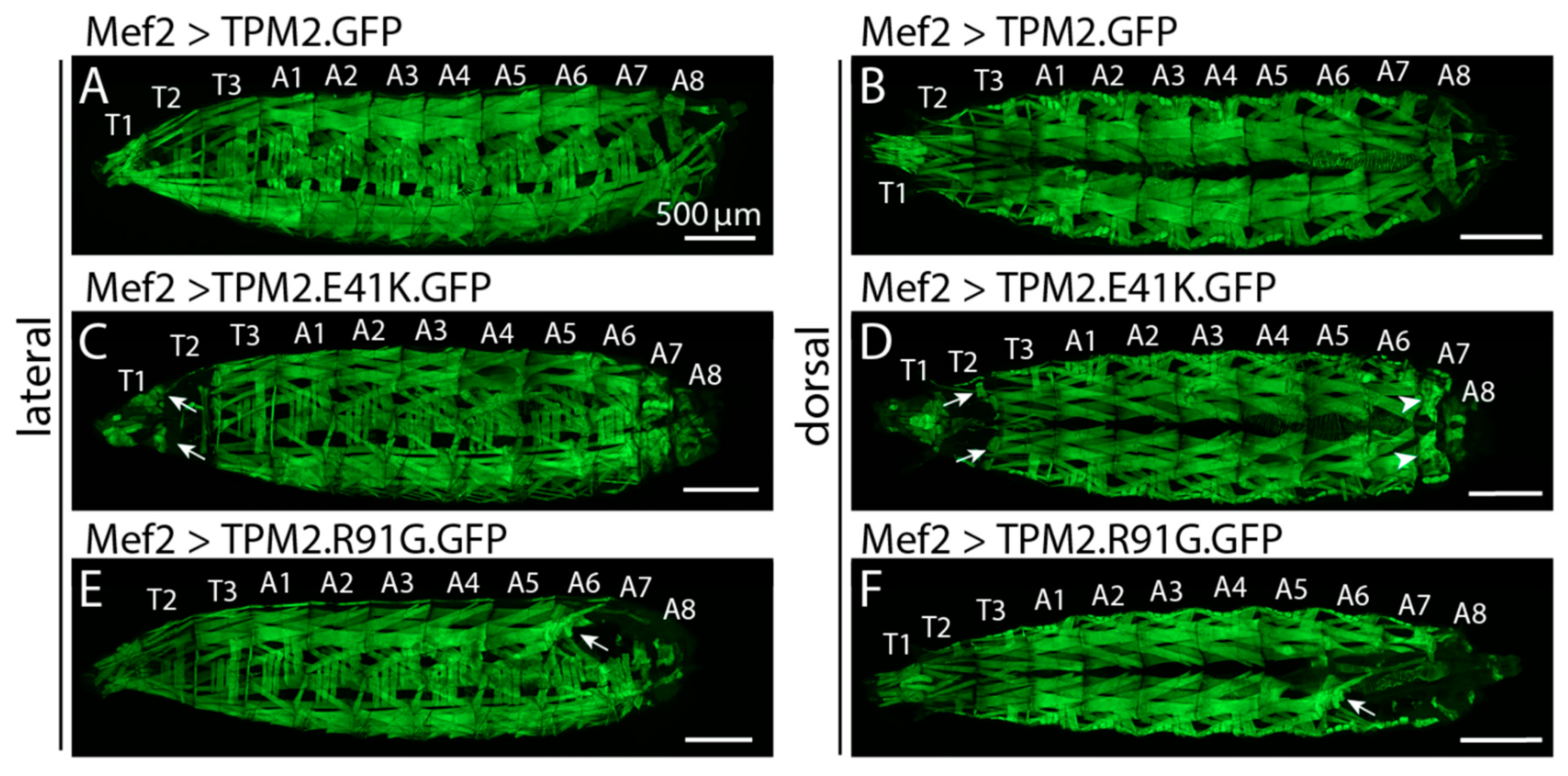
Figure 2.
