
Molecular Genetics and Complex Inheritance of Congenital Heart Disease


 , ,
, ,
Abstract
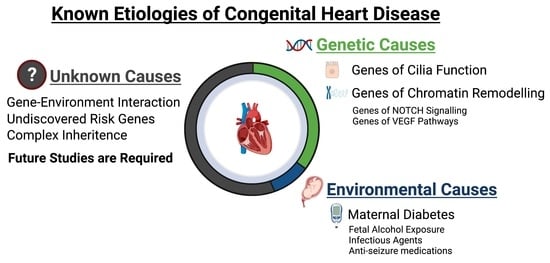
:
{kind=link}
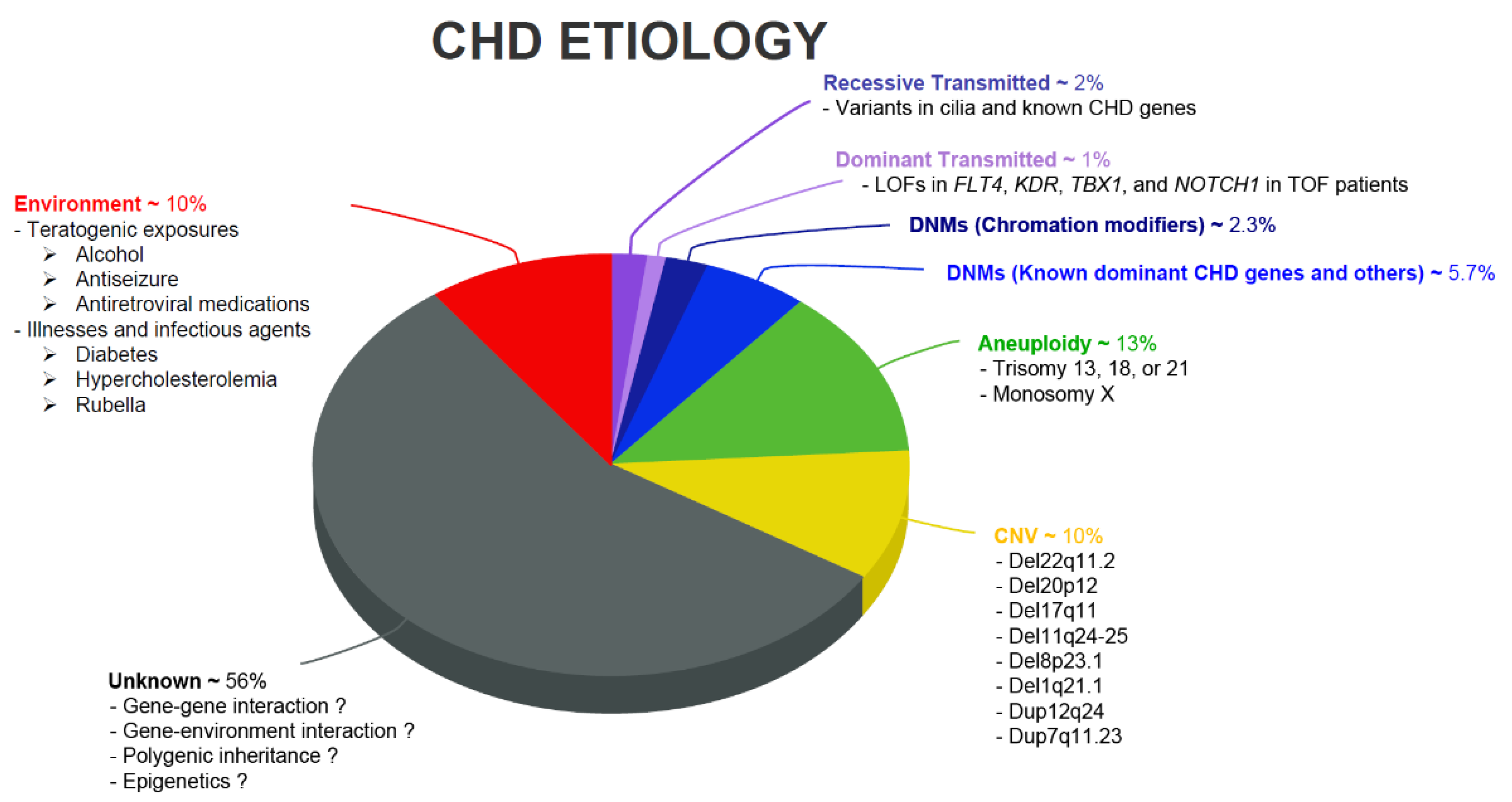{kind=link}
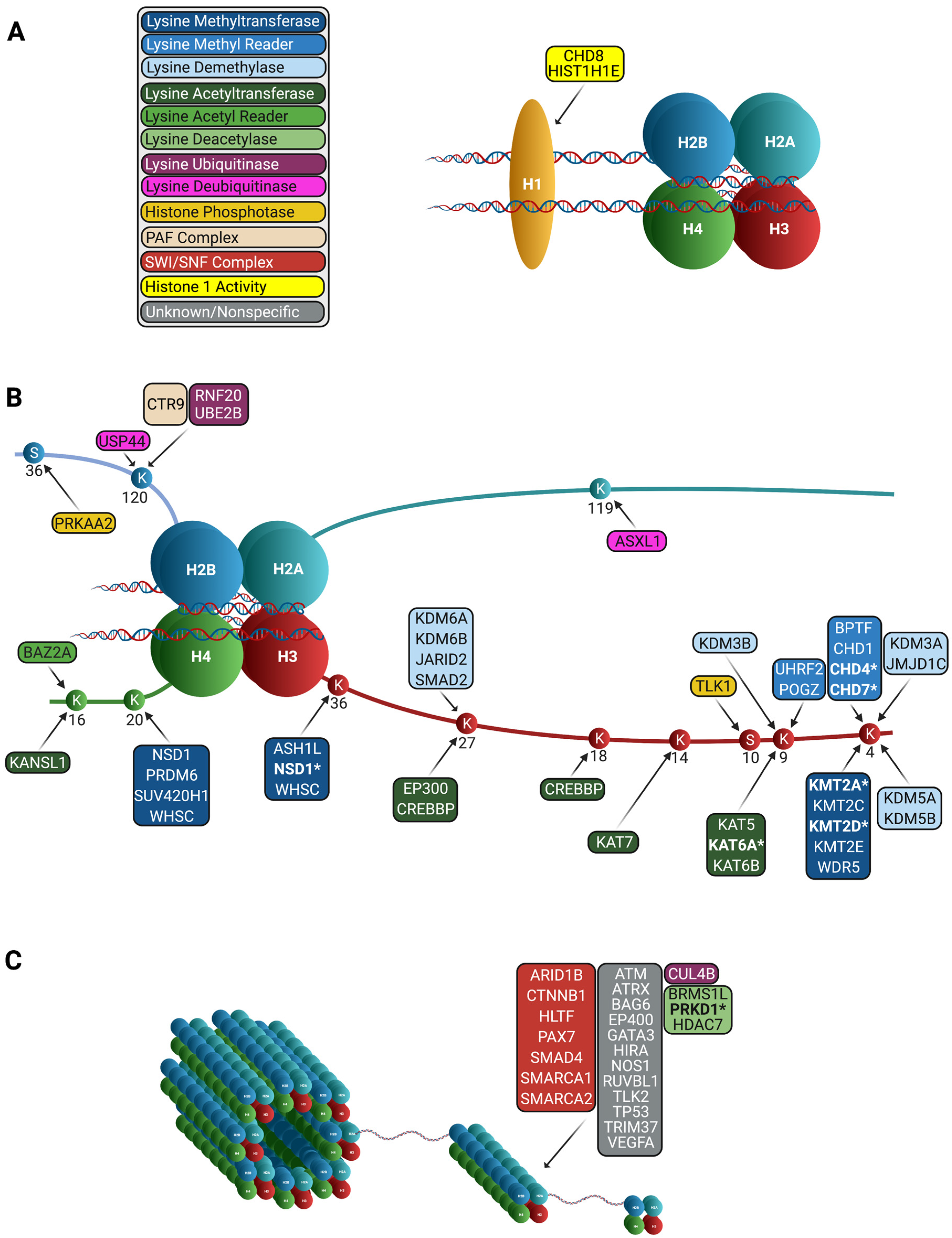{kind=link}
1. Introduction
2. Epidemiology and Risk Factors for CHD
2.1. Epidemiology: Prevalence and Comorbidities
2.2. Non-Genetic Risk Factors: Maternal Exposures, Illnesses, Infectious Agents
3. Genetic Risk Factors and Relevant Biological Pathways
3.1. Aneuploidies and Copy Number Variations
3.2. De Novo Mutations
3.3. Transmitted Mutations
3.4. Biological Pathways
4. Genetic Modifiers and Complex Genetic Inheritance
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Triedman, J.K.; Newburger, J.W. Trends in Congenital Heart Disease: The Next Decade. Circulation 2016, 133, 2716–2733. [Google Scholar] [CrossRef]
- Peacock, T.B. On Malformations, &c., of the Human Heart: With Original Cases; University of Michigan: Ann Arbor, MI, USA, 1981. [Google Scholar]
- Nora, J.J.; Dodd, P.F.; McNamara, D.G.; Hattwick, M.A.; Leachman, R.D.; Cooley, D.A. Risk to offspring of parents with congenital heart defects. JAMA 1969, 209, 2052–2053. [Google Scholar] [CrossRef]
- Oyen, N.; Poulsen, G.; Boyd, H.A.; Wohlfahrt, J.; Jensen, P.K.; Melbye, M. Recurrence of congenital heart defects in families. Circulation 2009, 120, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Whittemore, R.; Hobbins, J.C.; Engle, M.A. Pregnancy and its outcome in women with and without surgical treatment of congenital heart disease. Am. J. Cardiol. 1982, 50, 641–651. [Google Scholar] [CrossRef]
- Pediatric Cardiac Genomics Consortium; Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; et al. The Congenital Heart Disease Genetic Network Study: Rationale, design, and early results. Circ. Res. 2013, 112, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Firth, H.V.; Wright, C.F. The Deciphering Developmental Disorders (DDD) study. Dev. Med. Child Neurol. 2011, 53, 702–703. [Google Scholar] [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Watkins, W.S.; Hernandez, E.J.; Wesolowski, S.; Bisgrove, B.W.; Sunderland, R.T.; Lin, E.; Lemmon, G.; Demarest, B.L.; Miller, T.A.; Bernstein, D.; et al. De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat. Commun. 2019, 10, 4722. [Google Scholar] [CrossRef] [PubMed]
