
The Importance of Being PI3K in the RAS Signaling Network


Tumour-Stroma Signalling Laboratory, Centro de Investigación del Cáncer, Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Científicas (CSIC)-Universidad de Salamanca, Campus Miguel de Unamuno, 37007 Salamanca, Spain
*
Author to whom correspondence should be addressed.
Genes 2021, 12(7), 1094; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071094
Submission received: 21 June 2021
/
Revised: 6 July 2021
/
Accepted: 16 July 2021
/
Published: 19 July 2021
(This article belongs to the Special Issue RAS Signaling in Health and Disease)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Ras proteins are essential mediators of a multitude of cellular processes, and its deregulation is frequently associated with cancer appearance, progression, and metastasis. Ras-driven cancers are usually aggressive and difficult to treat. Although the recent Food and Drug Administration (FDA) approval of the first Ras G12C inhibitor is an important milestone, only a small percentage of patients will benefit from it. A better understanding of the context in which Ras operates in different tumor types and the outcomes mediated by each effector pathway may help to identify additional strategies and targets to treat Ras-driven tumors. Evidence emerging in recent years suggests that both oncogenic Ras signaling in tumor cells and non-oncogenic Ras signaling in stromal cells play an essential role in cancer. PI3K is one of the main Ras effectors, regulating important cellular processes such as cell viability or resistance to therapy or angiogenesis upon oncogenic Ras activation. In this review, we will summarize recent advances in the understanding of Ras-dependent activation of PI3K both in physiological conditions and cancer, with a focus on how this signaling pathway contributes to the formation of a tumor stroma that promotes tumor cell proliferation, migration, and spread.
1. Introduction
Ras proteins are the founding members of the Ras superfamily of GTPases, which in humans is composed of more than 150 members [1,2]. Ras proteins are membrane-bound small GTPases that act as molecular transducers, coupling cell surface receptors to intracellular effector pathways to regulate cellular processes such as cell proliferation, differentiation, migration, and apoptosis [3,4]. In humans, three Ras genes (H-ras, N-ras, and K-ras) encode four distinct Ras proteins: H-Ras, N-Ras, K-Ras4A, and K-RasS4B, the latter 2 resulting from alternative RNA splicing of the K-ras gen. These four Ras isoforms are ubiquitously expressed and are highly similar in primary sequence, structure, and biochemical properties [5,6]. They share 90% sequence identity in the G domain [7], being 100% identical in the N-terminus of this domain, termed the effector lobe, and sharing 82% of the sequence of the allosteric lobe [5,8]. In contrast, the C-terminal hypervariable region (HVR) shares little sequence similarity [7,9].
Alterations in Ras signaling are implicated in the development of different diseases, such as neurological disorders, developmental disorders, and autism. Additionally, Ras proteins are recognized as major oncogenes, as mutations in all three Ras genes occur in approximately 30% of human cancers [10]. Mutant Ras is a driver both in tumor initiation and tumor maintenance [5,7]. In general, K-Ras is the most frequently mutated isoform (accounting for 75% of Ras mutation in cancer), followed by N-Ras (17%) and H-Ras (7%) [10]. The frequency in which every isoform appears mutated varies by tissue type: a large percentage of adenocarcinoma of the lung (32%), pancreas (86%), and colon (41%) is driven by K-Ras mutations; 29% of melanomas are driven by mutations in N-Ras, while H-Ras mutations appears mutated in 5% of head and neck squamous cell carcinoma and 6% of bladder cancers [11].
2. Ras Activation and Downstream Signaling
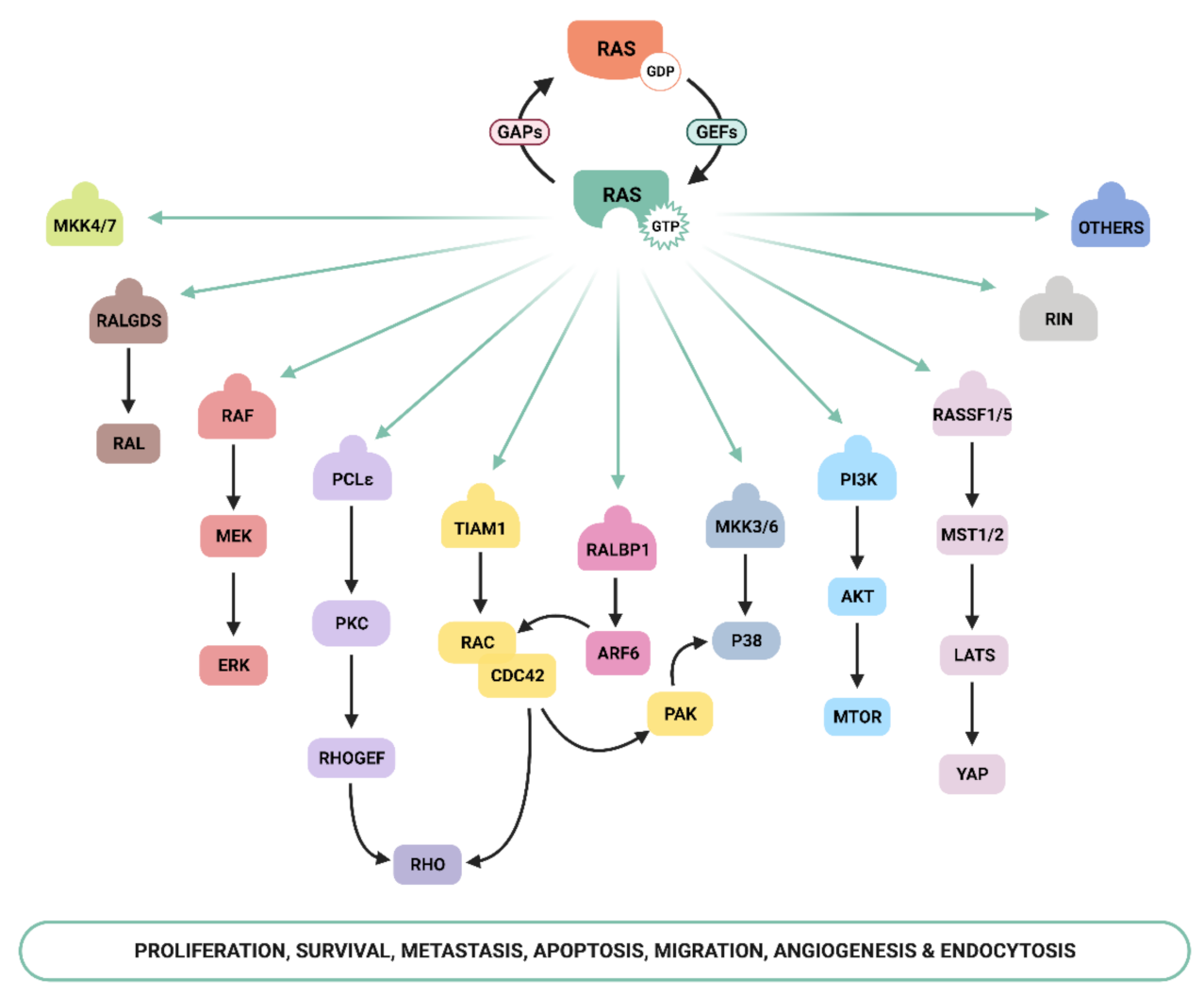
Ras signaling is activated by cellular receptors including receptor tyrosine kinases (RTKs), G-protein coupled receptors (GPCRs), cytokines receptors, and extracellular matrix receptors [12]. Ras proteins act as molecular switches that cycle between two conformational states: an active GTP-bound state and an inactive GDP-bound state. This GDP/GTP cycling is tightly controlled by two main classes of regulatory proteins: guanine-nucleotide-exchange factors (GEFs), which upon receptor stimulation promote the activation of Ras proteins by stimulating GDP for GTP exchange, and GTPase-activating proteins (GAPs), which stimulate Ras-mediated GTP hydrolysis and Ras inactivation [6,12,13]. GTP binding to Ras induces changes in conformation, mainly in two regions named switch I and switch II, that greatly increase the affinity of Ras for its downstream effectors [14,15].
Ras-GTP stimulates a wide range of downstream effectors, although the best characterized are mitogen-activated protein kinases (MAPK), phosphoinositide-3 kinase (PI3K) [16,17], and the Ral pathways [18] (Figure 1). The Raf–MEK–ERK signaling axis was the first Ras-effector that was described [19,20] and it regulates cell growth, differentiation, inflammation, and apoptosis [21]. Activated Ras promotes the translocation of the Raf serine/threonine kinase to the plasma membrane, where it is activated and phosphorylated by different protein kinases. Active Raf phosphorylates and activates the MEK1/2 kinase, which in turn phosphorylates ERK1/2 mitogen-activated protein kinase that eventually exert their function on a large number of downstream molecules [22]. The phosphatidylinositol 3 kinase (PI3K) pathway is the second best-characterized Ras effector and participates in the regulation of a wide range of cellular activities, including cell growth, proliferation, differentiation, migration, and apoptosis [23]. Genetic alterations in PI3K/AKT and Raf/MAPK/ERK pathway components leading to aberrant activation of signaling are frequent in cancer [21,24]. The functional importance of the Ras–PI3K pathway and its role in cancer will be extensively discussed in the next sections.
Apart from the above-mentioned effectors, an increasing number of molecules that specifically interact with Ras have been described, such as Ral-GEF, Rho-GTPases, Novel Ras effector 1A (NORE1A), Af6, phospholipase C (PLC), Ras and Rab interactor 1 (RIN1), T-cell lymphoma invasion and metastasis-inducing protein (Tiam), and growth factor receptor 14 (Grb1), although the precise physiological role is still not fully understood for many of them [25]. Recently, Ras signaling has been shown to interact with the Hippo-signaling effector YAP to facilitate tumorigeneses [26]. Thus, the list of Ras effectors continues to grow, and more interactors belonging to the family of Ras effectors are expected to be characterized in the next years [14,27,28,29,30].