DA associated TPM2 variants cause muscle phenotypes in Drosophila. Confocal micrographs of live L3 larva that express GFP-tagged TPM2 variants in skeletal muscles (body wall muscles). Mef2.Gal4 was used to activate UAS.TPM2 transgenes. Lateral and dorsal views are shown for each genotype. (A,B) Larva that express TPM2.GFP showed normal muscle histology. Larva that expresses TPM2.E41K.GFP (C,D) or TPM2.R91G.GFP (E,F). GFP have rounded myofibers that appear to result from internal tears (arrows; note affected muscles remain associated with tendons at segment boundaries) and shortened segments that could be due to hypercontractile muscles (arrowheads). Thoracic segments (T1–T3) and abdominal segments (A1–A8) are labeled. Scale bars, 500 mM. Previously unpublished data.
Figure 2.
DA associated TPM2 variants cause muscle phenotypes in Drosophila. Confocal micrographs of live L3 larva that express GFP-tagged TPM2 variants in skeletal muscles (body wall muscles). Mef2.Gal4 was used to activate UAS.TPM2 transgenes. Lateral and dorsal views are shown for each genotype. (A,B) Larva that express TPM2.GFP showed normal muscle histology. Larva that expresses TPM2.E41K.GFP (C,D) or TPM2.R91G.GFP (E,F). GFP have rounded myofibers that appear to result from internal tears (arrows; note affected muscles remain associated with tendons at segment boundaries) and shortened segments that could be due to hypercontractile muscles (arrowheads). Thoracic segments (T1–T3) and abdominal segments (A1–A8) are labeled. Scale bars, 500 mM. Previously unpublished data.


{kind=link}
{kind=link}
Table 1.
List of genes and associated conditions and models of distal arthrogryposis (DA) and lethal congenital contracture syndrome (LCCS) used for study. Autosomal dominant (AD), Autosomal recessive (AR) [9,11,12,14,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57].
Table 1.
List of genes and associated conditions and models of distal arthrogryposis (DA) and lethal congenital contracture syndrome (LCCS) used for study. Autosomal dominant (AD), Autosomal recessive (AR) [9,11,12,14,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57].
| Gene | Full Name | Disorder | Inheritance Pattern | Modeled in | Source Human | Models of Disease Source |
|---|---|---|---|---|---|---|
| ARTHROGRYPOSIS | ||||||
| MYH3 | Myosin, Heavy Polypeptide 3, Skeletal Muscle, Embryonic | DA1, DA2A, DA2B, DA8, Spondylocarpotarsal Syndrome | AD, AR | Zebrafish, Cell, Biochemical | Toydemir et al., 2006b [9]; Chong et al., 2015 [57]; Cameron-Christie et al., 2019 [21] | Racca et al., 2015 [12]; Walklate et al., 2016 [54]; Wang et al., 2019 [55]; Whittle et al., 2020 [16]; Guo et al., 2020 [14]; Das et al., 2019 [25] |
| TPM2 | Tropomyosin 2 | DA1, Cap Myopathy, Nemaline Myopathy | AD, AR | Drosophila, Biochemical | Sung et al., 2003 [50] | Williams et al., 2015 [56]; Borovikov et al., 2017 [20]; Matyushenko & Levitsky, 2020 [39]; |
| MYLPF | Myosin Regulatory Light Chain 2, Skeletal Muscle Isoform | DA1, DA2B | AD, AR | Zebrafish | Chong et al., 2020 [23] | Chong et al., 2020 [23] |