- Boskovski, M.T.; Homsy, J.; Nathan, M.; Sleeper, L.A.; Morton, S.; Manheimer, K.B.; Tai, A.; Gorham, J.; Lewis, M.; Swartz, M.; et al. De Novo Damaging Variants, Clinical Phenotypes, and Post-Operative Outcomes in Congenital Heart Disease. Circ. Genom. Precis. Med. 2020, 13, e002836. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.J.; Rouillard, A.D.; Fernandez, N.F.; Wang, Z.; Lachmann, A.; Shankaran, S.S.; Bisgrove, B.W.; Demarest, B.; Turan, N.; Srivastava, D.; et al. Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects. JACC Basic Transl. Sci. 2020, 5, 376–386. [Google Scholar] [CrossRef]
- Hsieh, A.; Morton, S.U.; Willcox, J.A.L.; Gorham, J.M.; Tai, A.C.; Qi, H.; DePalma, S.; McKean, D.; Griffin, E.; Manheimer, K.B.; et al. EM-mosaic detects mosaic point mutations that contribute to congenital heart disease. Genome Med. 2020, 12, 42. [Google Scholar] [CrossRef]
- Morton, S.U.; Shimamura, A.; Newburger, P.E.; Opotowsky, A.R.; Quiat, D.; Pereira, A.C.; Jin, S.C.; Gurvitz, M.; Brueckner, M.; Chung, W.K.; et al. Association of Damaging Variants in Genes with Increased Cancer Risk Among Patients with Congenital Heart Disease. JAMA Cardiol. 2020. [Google Scholar] [CrossRef]
- Richter, F.; Morton, S.U.; Kim, S.W.; Kitaygorodsky, A.; Wasson, L.K.; Chen, K.M.; Zhou, J.; Qi, H.; Patel, N.; DePalma, S.R.; et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 2020, 52, 769–777. [Google Scholar] [CrossRef]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Glessner, J.T.; Bick, A.G.; Ito, K.; Homsy, J.G.; Rodriguez-Murillo, L.; Fromer, M.; Mazaika, E.; Vardarajan, B.; Italia, M.; Leipzig, J.; et al. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ. Res. 2014, 115, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Alankarage, D.; Ip, E.; Szot, J.O.; Munro, J.; Blue, G.M.; Harrison, K.; Cuny, H.; Enriquez, A.; Troup, M.; Humphreys, D.T.; et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet. Med. 2019, 21, 1111–1120. [Google Scholar] [CrossRef]
- Fotiou, E.; Williams, S.; Martin-Geary, A.; Robertson, D.L.; Tenin, G.; Hentges, K.E.; Keavney, B. Integration of Large-Scale Genomic Data Sources with Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ. Genom. Precis. Med. 2019, 12, 442–451. [Google Scholar] [CrossRef]
- Li, A.H.; Hanchard, N.A.; Azamian, M.; D’Alessandro, L.C.A.; Coban-Akdemir, Z.; Lopez, K.N.; Hall, N.J.; Dickerson, H.; Nicosia, A.; Fernbach, S.; et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet. 2019, 27, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Hedermann, G.; Hedley, P.L.; Thagaard, I.N.; Krebs, L.; Ekelund, C.K.; Sørensen, T.I.A.; Christiansen, M. Maternal obesity and metabolic disorders associate with congenital heart defects in the offspring: A systematic review. PLoS ONE 2021, 16, e0252343. [Google Scholar] [CrossRef] [PubMed]