3. PI3K Family
PI3Ks are a family of lipid kinases with a key role in the regulation of cellular functions such as development, cell growth, metabolism, mobility, apoptosis, and proliferation [31]. Genetic deregulation of PI3K activity is associated with many human conditions such as allergy, inflammation, diabetes, neurological disorders, heart disease, or cancer [32,33].
PI3Ks are grouped into three different classes (Class I, II, and III) on the basis of protein structure, tissue expression, and substrate preference. These classes share four homologous regions, although the kinase domain is the most conserved [34]. Class I PI3K is the best-characterized family and the one most clearly implicated in human cancer [24], although much remains to be learned about their coupling to upstream signals and their relative functional output. Class I PI3K is composed of heterodimers formed by a catalytic subunit, which contains the kinase domain that catalyzes production of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), and a regulatory subunit, which interacts with the catalytic subunit to downregulate substrate access and kinase activity [35]. Class I PI3K is further subdivided into Class IA and Class IB PI3Ks. The catalytic subunit of Class IA consists of p110α, p110β, or p110δ, which in humans are encoded by Pik3ca, Pik3cb, and Pik3cd, respectively. It can bind any of five different p85-like regulatory subunits (p85α, p55α, p50α, p85β, and p55γ). Class IB PI3Ks feature the p110γ catalytic subunit, which differs from the Class IA PI3K in its extreme N-terminus (lacking a p85 binding site) and binds either a p84 or p101 regulatory subunit. P110α and p110β isoforms are commonly expressed in all tissues, p110δ is mainly expressed in immune cells, and p110γ is expressed in immune cells and at low levels in the heart, pancreas, liver, and skeletal muscle [32,34,36].
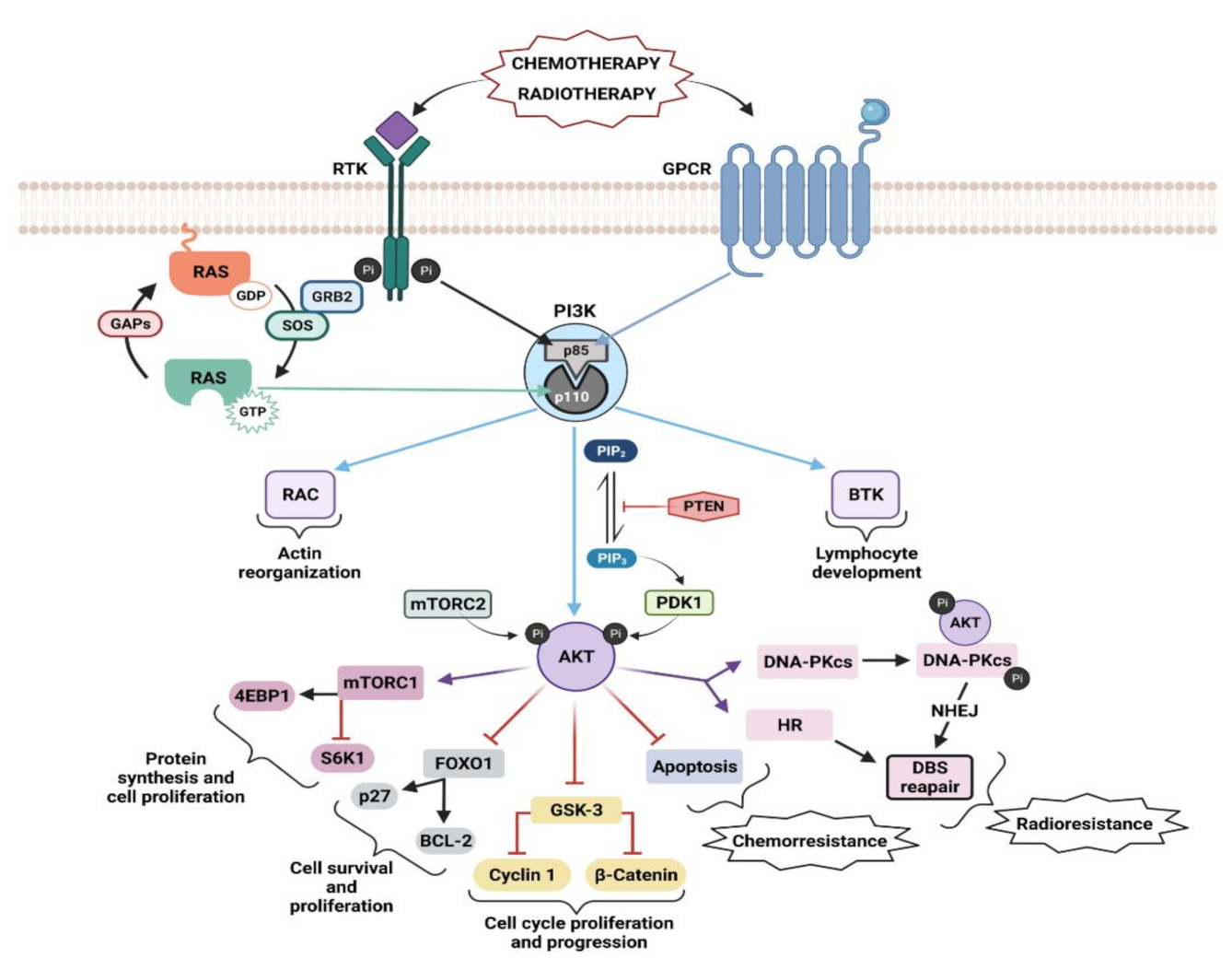
Class I PI3Ks are activated through different upstream mechanisms, such as RTKs, GPCRs, or GTPases, which induce recruitment of PI3K to the membrane (Figure 2) [23]. Engagement of the ligand to its receptor induces the dimerization and autophosphorylation at tyrosine residues of the RTKs and subsequent interaction with Src homology 2 (SH2) domain–containing molecules [37,38]. From here, PI3K could be activated in three different ways: (i) by direct binding of the regulatory subunit of PI3K, p85, to phospho-YXXM motifs (X indicates any amino acid) within the RTK [39], triggering activation of the p110 catalytic subunit of PI3K; (ii) by Growth Factor Receptor-bound Protein 2 (GRB2) mediated activation, in which GRB2 binds preferentially to phospho-YXN motifs of the RTK [40] and to the scaffolding protein GAB, which in turn can bind to p85; (iii) via binding to Ras: GRB2 binds and activates Son of Sevenless (SOS), which then activates Ras, and in turn activates p110 independently of p85. GRB2 can also exist in a large complex that contains SOS, Ras, and GAB or other scaffolding proteins, bringing these activators into close proximity to p110 PI3K [41]. It remains unclear which of these pathways predominates in different physiological situations as data show that GPCRs, RTKs, and Ras proteins exhibit a wide level of plasticity in terms of activating the various Class I PI3Ks [23]. Only p110α, p110δ, and p110γ isoforms are activated by the Ras subfamily of proteins, while p110β is activated by members of the Rho GTPase subfamily, specifically by Cdc42 and Rac1 [17,23,42,43]. PI3K isoforms also show differences when activated by direct binding to RTKs or GPCRs. In the case of RTKs-dependent, Ras-independent activation, the Src Homology 2 (SH2) domain of the regulatory subunits of PI3Kα, PI3Kβ, and PI3Kδ, can bind to phosphotyrosyl residues of RTK or adaptor proteins such as insulin receptor substrate (IRS1) [23,44]. GPCRs only activate PI3Kγ and PI3Kβ isoforms as a result of direct interaction of the regulatory subunit and the Gβγ subunit of the heterotrimeric G proteins, following activation of GPCRs [23,35,45,46,47]. The different mechanisms by which each isoform is activated may have a significant effect on the pathological and physiological processes it mediates. A rigorous review of upstream regulators of PI3K and their role in disease has been published by Wang and colleagues [35].
Once activated, Class I PI3K phosphorylates the 3-OH group of phosphatidylinositol 4,5-bisphosphate (PIP2), resulting in the generation of PIP3 [48] that acts as a second messenger and recruits proteins that contain a pleckstrin homology (PH) domain, such as serine–threonine kinase AKT and its activating kinase 3-Phosphoinositide-dependent protein kinase 1 (PDK1), to the cellular membrane [49,50]. PDK1 phosphorylates AKT at T308, leading to partial AKT activation. Phosphorylation of AKT at S473 in the carboxy-terminal hydrophobic motif by the mammalian target of rapamycin complex 2 (mTORC2), fully activates AKT [22,51,52]. Phosphorylated AKT subsequently phosphorylates a large number of downstream targets that play an important role in the regulation of apoptosis and cell survival (Figure 2).
Phosphorylated AKT activates the mammalian target of rapamycin (mTOR) via phosphorylation and inactivation of proline-rich AKT substrate of 40 kDa (PRAS40) and tuberous sclerosis protein 2 (TSC2) [53]. mTOR phosphorylates 4E binding protein 1 (4EBP1) and ribosomal protein S6 kinase (S6K1), responsible for ribosomal S6 protein (S6/RPS6) phosphorylation [37] to promote protein synthesis and cellular proliferation [54]. AKT exerts an inhibitory phosphorylation on the glycogen synthase kinase-3 (GSK-3), inducing the expression of cell-cycle regulators such as c-Myc, cyclin D1, and cyclin E and thus promote cell cycle progression [55]. Another important target of AKT is the proapoptotic forkhead box transcription factors (FOXO) FOXO1, FOXO3, and FOXO4. AKT phosphorylates them, blocking the transcription of target genes that promote apoptosis (such as BIM), cell-cycle arrest (such as p21 and p27), and metabolic processes (such as sestrin3 and G6PC) [55,56]. AKT controls apoptosis through the inhibition of the proapoptotic activity of the BAD and BAX factors, from the the B-cell lymphoma 2 (BCL-2) family [57,58,59]. The murine double minute 2 (MDM2) is also phosphorylated and activated by AKT. MDM2 negatively regulates p53, which promotes cell survival [60,61]. Importantly, AKT activates the IKK/NF-κB signaling pathway, promoting many steps of cancer initiation and progression [55,62,63,64].