| MYBPC1 | Myosin-Binding Protein C, Slow-Type | DA1, DA2, LCCS4 | AD | Zebrafish | Gurnett et al., 2010 [30]; Li et al., 2015 [58]; Ekhilevitch et al., 2016 [28]; Shashi et al., 2019 [49]; | Ha et al., 2013 [31] |
| MYBPC2 | Myosin-Binding Protein C, Fast-Type | DA (unspecified) | AD | Zebrafish | Bayram et al., 2016 [18] | Li et al., 2016 [37] |
| TNNT3 | Troponin T3, Fast Skeletal Type | DA2B | AD, AR | Mouse | Sung et al., 2003 [50]; Sandaradura et al., 2018 [48] | Ju et al., 2013 [34] |
| TNNI2 | Troponin I2, Fast Skeletal Type | DA2B | AD | Mouse, Drosophila | Sung et al., 2003 [50] | Zhu et al., 2014 [17]; Vigoreaux, 2001 [53] |
| PIEZO2 | Piezo Type Mechanosensitive Ion Channel Component 2 | DA3, DA5 | AR | Cell | McMillin et al., 2014 [40] | Coste et al., 2013 [24]; McMillin et al., 2014 [40] |
| ECEL1 | Endothelin Converting Enzyme Like 1 | DA5 (or DA5D) | AR | - | McMillin et al., 2013 [41] | - |
| MYH8 | Myosin, Heavy Polypeptide 8, Skeletal Muscle, Fetal | DA7 | AD | - | Toydemir et al., 2006a; [51] Veugelers et al., 2004 [52] | - |
| LETHAL CONGENITAL CONTRATURE SYNDROME | ||||||
| GLE1 | GLE1 RNA Export Mediator | LCCS1 | AR | Zebrafish, Cell, Biochemical | Jao et al., 2012 [33] | Folkmann et al., 2013 [29]; Jao et al., 2012 [33] |
| ERBB3 | ERB-B2 Receptor Tyrosine Kinase 3 | LCCS2 | AR | Mouse | Narkis et al., 2007 [44] | Riethmacher et al., 1997 [47] |
| PIP5K1C | Phosphatidylinositol 4-Phosphate 5-Kinase, type 1, gamma | LCCS3 | AR | Mouse | Narkis et al., 2007 [43] | DiPaolo et al., 2004 [26] |
| MYBPC1 | Myosin-Binding Protein C, Slow-Type | LCCS4, DA1, DA2 | AD, AR | Zebrafish | Markus et al., 2012 [11] | Ha et al., 2013 [31] |
| DNM2 | Dynamin, 2 | LCCS5, Centronuclear Myopathy, CMT2M, CMT Intermed | AD, AR | Mouse | Koutsopoulos et al., 2013 [35] | Durieux et al., 2010 [27]; Koutsopoulos et al., 2013 [35] |
| ZBTB42 | Zinc finger-and BTB Domain-containing Protein 42 | LCCS6 | AR | Zebrafish | Patel et al., 2014 [45] | Patel et al., 2014 [45] |
| CNTNAP1 | Contactin-associated protein 1 | LCCS7, Congenital Hypomyelinating Neuropathy | AR | Mouse | Laquerriere et al., 2014 [36] | Bhat et al., 2001 [19] |
| ADCY6 | Adenylyl cyclase 6 | LCCS8 | AR | Zebrafish | Laquerriere et al., 2014 [36] | Laquerriere et al., 2014 [36] |
| ADGRG6 | Adhesion G-protein coupled receptor G6 or GPR126 | LCCS9 | AR | Zebrafish | Ravenscroft et al., 2015 [46] | Monk et al., 2009 [42] |
| NEK9 | Nima-related kinase 1 | LCCS10 | AR | - | Casey et al., 2016 [22] | - |
| GLDN | Gliomedin | LCCS11 | AR | - | Maluenda et al., 2016 [38] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Whittle, J.; Johnson, A.; Dobbs, M.B.; Gurnett, C.A. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes 2021, 12, 943. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060943
AMA Style
Whittle J, Johnson A, Dobbs MB, Gurnett CA. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes. 2021; 12(6):943. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060943
Chicago/Turabian StyleWhittle, Julia, Aaron Johnson, Matthew B. Dobbs, and Christina A. Gurnett. 2021. "Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome" Genes 12, no. 6: 943. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060943
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.