- Cavadino, A.; Sandberg, L.; Öhman, I.; Bergvall, T.; Star, K.; Dolk, H.; Loane, M.; Addor, M.C.; Barisic, I.; Cavero-Carbonell, C.; et al. Signal Detection in EUROmediCAT: Identification and Evaluation of Medication-Congenital Anomaly Associations and Use of VigiBase as a Complementary Source of Reference. Drug Saf. 2021, 44, 765–785. [Google Scholar] [CrossRef]
- Zhang, T.N.; Wu, Q.J.; Liu, Y.S.; Lv, J.L.; Sun, H.; Chang, Q.; Liu, C.F.; Zhao, Y.H. Environmental Risk Factors and Congenital Heart Disease: An Umbrella Review of 165 Systematic Reviews and Meta-Analyses with More Than 120 Million Participants. Front. Cardiovasc. Med. 2021, 8, 640729. [Google Scholar] [CrossRef] [PubMed]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Y: Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Van der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.J.; Mulder, B.J. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2011, 8, 50–60. [Google Scholar] [CrossRef]
- Costain, G.; Silversides, C.K.; Bassett, A.S. The importance of copy number variation in congenital heart disease. NPJ Genom. Med. 2016, 1, 16031. [Google Scholar] [CrossRef] [Green Version]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Akerberg, B.N.; Pu, W.T. Genetic and Epigenetic Control of Heart Development. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef]
- Chang, C.P.; Bruneau, B.G. Epigenetics and cardiovascular development. Annu. Rev. Physiol. 2012, 74, 41–68. [Google Scholar] [CrossRef]
- Han, P.; Hang, C.T.; Yang, J.; Chang, C.P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 2011, 108, 378–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, A.; Makova, S.Z.; Barish, S.; Zaidi, S.; Mehta, S.; Drozd, J.; Jin, S.C.; Gelb, B.D.; Seidman, C.E.; Chung, W.K.; et al. Histone H2B monoubiquitination regulates heart development via epigenetic control of cilia motility. Proc. Natl. Acad. Sci. USA 2019, 116, 14049–14054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.Y.; Uebersohn, A.; Spencer, C.I.; Huang, Y.; Lee, J.E.; Ge, K.; Bruneau, B.G. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development 2016, 143, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, P.; Kunderfranco, P.; Greco, C.; Guffanti, A.; Stirparo, G.G.; Rusconi, F.; Rizzi, R.; Di Pasquale, E.; Locatelli, S.L.; Latronico, M.V.; et al. DOT1L-mediated H3K79me2 modification critically regulates gene expression during cardiomyocyte differentiation. Cell Death Differ. 2016, 23, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [Green Version]