TEC family tyrosine kinases such as Bruton’s tyrosine kinase (BTK) and Targeting Interleukin-2-Inducible T-cell Kinase (ITK) are also PI3K effectors with a key role in lymphocytes [65]. BTK has a critical role in B-cell function and malignancy [66]. BTK is involved in the regulation of the developmental progression of pre-B-cells [67], and it is essential for B-cell receptor (BCR)-mediated proliferation and survival of mature B-cells [68,69]. Mutation of genes encoding BTK are implicated in human immunodeficiency disease X-linked agammaglobulinemia (XLA) [70,71], while BTK is abundantly expressed in B-cell leukemias and lymphomas [72]. The initial link between PI3K, BTK, and B-cells was provided by mouse knockout studies showing that the deletion of Pik3r1 or Pik3cd caused defects in B-cell development and survival similar to those in mice lacking BTK [73]. Furthermore, pharmacological inhibitors of BTK and p110δ have shown a strong convergence of clinical activity in cancer, with best responses in malignancies of mature B-cells [74,75,76,77]. PI3K-dependent BTK signaling is a key example of a PI3K network that has emerged as an effective therapeutic target in cancer [78].
PI3K also targets GEFs involved in the activation of Rho/Rac/cdc42 GTPases family proteins [3,82]. For example, both pharmacological and genetic PI3K-α inhibition in endothelial cells reduced RhoA activation, which correlated with a migration and tail retraction defect [83]. In epithelial cells, growth factors stimulate the PI3K-dependent activation of Rac, leading to disruption of the actin cytoskeleton, release of filamentous actin-bound aldolase A, and an increase in aldolase activity [84].
PI3K pathway activity is negatively regulated by the phosphatase and tensin homolog (PTEN), a phosphatase that dephosphorylates and hydrolyses the secondary messenger PIP3, converting it back to PIP2. Loss of function mutations in PTEN result in enhanced PI3K signaling, which is associated with oncogenic cellular transformation and cancer [23,85]. Similar to PTEN, the SH2 domain-containing enzyme inositol 5′-phosphatase (SHIP) is also a negative regulator of PI3K, which specifically hydrolyses the 5-phosphate group from PIP3 [35]. A tumor suppressor role of SHIP1 has only been described in a single murine B-cell lymphoma model driven by oncogenic c-Myc and in lymphatic metastasis of breast cancer; no additional studies have demonstrated a tumor suppressor role of SHIP1 in other spontaneous malignancies in humans [86,87].
4. Ras–PI3K Interaction
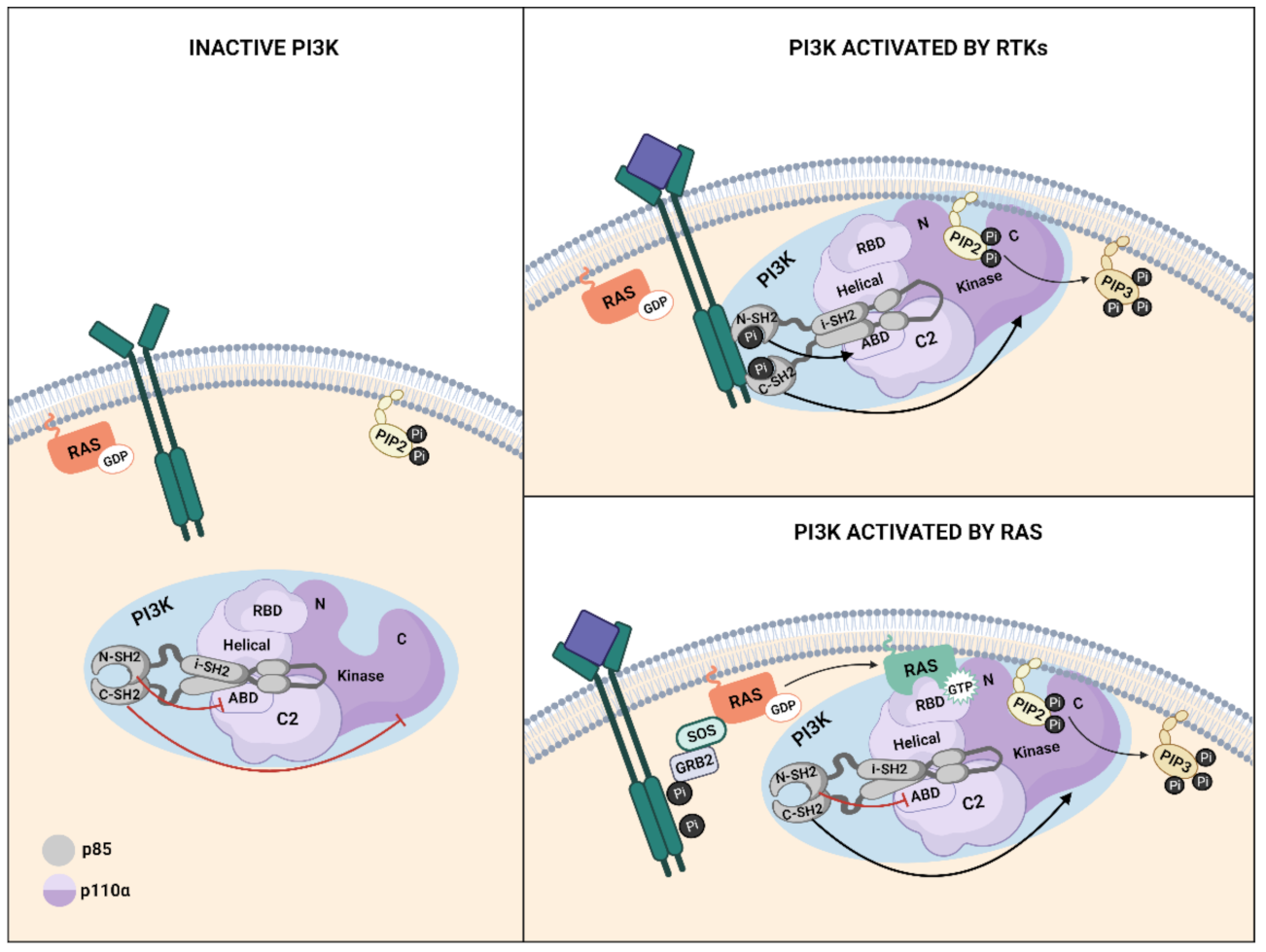
Ras-mediated PI3K activation in response to growth factors requires two steps, the first of which is phosphorylation of the RTK and, in some cases, adaptor proteins, and secondly, activation of Ras small GTPases. Interaction between Ras and Class I PI3K is mediated by the binding of the Ras binding domain (RBD) present in the catalytic subunit of PI3K and the Ras switch region I (SW1) and II (SW2) that form part of the effector lobe of Ras [17,88]. Upon GTP loading of Ras, both switch regions suffer significant structural changes, increasing the affinity of Ras for effector targets [49,89]. Direct interaction between GTP-bound Ras and p110α augments the activity of p110α, possibly by inducing a conformational change at the substrate binding site, stimulating its catalytic activity [23] or by mediating a closer interaction with the plasma membrane [17,90]. Recent studies suggest that, at least for the RTK-induced activation of mammalian PI3K, Ras mostly acts to stabilize PI3K at the plasma membrane, whereas PI3K is allosterically activated by the RTK [91,92].
The multidomain architecture in PI3K regulatory and catalytic subunits contributes to the mechanisms through which PI3Kα activity is controlled as interactions between both subunits stabilize the overall structure and keep PI3K in the inactive state [93,94] (Figure 3). The N-terminal-SH2 (nSH2) and the Inter-SH2(iSH2) domains of p85 are required for full inhibition of p110α lipid kinase activity [95,96]. When the ligand binds to RTK, the phosphorylated tyrosine (pY) motifs in the receptors bind to the nSH2 domain of p85, thus disrupting the inhibitory contact between the nSH2 and the catalytic subunit and inducing a conformational change in p110α that leads to its activation [95]. Specifically, after nSH2 release, the C-lobe of the kinase domain moves away from the C2 domain, exposing the p110α membrane binding surface to membrane interactions. The activation loop in the kinase domain becomes more flexible and approaches ATP, generating an active PI3Kα conformation for substrate catalysis [36] (Figure 3). For Ras-mediated PI3K activation, the inhibition exerted by p85 is relieved through dissociation of p110 and p85, with the nSH2 domain of p85 playing a key role [96,97] (Figure 3). Recently, computational analysis of K-Ras4B-mediated activation of p110α has shown that K-Ras4B/RBD binding disrupted the interactions along the p110/p85 interface through the induction of overall structural displacement of both nSH2 and iSH2 in p85 from the neighboring functional domains in p110 including the C2, helical, and kinase domains. This results in the exposure of the kinase domain and facilitates the full activation of PI3Kα [98].
Ras-mediated regulation of PI3K plays a role in many cellular processes involved in both normal physiology and disease [61]. In all these contexts, Ras-stimulated production of PIP3 results in membrane recruitment and, in many cases, activation of PH domain-containing proteins, including PDK1 and AKT kinases, GEFs for Rac small GTPases, and BTK. The best-characterized roles of Ras–PI3K signaling in eukaryotes are the regulation of AKT function in cell survival and growth, as well as the remodeling of the actin cytoskeleton [99,100]. The Ras–PI3K pathway can also regulate the actin cytoskeleton by promoting Ras signaling in various cellular contexts [91,101].
4.1. Ras–PI3K Interaction in Cell Growth and Apoptosis
Significant efforts have been made to understand the role of Ras and PI3K in the regulation of different cellular events and its involvement in cancer since the discovery of their interaction [102]. PI3K activation by different upstream receptors makes it difficult to uncover the specific role of Ras-dependent activation of PI3K in the control of physio-pathological processes. Adding to this complexity, Ras can activate a large number of effectors, each of them contributing to the regulation of a specific cellular event. Thus, fully understanding of the specific roles played by this interaction has been challenging.
Different approaches have been used to examine the functional relevance of Ras-dependent activation of PI3K. Traditionally, the Ras–PI3K signaling pathway was studied using genetic animal models or cell lines in which Ras was constitutively activated or silenced [103,104,105,106], and AKT phosphorylation was used as a readout for Ras-dependent PI3K activation. These tools were usually complemented with pan- or isoform-specific PI3K and ERK small molecule inhibitors to allow the evaluation of the individual roles of Ras effectors in the final physiological response [107,108]. The general conclusion drawn from these studies was that Ras requires PI3K activation to regulate cell viability, through the control of proliferation and apoptosis [65,104,109], and to induce transformation of epithelial cells [99,103]. Thus, it was concluded that dysregulation of the processes controlled by the Ras–PI3K pathway or mutation on any of its components leads to oncogenesis.
The use of PI3K inhibitors to study Ras-downstream signaling has also been used to determine the functionality of this effector pathway. However, these inhibitors block PI3K activity independently of how it is activated. To circumvent this issue, Rodriguez-Viciana et al. designed different Ras mutants that are specifically unable to interact with each of its effectors [100]. This study showed that mutations in different residues of the effector-binding domain of Ras altered the transformation capability of oncogenic Ras and that both Raf and PI3K activation are critical to induce fibroblast transformation by Ras. Additionally, it demonstrated that PI3K, but not Raf, modulates membrane ruffling and cytoskeletal reorganization. Subsequent studies using these mutants showed that Ras–PI3K interaction protected fibroblasts from c-Myc-induced apoptosis as effectively as constitutive activation of p110α, and this protection was abolished by the treatment of fibroblasts with a PI3K inhibitor [109].
Important advances in the understanding of the specific role of Ras-dependent activation of PI3K were achieved by the generation of different mouse models in which Ras binding to the catalytic unit of PI3K was prevented. Suire et al. generated a mouse model in which five residues of the Ras-binding domain of p110γ were altered (T232D, K251A, K254S, K255A, and K256A). Using this model, they established that Ras interaction with p110γ is not necessary for mouse development, since mutant mice were viable, fertile, of normal size, and with generally normal blood counts. However, the study showed that neutrophils in which p110γ cannot interact with Ras showed reduced PIP3 accumulation, AKT phosphorylation, chemokinetic responses, and reactive oxygen species (ROS) production in response to GPCR agonists [101].
Studies in Drosophila showed that Ras–PI3K interaction is essential during development [110,111]. Substitution of four amino acids in the Ras binding domain of Dp110 (the only Class I PI3K in Drosophila) (T231D, K250A, R253A, and K257A) prevented its interaction with Ras. The resulting mutant flies were viable, but smaller in size. Female flies were less fertile than wild-types (WTs), pointing to a role of Ras/PI3K interaction in the response to growth-stimulating stimulus. Furthermore, AKT activation was diminished in response to insulin in the brain and imaginal discs, and basal AKT activation was downregulated in the ovaries [110]. Further studies using this model showed that Ras activation of PI3K is also required to decrease motor neuron excitability, but not for the PI3K-dependent increase in nerve terminal growth [112].
Generation of a mouse model in which the binding of Ras to the p110α subunit of PI3K was prevented by the introduction of two point mutations (T208D and K227A) in the RBD domain of the endogenous Pik3ca gen revealed additional roles of Ras-dependent activation of PI3K. In these mice, RBD mutations did not affect the basic enzymatic activity of p110α, coupling to the p85 regulatory subunit, expression of PI3K components (p85, p110α and p110β), or interaction with other Ras effectors. However, the homozygous mutant mice died shortly after birth due to deficient lymphatic vasculature development, and the few surviving mutant adults exhibited reduced body weight compared to WT [113]. The importance of a functional p110α subunit in vascular development during embryogenesis was also shown with a mouse model where ubiquitous or endothelial cell-specific inactivation of p110α led to embryonic lethality at mid-gestation, associated with severe defects in the vascular remodeling [83]. Conversely, mice with inactive p110β or p110γ were viable and fertile, without obvious vascular defects, confirming that p110α plays a unique role among Class IA PI3K isoforms in embryonic vascular morphogenesis and remodeling [83] and that this effect may be mediated by Ras. These results also suggested that cellular responses during embryogenesis and adulthood require different growth signals, and Ras activation of PI3K may have a function in the regulation of cell responses to different growth stimulus. This idea is reinforced by the observation that, in fibroblasts lacking Ras–PI3K interaction, epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF2) fail to properly induce AKT activation, whereas platelet-derived growth factor (PDGF) does not [113,114], and migration is impaired in response to EGF, FGF2, hepatocyte growth factor (HGF), and insulin but not in response to PDGF [115]. Furthermore, insulin-induced activation of AKT and ERK is also reduced in hepatocytes in which H-ras is silenced [116], indicating that different Ras isoforms may also differentially regulate growth factor-induced signaling pathways.
It is well established that PI3K is one of the principal effectors of Ras in the regulation of the cell cycle, being the main effector promoting transition from G0 to G1 phase in quiescent cells [117,118,119], as well as entrance of cells into S phase [117]. In parallel, Ras regulates pro-survival signals via PI3K activation of AKT. Phosphorylated AKT, in turn, phosphorylates a number of substrates that play a role in the regulation of apoptosis, such as BAD [57,120,121], NF-kB [122,123,124], or FOXO [125]. Thus, by stimulating proliferation and preventing apoptosis, Ras–PI3K signaling supports tumor growth.
The role of PI3K signaling in apoptosis inhibition is not restricted to the classical Ras isoforms. R-Ras is a Ras family member that functionally differs from H-Ras, N-Ras, and K-Ras [126] and is highly expressed in the vascular endothelial cells [127], where it promotes lumenogenesis, supports the lumen structure with the stabilized microtubules [128], and promotes vessel maturation by endothelial barrier stabilization and pericyte association [129,130]. Using human umbilical vein endothelial cells (HUVEC) cells, Takino et al. described that Ras guanyl nucleotide releasing protein 2 (Ras-GRP2), a Ras GEF, inhibits apoptosis by inhibiting BAX activation in a PI3K-dependent way [131]. The authors demonstrated that Ras-GRP2 suppresses BAX activation-induced apoptosis by promoting HK-2 translocation to mitochondria via R-Ras/PI3K/AKT signaling pathway. The BAX pathway is involved in apoptosis in endothelial cells in conditions of hyperglycemia and high methylglyoxal levels as a trigger of atherosclerosis and in lipopolysaccharide-induced apoptosis caused by inflammation [132,133]. Therefore, inhibition of BAX translocation by Ras-GRP2/R-Ras/PI3K/AKT/HK-2 may result in a survival benefit in these conditions.
4.2. Ras–PI3K Signaling in the Immune System
Most studies on the role of PI3K in the immune system focus on PI3Kγ and PI3Kδ isoforms, while the function of PI3Kα in immune cells is less understood. The importance of PI3Kγ and PI3Kδ isoforms for the normal activity of the immune system has been revealed in mice lacking functional versions of PI3Kγ [47,134] or PI3Kδ [135,136], which are viable but show significant defects in both innate and adaptive immunity [137]. Mice lacking 110δ exhibit impaired B-cell development and humoral immune responses, reduced T-independent antibody responses, and relatively normal numbers of thymocytes [135,136,138], while p110γ-deficient mice have a modest reduction in thymic cellularity and no alterations in development or function of B-cells [134].
The contribution of Ras signaling to 110δ or p110γ in the maturation of the immune system during development has not been determined; however, results from studies using N-Ras knockout (KO) mice overlap with those obtained with p110γ deficient mice [134,139]: both N-Ras and p110γ regulate proliferation and IL-2 production of T-cells and promote an inflammatory response after stimulation with an infectious agent.
PI3Kγ can operate as a p110γ/p84 or p110γ/p101 complex, which confers PI3Kγ signal-specificity [140]. These complexes show some differences in tissue distribution. The p84 subunit is highly expressed in the immune system but also in other tissues and organs, such as the heart, pancreas, and the central nervous system, while p101 shows a more restricted expression profile in the immune system but was below the detection limit in other body compartments [141,142,143]. Such differences of expression and distribution in human tissues point to different cellular functions of both enzymes. In fact, p84 and p101 regulatory subunits show a distinct function in specific cell types, such as mast cells, neutrophils, and cardiomyocytes [142,143,144,145]. For example, neutrophils lacking p84 have selective defects in p110γ-dependent oxidase activation, but neutrophils lacking p101 show selective defects in p110γ-dependent motility [145]. Given the different role that p84 and p101 can play in the control of cellular functions, it seems indispensable to understand the mechanisms that drive their activation.
The PI3Kγ isoform can be activated by GPCRs or by Ras-dependent mechanisms [35], but it is unclear which of the two mechanisms is more relevant [146]. There is a general agreement that p101/p110γ is more sensitive to activation by Gβγ than p84/p110γ, which may be due to a decrease in the affinity of p84 to Gβγ [142,147]. In agreement with this, studies in HEK cells have suggested that Ras is an indispensable co-regulator of p110γ/p84, which requires Ras co-stimulation for full activation, whereas p110γ/p101 activation is mainly mediated by interaction with the Gβγ subunit of GPCRs, without any input from Ras [140]. In apparent contradiction to this, data obtained with primary neutrophils bearing mutations that prevent interaction of Ras with the RBD domain of p110γ show that Ras-GTP is an important regulator of p101/p110γ and not only of p84/p110γ [101,145]. These data illustrate that p84 and p101 variants of PI3Kγ are stimulated by Ras and Gβγ to different degrees depending on cell type, stimulus, and Ras isoform involved and that these differences shape cellular responses.