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019, 21, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Manshaei, R.; Merico, D.; Reuter, M.S.; Engchuan, W.; Mojarad, B.A.; Chaturvedi, R.; Heung, T.; Pellecchia, G.; Zarrei, M.; Nalpathamkalam, T.; et al. Genes and Pathways Implicated in Tetralogy of Fallot Revealed by Ultra-Rare Variant Burden Analysis in 231 Genome Sequences. Front. Genet. 2020, 11, 957. [Google Scholar] [CrossRef]
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Buijtendijk, M.F.J.; Barnett, P.; Van den Hoff, M.J.B. Development of the human heart. Am. J. Med. Genet. C Semin Med. Genet. 2020, 184, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Klena, N.T.; Gibbs, B.C.; Lo, C.W. Cilia and Ciliopathies in Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, G.C.; Young, C.B.; Lo, C.W. Role of cilia in the pathogenesis of congenital heart disease. Semin. Cell Dev. Biol. 2021, 110, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, M. Heterotaxia, congenital heart disease, and primary ciliary dyskinesia. Circulation 2007, 115, 2793–2795. [Google Scholar] [CrossRef] [Green Version]
- Icardo, J.M.; Sanchez de Vega, M.J. Spectrum of heart malformations in mice with situs solitus, situs inversus, and associated visceral heterotaxy. Circulation 1991, 84, 2547–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.L.; Liu, J.C. Role of Notch signaling in the mammalian heart. Braz. J. Med. Biol. Res. 2014, 47, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chapman, G.; Moreau, J.L.M.; Ip, E.; Szot, J.O.; Iyer, K.R.; Shi, H.; Yam, M.X.; O’Reilly, V.C.; Enriquez, A.; Greasby, J.A.; et al. Functional genomics and gene-environment interaction highlight the complexity of congenital heart disease caused by Notch pathway variants. Hum. Mol. Genet. 2020, 29, 566–579. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, D.; Devriendt, K.; Driscoll, D.A.; Goldmuntz, E.; Gewillig, M.; Vlietinck, R.; Collen, D.; Carmeliet, P. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: A family based association study. J. Med. Genet. 2005, 42, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Waldner, M.J.; Neurath, M.F. Targeting the VEGF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 5–13. [Google Scholar] [CrossRef]