Ras–PI3K signaling has been shown to regulate different functions in neutrophils. Neutrophils in which PI3Kγ cannot bind to Ras (p110γDASAA/DASAA) and p101−/− neutrophils (used as a specific mean of assessing the role of Gβγs in the activation of PI3Kγ) exhibited a reduction of PIP3 accumulation, AKT activation in response to GPCR agonists, and reduced migration. However, loss of Ras binding to PI3Kγ specifically reduced neutrophils’ ability to produce ROS after stimulation with the peptide N-formyl methionyl–leucyl–phenylalanine (fMLP) or the complement fragment (C)5a (C5a) [101]. Further investigation confirmed that, at least in neutrophils, Ras induces ROS formation via two Ras-GEFs: Ras guanyl releasing protein 4 (Ras-GRP4) was required for fMLP-stimulated ROS formation in unprimed neutrophils, while SOS1/2 have a role in primed neutrophils [148]. In vivo studies demonstrated that Ras signaling in the absence of GPCR stimulation is not sufficient to activate PI3Kγ, and Ras interaction with p110γ is required for full GPCR stimulation of PI3Kγ, suggesting that Ras combines synergistically with inputs from Gβγ to determine PI3Kγ activity [149].
Ras signaling to PI3K is also relevant for the control of allergies. Mast cells are key effectors in the pathology of allergic disease [150]. In mast cells, the pharmacological inhibition of Ras with a farnesyltransferase inhibitor (FTI-227) reduced AKT phosphorylation upon activation with adenosine. Functionally, FTI-277 attenuated degranulation of IgE/antigen-activated mast cells upon co-stimulation with adenosine, reduced expression of tumor necrosis factor α (TNF-α) and IL-6, and impaired cell migration [146]. These data suggest that the development of specific p110γ/p84 targeting strategies for mast cell-related diseases will presumably have limited effects on p101-dominated immune cells, thus reducing side effects.
5. Ras–PI3K Interaction in Cancer
Mutations in the Ras genes were first reported in cancer over 30 years ago and numerous studies have since validated mutant Ras as a driver of tumor initiation and maintenance [151]. Activating point mutations in the genes encoding the Ras subfamily are found in approximately 27% of all human cancers, with 98% of the mutations occurring at one of three mutational hotspots (G12, G13, and Q61), regardless of the isoform involved [5,10]. These mutations disrupt GAP-mediated GTP hydrolysis, which results in an accumulation of constitutively GTP-bound Ras in cells and an exacerbated activation of effector pathways even in the absence of upstream stimulation [10,11]. Additional mechanisms of Ras activation in cancer include (i) perturbation in GDP–GTP regulation; (ii) loss of GAPs, such as neurofibromin (NF1), and (iii) persistent RTK-mediated activation of GEFs [5].
PI3K also plays a critical role in cancer. Mechanisms that enhance PI3K activity include mutations in proteins upstream of PI3K (such as Ras or RTKs), mutational activation of PI3K itself (mainly mutations in the Pik3ca gen), or loss of function of its inhibitor PTEN [23]. In particular, mutations in Pik3ca are the second most common alteration in human tumors [35,152]. Gene insertions, deletions, and somatic missense mutations in Pik3ca are frequent in colon, breast, brain, liver, stomach, and lung cancers [153]. There are two common “hotspots” for mutations in Pik3ca, H1047R, which enhances the interaction of the kinase domain with membranes and bypasses the requirement for association with Ras [154], and E525/E545K, which disrupts the inhibitory interface with the n-SH2 domain of the regulatory subunit [23,95,155]. Other mutations that activate PI3K occur in the Pik3r1 and Pik3r2 genes, coding for the regulatory subunits p85α and p85β, respectively. Mutations in these genes reduced the ability to interact with and stabilize the p110 catalytic subunit in type IA PI3Ks [156]. Aberrant PI3K activation can also occur due to loss-of-function mutations and deletions in PTEN, which is the third most frequent alteration in human tumors [35]. Ras and Pik3ca mutations are mutually exclusive in endometrial and breast cancers, but co-exist in colorectal and lung tumors [31,157,158,159], suggesting that the acquisition of both mutations may confer additional characteristics that are beneficial for the tumor. For example, in colorectal cancer, coexistant K-Ras and PI3K mutations decrease sensitivity to PI3K and mTOR inhibition [158], and in lung cancer, the addition of a constitutively active PI3K mutant to epidermal growth factor receptor (EGFR) mutant tumors confers gefitinib resistance in vitro [160]. Thus, determining Pik3ca mutation status and its coexistence with other gene mutation may be helpful to predict response to targeted therapy.
The initial indications that Ras interaction with PI3K could regulate tumorigenesis were obtained by in vitro studies showing its role in oncogenic cell transformation and cell survival [65,100,103,161]. Since then, a large number of studies have shown that PI3K activation regulates different aspects of Ras transformation [162,163]. To provide some specific examples, one study showed that activation of PI3K/AKT pathway is involved in intestinal epithelial cell transformation following induction of oncogenic Ha-RasVal12: pharmacological inhibition of PI3K activity exerted an anti-neoplastic effect on Ras-expressing intestinal epithelial cells, which included stimulation of apoptosis, reversal of morphological transformation, and accumulation of cells in the G1 phase of the cell cycle [103]. PI3K is also required for Ras transformation of fibroblasts and acts upstream of Rac GTPase to induce cortical actin rearrangement, membrane ruffling, and lamellipodia formation in oncogenic Ras-transformed cells [100]. Using a dominant negative mutant of Ras and PI3K inhibition, it was demonstrated that Ras and PI3K activation participates in ghrelin-caused proliferation of human colon cancer cells through the activation of the AKT/mTOR pathway [65]. Moreover, Ras–PI3K signaling regulates prostaglandin E2 inhibition of apoptosis in cancer cells [161].
In vivo, Ras activation of PI3K signaling is essential for lung carcinogenesis driven by oncogenic mutation of K-Ras and the formation of skin tumors induced by activating mutations of H-Ras [113]. However, once tumors are established, they are less dependent on RAS-PI3K signaling and disruption of their interaction only leads to partial tumor regression, followed by long term stasis [164]. Similar results were obtained in lung tumors developed in response to K-Ras activation with normal PI3K signaling, in which established tumors did not reduce in size when mice were treated with NVP-BEZ235, a dual pan-PI3K/mTOR inhibitor, even though tumors driven by expression of activated PI3K did regress upon treatment with this drug [160]. These data suggest that additional K-Ras activated pathways cooperate with Ras to promote tumor maintenance and development. Combination of PI3K and MEK inhibition caused impressive tumor regression, albeit accompanied by significant toxicity [164,165]. Similarly, subcutaneous tumors formed by tumor cells in which Ras preferentially binds to PI3K continue to grow even in the absence of oncogenic Ras signaling. However, if tumors are formed by cells in which Ras preferentially binds to Raf or RalGEF, they stop growing or even regress in some instances. In this setting, the activation of the three pathways was required for Ras-induced tumor initiation, suggesting that Ras-dependency of downstream pathway activation may vary with tumor stage and tumor type [166]. Ras–PI3K interaction is also required for tumor progression of WT Ras cancers since removal of Ras–PI3K interaction in an EGFR mutant lung cancer model caused important tumor regression [114]. These results suggest that direct targeting of the Ras-binding domain of p110α may be effective in K-Ras-driven lung tumors and in WT-mutant lung tumors. According to the observation that PI3K mediates oncogenic signaling in Ras-WT cancers, Molina-Arcas and colleagues showed that PI3K is critical for survival of both K-Ras mutant and K-Ras WT NSCLC cells, as PI3K inhibition induced loss of cell viability irrespective of the genotype [167]. However, opposite results on the role of oncogenic Ras on PI3K activation were obtained in colorectal cancer cells carrying mutations in K-Ras, where knockdown of K-Ras did not suppress AKT phosphorylation and PI3K/AKT pathway required insulin-like growth factor I receptor (IGF-IR)-induced activation. This is therapeutically important as, while suppression of mutant K-Ras is not sufficient to downregulate PI3K/AKT, IGF-IR inhibition could suppress PI3K signaling in K-Ras mutated colorectal cancers [168]. In summary, PI3K is not under the sole control of mutant K-Ras, and consequently, to discern the mechanism of PI3K activation in different cancers seems to be essential for the development of new drugs.
p110α is also critical for K-Ras-driven pancreas carcinogenesis as p110α inactivation prevents mouse lethality and the appearance of all types of pancreatic lesions induced by mutated K-Ras [169]. Supporting these data, Eser et al. demonstrated that oncogenic Pi3kca mouse models show similar patterns of acinar-to-ductal metaplasia (ADM), pancreatic cancer progression, and activation of key downstream effectors of PI3K to the K-Ras G12D model, indicating that PI3K signals downstream of mutated K-Ras in pancreatic cancer [170]. Further, PDK1, AKT, or PI3K inhibition resulted in normal life expectancy and inhibition of pancreas cancer development in the K-Ras G12D model.
The array of proliferative signals generated by oncogenic Ras culminates with the upregulation of several transcription factors that are required for cell cycle entry and progression, including FOS, serum response factor (SRF), the leucine zipper protein JUN, the ETS domain-containing transcription factor ELK1, activating transcription factor 2 (ATF2) and NF-κB [171,172,173,174,175,176]. In turn, these factors trigger the expression of cyclin D1 [177,178,179]. Although initial studies attributed the stimulation of cyclin D1 transcription by Ras to the activation of the Raf–MAPK pathway, it has become evident that additional levels of control are achieved through the Ras activation of PI3Ks, the RHO family of GTPases Rac1, and the Ral-guanine nucleotide dissociation stimulator (GDS) family of GEFs [19,177,178,180,181,182,183]. In addition to stimulating cyclin D1 gene transcription, oncogenic H-Ras regulates the metabolic stability of the cyclin D1 protein through PI3K-dependent inhibition of GSK3β, the kinase that is responsible for the phosphorylation and the consequent ubiquitylation and proteasomal degradation of cyclin D1 [184].
Numerous molecular abnormalities that result in constitutive activation of the PI3K pathway have been reported in hematologic malignancies, demonstrating the importance of targeting PI3K in leukemia and lymphomas [185]. However, there are few data describing the requirement of PI3K activation in Ras-mutated hematological malignancies. A possible link between Ras-p110γ interaction and the development of T-cell acute lymphoblastic leukemia (T-ALL) was shown by Janas et al. [186]. The authors demonstrated that thymocytes harboring a mutation of p110γ that blocked its interaction with Ras have a reduced proliferation rate in response to CXC Motif Chemokine Receptor 4 (CXCR4, a receptor implicated in β-selection of thymocytes) and consequently were defective when undergoing β-selection in comparison with WT counterparts. Thus, in thymocytes, p110γ is a key target of Ras signaling that is required for an optimal proliferative response at β-selection, and therefore this pathway may be implicated in the development of T-ALL, which typically presents the expansion and dissemination of cells arrested between β-selection and double positive stages. p110α may also have a role in Ras-driven leukemia, since the loss of p110α caused a significant improvement in survival in a murine model of leukemia induced by oncogenic K-Ras mutations and prolonged the latency of the myeloproliferative neoplasm [105].
As metabolic disorder was pointed out as a hallmark of cancer, great attention was paid to the crucial role of oncogenic Ras in tumorigenesis, where it orchestrates a metabolic reprogramming of tumors [187]. Oncogenic Ras plays an important role in regulating cancer cell metabolism by triggering several main metabolic changes [188]. Activation of Ras upregulates growth-promoting pathways controlled by PI3K-mTOR, and growing evidence suggests that there is a tight correlation between metabolic rewiring in cancer and these pathways. For example, Ras mutant cells are dependent on sufficient glucose uptake and PI3K can increase glycolysis through activation of AKT and stabilization of Hypoxia-inducible factor 1 (HIF-1) [189,190]. Additionally, one of the effectors of PI3K, mTORC1, can drive a complex pathway to support glycolysis, the pentose phosphate pathway (PPP), and nucleotide biosynthesis [191,192]. Analysis of different genes with driver mutations that were linked to the regulation of metabolic pathways in K-Ras-driven cancers, such as Pik3ca [49], KMT2D [193], PTEN [194], and IDH1 [195], suggested a theoretical broader transcriptional and signaling circuit coordinated by Ras together with p110α, NF1, and PTEN for the tight regulation of metabolic pathways [188]. Given the relevance of Ras–PI3K signaling in metabolic rewiring, the different levels of interplay between the PI3K–AKT and K-Ras–MAPK signaling cascades [164] should be considered, including for potential therapies. Additional studies are needed to understand how the cross-talk between these two pathways contribute to the metabolic rewiring needed to sustain uncontrolled proliferation in cancer [196].
5.1. Ras–PI3K Signaling in Migration and Metastasis
The metastatic spread of the primary tumor accounts for over 90% of cancer deaths [197]. Metastasis formation requires complex interactions between cancer cells and multiple microenvironments [198]. The epithelial-to-mesenchymal transition (EMT) process is a crucial mechanism in the progression of tumor metastasis and is characterized by the loss of cell–cell adhesions and the acquisition of cell–matrix interactions [199], which allow cancer cells to become mobile and invasive, enabling metastasis and chemotherapy resistance [197]. Both Ras and PI3K/AKT pathways are well documented to drive EMT, invasion, and metastasis [200,201,202,203,204,205]. However, the importance of Ras–PI3K interaction in Ras-induced EMT is not clear, and the signaling pathways by which Ras contributes to EMT are controversial [206]. On one hand, K-Ras silencing inhibited EMT and proliferation of breast cancer cells, while promoting apoptosis, but these effects were reversed when mutant cells were treated with a PI3K/AKT agonist [99]. Furthermore, PI3K-AKT signaling was suggested to be required for transforming growth factor β (TGF-β) induced EMT and cell migration in mammary epithelial cells [207]. In the same line, another study demonstrated that H-Ras-driven tumors induce PI3K/AKT-dependent ß-catenin activation and that this is associated with increased cell proliferation, EMT, and cell invasion [208]. These data would suggest that PI3K acts downstream of Ras to regulate EMT, proliferation, and apoptosis. Additional supporting evidence of the role of PI3K as a mediator of Ras-induced EMT was obtained in esophageal cancer cells, where depletion of Ras-GAP SH3 domain binding protein (G3BP1, a protein that modulate the Ras signaling pathway via interacting with the SH3 domain of RasGAP and found overexpressed in some cancers [209,210,211]) represses invasion and migration potential via EMT suppression. This was accompanied by a significant decrease in the levels of p-AKT and p-GSK3β. The overexpression of G3BP1 had the opposite effect, which was revered by pharmacological inhibition of PI3K [212]. On the other hand, other studies have established that PI3K is not required for Ras-induced EMT. Janda and colleagues demonstrated that while the PI3K pathway is required to induce rapid tumor growth and enhanced proliferation of mammary epithelial cells expressing oncogenic H-Ras in collagen gels, it fails to cause EMT in vitro and in vivo and EMT induction is mediated by MAPK-pathway activation [213,214]. According to these data, Fischer and colleagues showed that ERK/MAPK activity is necessary and sufficient for the cooperation with TGF-β to regulate proliferation and survival of hepatocytes during EMT and to promote hepatocellular tumor progression [206]. All together, these data suggest that the contribution of Ras effectors in the control of EMT may depend on the cell type, the cellular context, and the Ras isoform involved.
Acquisition of a migratory phenotype and extensive reorganization of the actin cytoskeleton is one of the first requirements for metastasis formation. Oncogenic activation of RAS has been implicated in facilitating almost all aspects of the malignant phenotype, and metastasis is not an exception [215,216]. Oncogenic Ras induces alterations in cell–cell and cell–matrix interactions and the acquisition of a migratory phenotype. The perturbation of cell–cell contacts by oncogenic RAS is accomplished through the targeting of the molecular machinery that maintains intercellular adhesion junctions, including the E-cadherin receptor and its associated cytoplasmic protein β-catenin [217,218]. Additionally, oncogenic RAS directly contributes to the enhanced motility of cancer cells by causing pronounced changes in the polymerization, organization, and contraction of actin, the polymerization and/or stability of microtubules, and the transcriptional regulation of mitogenic gene products [215,219]. Collectively, these changes establish the front–rear asymmetry that is required for cell migration.
The existence of several functional links between the Ras, PI3K, and Rho-mediated signaling pathways must be considered when assessing oncogenic Ras in metastasis [82,220,221,222]. Ras regulates the actin cytoskeleton through Rac, a process that is entirely dependent on normal PI3K function acting upstream of Rac [100]. Rac and PI3K are linked by a feedback loop that is critically involved in the establishment and maintenance of cell polarity. Ras-GTP activates PI3K and the production of PIP3 at the leading edge of migrating cells. This increase in PIP3 induces Rac activation [223], which in turn sustains the formation of directional protrusions. Active Rac binds to p85 and further stimulates the activation of PI3K, thus amplifying Rac activation and migration induction [224]. PI3K also activates Rac through interaction with the Rac-specific GEF Tiam1 [225], and PTEN−/− cells are more motile and contain higher levels of Rac-GTP and Cdc42-GTP than WT cells [226]. In some studies, it was observed that even though PI3K is the main mediator, different Ras isoforms activate different Rho family members. For example, in Caco2-cells, oncogenic K-Ras enhances cell migration and filopodia formation through Cdc42, and oncogenic H-Ras induces EMT characteristics and promotes migration and invasion through Rac1 [227].
Inhibition of PI3K activity halts Ras induction of membrane ruffling, while activated PI3K is sufficient to induce membrane ruffling, acting through Rac [228]. Disruption of Ras binding to PI3K impairs migration in MEFs in response to EGF, FGF2, insulin, and oncogenic Ras, but not in response to PDGF, which has a diminished ability to activate Rac [115]. Furthermore, in response to EGF, cells deficient in Ras–PI3K interaction migrated in a disorderedly manner, suggesting a defect in polarization, and showed reduced invasion ability. The Ras–Raf scaffold protein Sur8 plays an important role in mediating motility and invasive potential of cells predominantly through the PI3K pathway via activation of Rac and MMPs, with a minor contribution of the Erk pathway [229]. Although PI3K activation is mainly linked to Rac activation, it has been reported that PI3K can also activate RhoA [83,230]. For example, endothelial cells with reduced PI3Kα activity have defects in migration, correlating with reduced levels of active RhoA [83].
Ras–PI3K interaction also regulates non-cell-autonomous cancer cell motility. Co-culture of Ras-transformed epithelial cells and normal epithelial cells showed that the majority of Ras-transformed cells are extruded towards the basal side of the surrounding normal cells, which is concomitant with enhanced motility [231]. The basally extruded Ras cells exhibited motility when surrounded by normal cells, but were non-motile when cultured alone, indicating that a non-cell-autonomous mechanism is at play. Inhibition of PI3K suppresses basal extrusion of oncogenic Ras cells, suggesting that the PI3K/AKT pathway is involved in the extrusion of oncogenic Ras cells to the basal side of normal epithelial cells. This data indicates that the role of Ras–PI3K in the regulation of metastasis is more complex than initially thought and therefore a complete characterization of the paracrine effects of K-Ras-mutant cancer cells would be valuable to fully understand its role in this process and to identify tumor specificities.