- Peters, T.H.; Sharma, V.; Yilmaz, E.; Mooi, W.J.; Bogers, A.J.; Sharma, H.S. DNA microarray and quantitative analysis reveal enhanced myocardial VEGF expression with stunted angiogenesis in human tetralogy of Fallot. Cell Biochem. Biophys. 2013, 67, 305–316. [Google Scholar] [CrossRef]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Guo, X.; Zhao, B.; Liu, J.; Da, M.; Wen, Y.; Hu, Y.; Ni, B.; Zhang, K.; Yang, S.; et al. Association analysis identifies new risk loci for congenital heart disease in Chinese populations. Nat. Commun. 2015, 6, 8082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Shi, Y.; Mo, X.; Xu, J.; Zhao, B.; Lin, Y.; Yang, S.; Xu, Z.; Dai, J.; Pan, S.; et al. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat. Genet. 2013, 45, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Bentham, J.; Topf, A.; Zelenika, D.; Heath, S.; Mamasoula, C.; Cosgrove, C.; Blue, G.; Granados-Riveron, J.; Setchfield, K.; et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 2013, 45, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Lahm, H.; Jia, M.; Dressen, M.; Wirth, F.; Puluca, N.; Gilsbach, R.; Keavney, B.D.; Cleuziou, J.; Beck, N.; Bondareva, O.; et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Agopian, A.J.; Goldmuntz, E.; Hakonarson, H.; Sewda, A.; Taylor, D.; Mitchell, L.E.; Pediatric Cardiac Genomics, C. Genome-Wide Association Studies and Meta-Analyses for Congenital Heart Defects. Circ. Cardiovasc. Genet. 2017, 10, e001449. [Google Scholar] [CrossRef] [Green Version]
- Cordell, H.J.; Topf, A.; Mamasoula, C.; Postma, A.V.; Bentham, J.; Zelenika, D.; Heath, S.; Blue, G.; Cosgrove, C.; Granados Riveron, J.; et al. Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of Fallot. Hum. Mol. Genet. 2013, 22, 1473–1481. [Google Scholar] [CrossRef]
- Bjornsson, T.; Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Norddahl, G.L.; Helgadottir, A.; Gretarsdottir, S.; Magnusdottir, A.; Danielsen, R.; Sigurdsson, E.L.; et al. A rare missense mutation in MYH6 associates with non-syndromic coarctation of the aorta. Eur. Heart J. 2018, 39, 3243–3249. [Google Scholar] [CrossRef]
- Helgadottir, A.; Thorleifsson, G.; Gretarsdottir, S.; Stefansson, O.A.; Tragante, V.; Thorolfsdottir, R.B.; Jonsdottir, I.; Bjornsson, T.; Steinthorsdottir, V.; Verweij, N.; et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat. Commun. 2018, 9, 987. [Google Scholar] [CrossRef] [Green Version]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yagi, H.; Onuoha, E.O.; Damerla, R.R.; Francis, R.; Furutani, Y.; Tariq, M.; King, S.M.; Hendricks, G.; Cui, C.; et al. DNAH6 and Its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. PLoS Genet. 2016, 12, e1005821. [Google Scholar] [CrossRef] [Green Version]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol. Cytogenet. 2020, 13, 16. [Google Scholar] [CrossRef]