5.2. Ras–PI3K Signaling in Therapy Resistance
Drug resistance is the main limiting factor for achieving cures in cancer patients [232]. Activation of the AKT pathway is one of the major mechanisms involved in intrinsic and acquired resistance to radiotherapy and chemotherapy agents [233,234] (Figure 2). Due to the central role of AKT in cell survival, protein synthesis, and proliferation, an increase in AKT activity can evade the cytotoxic effect of chemotherapeutic agents, leading to chemoresistance [79]. This may be due to protecting cells from drug-induced apoptosis through the intrinsic pathway (e.g., by inactivating BAD and caspase 9 and stimulating anti-apoptotic proteins MCI-1) or through upregulation of survivin as an inductor of apoptosis [235,236].
Radiotherapy induces phosphorylation of AKT through PI3K activation [237,238,239], and the level of phosphorylation is radiation-dose dependent [240]. Double strain breaks (DSBs) are the major cause of IR-induced cell death in radiotherapy, and clinical data exists indicating the prognostic value of AKT activation levels for radiotherapy response in some tumor types [241,242]. Activated AKT stimulates the repair of radiation-induced DSBs [80,81,238,243] and the C-terminal domain of AKT interacts with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), the major component in non-homologous end-joining repair of DSB, and phosphorylates it, stimulating function of DNA-PKcs in DSB repair. Hyperactivation of AKT due to deregulation of RTKs, PI3K, Ras, or PTEN leads to efficient DSB repair and the appearance of radiotherapy resistance [244,245,246,247,248].
Although the specific contribution of Ras in the activation of PI3K/AKT leading to therapy resistance is not always addressed, in some cases it is known to play a relevant role [249]. For example, in lung carcinoma cell lines A549 and H460, targeting EGFR, PI3K [250], and AKT [244] enhances repair of DNA-DSBs and induces DNA-PKcs–dependent radiosensitization. These reports are supported by data from Choi et al., who showed that in the same cell lines, silencing the expression of K-RasK-Ras induces radiosensitization due to a reduction of IR-induced phosphorylation of DNA-PKcs and impaired repair of DNA-DSBs in a PI3K-AKT–dependent manner [251] due to enhanced autocrine production of EGFR ligands and activation of the Ras/PI3K axis [247]. This effect was not observed in K-RasK-Ras wild type cells.
Platinum-based agents, such as cisplatin, carboplatin, and oxaliplatin, are used in the treatment of a variety of cancers [79,252,253]. They are DNA-intercalating agents that interfere with DNA replication and RNA transcription through the crosslinking of DNA. Cisplatin also induces mitochondrial ROS, further increasing DNA damage and the cytotoxic properties of the drug. This results in the formation of DNA adducts, driving tumor cells to apoptosis [254,255]. There are many resistance mechanisms associated with cisplatin, including the involvement of oncogenicK-RasK-Ras mutations [256,257,258,259] and the hyperactivation of the PI3K-AKT pathway. K-RasK-Ras mutations induce NRF2 transcription and pathway upregulation, resulting in the overactivation of the anti-oxidative stress pathway, rendering tumor cells resistant to cisplatin-induced ROS [257,260]. In lung cancer cell lines, SHP2 mediates cisplatin-resistance-related phosphatase in a process that involves the inhibition of apoptosis by the activation of Ras/PI3K/AKT/survivin signaling pathway, and, consequently, the inhibition of SHP2 was associated with reduced expression of Ras, AKT, AKT activation, and survivin [261]. In ovarian cancer cell lines, crosstalk between STAT3 and Ras/p53 activated PI3K and ERK pathways, which in turn inhibited endoplasmic reticulum stress (ERS)-associated molecules, blocking cellular autophagy and inducing cisplatin resistance [262].
ERK activation is a common feature of tumors with a K-Ras, N-Ras, or B-Raf mutation [263], and the inhibition of the Raf/MEK/ERK pathway was supposed to be effective in cancers harboring mutations in these genes [264]. However, a portion of patients developed drug resistance mechanisms and no longer responded to Raf or MEK inhibitors [265]. Upregulation of PI3K pathway was found to be a major mechanism of resistance to Raf and MEK inhibitors [266,267,268]. The activation of PI3K after ERK pathway inhibition comes from different mechanism that include RTK reactivation [268,269], activating mutations in Pi3kca or loss of PTEN [270], or activating a positive feedback loop composed of GAB1, Ras, and PI3K, which induces the bypass of the ERK signal to the PI3K signal [271].
Recently, small molecules against K-Ras G12C mutations have been developed, and the first drug targeting this mutation has been approved by FDA [272]. However, therapeutic resistance to K-Ras G12C inhibition has been observed in preclinical tumor models and also in the clinic [273,274,275]. The PI3K pathway may be implicated in the resistance to K-Ras G12C inhibitors [276] as preclinical studies have shown failure to inactivate the PI3K signaling pathway after treatment with G12C inhibitors [274,277,278]. Importantly, combination of G12C and PI3K pathway inhibitors was effective in vitro and in vivo on models that are resistant to single-agent G12C inhibitor [274], or significantly improved antitumor activity of G12C inhibitors [277,279], which could be explained by a concomitant inhibition of p-ERK (due to G12C inhibition) and p-AKT [274,277]. This combination could avoid the toxicity associated with the inhibition of MEK and PI3K, while the efficacy of inhibiting both pathways in tumor cells is maintained [279].
Tyrosine kinase inhibitors (TKIs) are used in a variety of cancers. Upregulation of Ras-mediated pathways is a common mechanism of resistance to TKIs [280,281,282,283,284,285], such as those targeting RTKs including FMS-like tyrosine kinase 3 (FLT3) and EGFR. Jacobsen et al. [286] showed that AKT pathway activation is a convergent feature in EGFR-mutant PC9 NSCLC cells with acquired resistance to EGFR TKI. They found that combining an EGFR TKI with an AKT inhibitor induced significant growth inhibition in vitro and in vivo. They also examined clinical samples and found that activated AKT was increased in the majority of EGFR-mutant patients after progression on EGFR TKIs. Moreover, the high levels of phosphorylated AKT in patients prior to EGFR TKI treatment correlate with significantly worse progression free survival (PFS) and overall survival (OS) after first-line EGFR TKI treatment. Lee and colleagues investigated possible mechanisms responsible for acquired resistance to HER2-targeted therapy in gastric cancer. They found that lapatinib (a dual EGFR and HER2 TKI)-resistant HER2-posititve gastric cancer cells upregulated phosphorylation of EGFR/HER2, and MET appeared to be closely related to the activation of PI3K/AKT and ERK1/2. Resistance to TKIs can also appear through mutations within the RTK or mutations within downstream pathways [282,284,285,287]. Given that Ras is activated downstream of these receptors, any mutations within Ras or its effector pathways will render the cell resistant to the TKI. Different studies have shown that K-RasK-Ras mutations render patients resistant to Gefitinib, which is used to treat NSCLC patients [288,289]. Similarly, treatment of colorectal cancer with anti-EGFR monoclonal antibodies cetuximab or panitumumab is only successful in a subset of patients [290] due to the appearance of Ras mutations and variations in the EGFR extracellular domain, which reduce antibody binding efficiency, initiating relapse [285]. Thus, combination treatment with PI3K inhibitors may delay the onset of resistance appearance.
Arginine (Arg) auxotrophy occurs in certain tumor types and is usually caused by the silencing of argininosuccinate synthetase 1 or arginine lyase genes. Such tumors are often associated with an intrinsic chemoresistance and thus a poor prognosis [291]. Arginine auxotrophy, however, renders these tumors vulnerable to treatment with arginine-degrading enzymes. Pegylated arginine deiminase (ADI-PEG20), which converts Arg to citrulline and ammonia, resulting in Arg deprivation, is used for the treatment of melanoma, liver cancer, and other types of cancer. ADI-PEG20 has been shown to activate Ras signaling and, therefore, ERK and PI3K/AKT/GSK-3β kinase cascades, resulting in the phosphorylation and stabilization of c-Myc by the attenuation of its ubiquitin-mediated protein degradation mechanism, which induces ADI resistance [292]. PI3K inhibitors suppress c-Myc induction and enhance ADI-mediated cell killing and in animal models of argininosuccinate synthetase (AS, the rate-limiting enzyme for arginine biosynthesis)-negative melanoma, combination therapy using a PI3K inhibitor together with ADI-PEG20 yielded additive antitumor effects as compared with either agent alone.
Many different signaling pathways and associated factors play a critical role in mediating drug resistance through Ras and PI3K pathways. Furthermore, not only can these pathways contribute to an increased cellular proliferation rate in bulk cancer, but they may also support the self-renewal and stemness properties of cancer stem cells. Whilst it seems inevitable that cancer therapy resistance will remain an issue, better targeting of its potential causes is an imperative, which requires a greater understanding of resistance mechanisms.
6. Ras–PI3K Signaling in the Tumor Microenvironment
Cancer cells dramatically alter their local tissue environment (TME). The clinical relevance of Ras mutations is due not only to their impact on cancer cells’ autonomous mechanisms but also to their ability to alter the way tumor cells interact and influence different components of the TME, from the extracellular matrix (ECM) to endothelial cells, cancer-associated fibroblasts (CAFs), and inflammatory/immune cells. Neo-angiogenesis, tumor inflammation, and matrix remodeling are interconnected processes that alter the expression profile of cancer cells, increasing their aggressiveness, altering their expression profile, and eventually contributing to the end-stage of tumorigenesis represented by tumor dissemination [293].