- Manheimer, K.B.; Richter, F.; Edelmann, L.J.; D’Souza, S.L.; Shi, L.; Shen, Y.; Homsy, J.; Boskovski, M.T.; Tai, A.C.; Gorham, J.; et al. Robust identification of mosaic variants in congenital heart disease. Hum. Genet. 2018, 137, 183–193. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Trevino, C.E.; Holleman, A.M.; Corbitt, H.; Maslen, C.L.; Rosser, T.C.; Cutler, D.J.; Johnston, H.R.; Rambo-Martin, B.L.; Oberoi, J.; Dooley, K.J.; et al. Identifying genetic factors that contribute to the increased risk of congenital heart defects in infants with Down syndrome. Sci. Rep. 2020, 10, 18051. [Google Scholar] [CrossRef]
- Stone, N.R.; Gifford, C.A.; Thomas, R.; Pratt, K.J.B.; Samse-Knapp, K.; Mohamed, T.M.A.; Radzinsky, E.M.; Schricker, A.; Ye, L.; Yu, P.; et al. Context-Specific Transcription Factor Functions Regulate Epigenomic and Transcriptional Dynamics during Cardiac Reprogramming. Cell Stem Cell 2019, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; Del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Sohlmer, J.; Zhou, C.; Witman, N.; Leung, C.Y.; Mononen, M.; Bylund, K.; Gruber, P.; Chien, K.R. Population and Single-Cell Analysis of Human Cardiogenesis Reveals Unique LGR5 Ventricular Progenitors in Embryonic Outflow Tract. Dev. Cell 2019, 48, 475–490. [Google Scholar] [CrossRef] [Green Version]
- Mononen, M.M.; Leung, C.Y.; Xu, J.; Chien, K.R. Trajectory mapping of human embryonic stem cell cardiogenesis reveals lineage branch points and an ISL1 progenitor-derived cardiac fibroblast lineage. Stem Cells 2020, 38, 1267–1278. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diab, N.S.; Barish, S.; Dong, W.; Zhao, S.; Allington, G.; Yu, X.; Kahle, K.T.; Brueckner, M.; Jin, S.C. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes 2021, 12, 1020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071020
Diab NS, Barish S, Dong W, Zhao S, Allington G, Yu X, Kahle KT, Brueckner M, Jin SC. Molecular Genetics and Complex Inheritance of Congenital Heart Disease. Genes. 2021; 12(7):1020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071020
Chicago/Turabian StyleDiab, Nicholas S., Syndi Barish, Weilai Dong, Shujuan Zhao, Garrett Allington, Xiaobing Yu, Kristopher T. Kahle, Martina Brueckner, and Sheng Chih Jin. 2021. "Molecular Genetics and Complex Inheritance of Congenital Heart Disease" Genes 12, no. 7: 1020. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071020