6.1. Ras–PI3K Interaction in Angiogenesis
Angiogenesis is essential for tumor growth and metastasis to guarantee sufficient blood supply. The TME is composed of different signaling molecules and pathways that orchestrate the intricate process of angiogenesis, and tumor cells can tilt the balance toward proangiogenic factors to stimulate vascular growth. Furthermore, the TME comprises a plethora of cell types that contribute to angiogenesis. Tumor cells exploit those cells by releasing cytokines, chemokines, and growth factors to attract them into the TME. Those recruited cell types, such as myeloid cells and fibroblasts, in turn, release their stores of proangiogenic factors to facilitate angiogenesis [294].
Ras and PI3K are expressed in both tumor cells and non-malignant cells from the TME, thereby playing a multifaceted role in the process of angiogenesis. Ras promotion of tumor-associated angiogenesis has been described a long time ago [295]. Nonetheless, evidence supporting the different mechanisms underlying Ras stimulation of endothelial cells and its role on the molecular basis of angiogenesis continue to accumulate.
The mechanisms by which Ras activation initiates and sustains pro-angiogenic processes are complex, impinge on the modulation of levels of endothelial growth factors, and also increase local inflammation and stromal remodeling. The impact of K-Ras on the regulation of the most potent angiogenesis inducer vascular endothelial growth factor (VEGF) has been extensively studied in different models [296,297,298]. Oncogenic Ras-mediated upregulation of VEGFA involves the activation of multiple signaling cascades that eventually culminate in the stabilization of HIF-a, boosting its transactivation potential at the VEGFA promoter [299,300,301]. Tumor cells bearing Ras mutations can promote tumor-associated angiogenesis through other mechanisms apart of VEGF secretion, such as secretion of different proinflammatory, angiogenic cytokines, such as IL-8 [302,303,304,305], IL-6, and GRO1 (also known as CXC Motif Chemokine Ligand 1, CXCL1) [306] or repression of antiangiogenic factors such as thrombospondin-1 (TSP-1) and TSP-2 [307,308]. Under hypoxic conditions, PI3K is a critical downstream effector of Ras in the induction of VEGF [309,310,311] and IL8 expression [304] to induce angiogenesis. Once produced, the proinflammatory cytokines recruit immune cells such as neutrophils and macrophages, which produce angiogenic growth factors [302,312,313].
Ras can also upregulate VEGFA via the angiogenic enzyme cyclooxygenase 2 (COX2), which through the production of prostaglandins leads to the enhancement of the cyclic AMP-dependent transcription of VEGF [295]. COX2 can increase the levels of a plethora of other endothelial growth factors, such as FGF2 and PDGF, and is required for integrin-mediated endothelial cell spreading and migration [314,315]. Inhibition of COX2 using celecoxib inhibits COX2-dependent angiogenesis via the inhibition of the PI3K/AKT/HIF-1α axis in a murine hepatocarcinoma model [316], suggesting that, at least in some cases, Ras activation of COX to induce angiogenesis might be mediated by activation of PI3K pathway.
Ras–PI3K signaling in the microenvironment is capable of orchestrating changes that promote angiogenesis and survival. Once VEGFR is activated, PI3K participate in the downstream intracellular signal transduction that induces proliferation of endothelial cells and increases vessels permeability [317,318,319]. Subcutaneous tumors formed by B16F10 or LLC1 cells in a Ras–PI3K deficient host grew more slowly than those formed in the WT host and presented a marked reduction in the amount of blood vessels [320]. The decrease in vessel density and vessel area in tumors was accompanied by an increase in necrotic areas of the tumor. The authors demonstrated that endothelial cells lacking Ras-binding to PI3K were not able to respond to VEGF or FGF-2 and concluded that intact Ras–PI3K interaction is needed in endothelial cells for new blood vessel formation induced by either VEGFA or FGF-2 and that disruption of this interaction results in deficient cellular signaling that translates into a reduced capability to form new vessels. However, since this model lacks Ras–PI3K interaction in all host tissues, additional mechanisms driven by other cellular components of the TME cannot be discarded in the global angiogenic defect. In fact, tumor-associated macrophages (TAMs) extracted from the tumors of mice lacking Ras–PI3K interaction had a reduced expression of VEGFA and TGF-β, two important angiogenic factors [320]. Soler et al. described a similar growth impairment and angiogenic defects on transplanted tumors in a mouse model heterozygous for expression of a kinase-dead Pik3ca in all host tissues, although this model presented an increase in the number of vessels, with reduced lumen size in the mutant [321].
6.2. Ras–PI3K Interaction in Tumor Immune Infiltration
Interplay between cancer cells and the immune microenvironment is important for cancer progression [322]. K-Ras influences the composition of the immune microenvironment and promotes the switch from an antitumorigenic to a protumorigenic response through different mechanisms that include the expression of several inflammatory cytokines (such as IL-6, IL-8, IL-17, and IL-22) [305,323,324], chemokines (CCL2, CXCL2, or CCL1) [325], and activation of signaling pathways such as NF-κB, which has been considered fundamental in K-Ras-induced inflammation in solid tumors. The regulation of these processes is important for the recruitment, activation, and differentiation of immune cells that eventually promote a protumorigenic environment and induce cancer cell evasion from anticancer therapies [293,326,327,328]. Although the specific role played by Ras–PI3K interaction in the regulation of these processes is not clear, PI3K controls activation of such proinflammatory pathways. For example, PI3K/AKT regulates the expression of proinflammatory cytokines such as IL-6, IL-8, IL-17, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [329,330]. According to these studies, PTEN knock-down in lung epithelial cells potentiated AKT phosphorylation and enhanced production of IL-6, CXCL8, CXCL10, CCL2, and CCL5 [331]. Importantly, PI3K-driven cancer activates the NF-κB-dependent transcriptional profile, increasing expression and secretion of cytokines and chemokines, especially IL-6, which helps to generate a pro-tumor microenvironment and facilitate tumor progression [332]. Based on these results, it is possible that PI3K acts downstream of oncogenic Ras to promote a proinflammatory microenvironment.
K-Ras tumors are usually infiltrated by immune cells. CD8+ T lymphocytes and natural killer cells (NK) generally have antitumor functions, whereas regulatory T-cells (Tregs), myeloid-derived suppressor cells (MDSCs), neutrophils, and macrophages are usually pro-tumorigenic [327]. The presence of TAMs is generally associated with tumor progression, and this is also valid in K-Ras-induced lung tumors [325,333]. Although the molecular mechanism is unknown, different studies have established that Ras signaling to PI3K may regulate macrophage infiltration in cancer. Disruption of Ras binding to PI3K, either constitutively or just in host tissues, severely impairs the recruitment of F4/80+ macrophages to tumors in K-Ras-driven lung tumors [164,320], and the few TAMs present in the tumors are closer to an M1 phenotype, which is associated with good prognosis in context of cancer [320]. Macrophage reliance on PI3K signaling for tumor recruitment is not limited to the p110α isoform. PI3Kδ-KO macrophages transferred to tumor-bearing NSG mice (NOD scid γ mouse) reduced tumor growth compared to the WT macrophages [334]. Furthermore, the blockade of PI3Kγ in pancreatic ductal adenocarcinoma (PDAC)-bearing mice reprograms TAMs to stimulate CD8+ T-cell-mediated tumor suppression and to inhibit tumor cell invasion, metastasis, and desmoplasia [335]. The importance of PI3Kγ for protumorigenic macrophages function was revealed by Kaneda et al., who showed that PDAC tumor growth and metastasis depend on macrophage PI3Kγ [335].
PI3K has been found to mediate immune evasion by downregulating major histocompatibility complex Class I (MHCI) antigen presentation at the cell surface, therefore decreasing cancer cell recognition by CD8+ cytotoxic T-cells [336,337]. Activation of PI3K signaling repressed the induction of MHCI and MHCII in oral carcinoma cells [338], and PI3K inhibition improved MHCI presentation and rendered tumor cells sensitive to recognition by CD8+ T-cells [339]. This effect may be, at least partially, regulated by Ras activation of PI3K, since the inactivation of oncogenic Ras increased MHCI expression in human colorectal cell lines [340], and a murine model of oncogenic Ras has been associated with the downregulation of MHCI surface expression [341]. Another mechanism by which Ras participates in immune escape is the stimulation of Treg development [342,343]. Tregs may be required for K-Ras-mediated lung tumorigenesis [344], and oncogenic K-Ras increases Treg production through the induction of the expression of IL10 and TGFβ1 after the activation of the MEK–ERK–AP-1 pathway [345]. Although the PI3K pathway was not directly evaluated, considering that TGFβ is the main cytokine implicated in the Treg induction phenotype [346] and that PI3K plays a pivotal role in regulation of the TGF-β1 expression [347,348,349], it is likely that PI3K signaling downstream of Ras participates in Treg cell generation. It is also important to consider that the Ras–PI3K pathway in non-oncogenic cells could modulate Tregs, as PI3K/AKT signals are necessary for Treg development and function [350]. As an example, PI3Kδ genetic ablation or pharmacological inhibition reduce Treg infiltration in preclinical mouse tumors and peripheral tissues [351,352], while PI3Kα/δ inhibition directly promotes durable anti-tumor responses and enhances cytotoxic T-cell function in vivo [353].
Neutrophils are also described as key drivers of cancer progression [354]. One of the mechanisms by which tumor-associated neutrophils (TANs) promote tumor progression is through the generation of ROS that mediates the inhibition of T-cell proliferation, creating an immunosuppressive environment [355]. Analysis of data from uterine corpus endometrial carcinoma (UCEC) patients showed that high expression levels of Pik3ca correlate with the neutrophil-related pathway Mac1 Integrin signaling, which is the most abundant integrin on neutrophils and is substantially up-regulated on the cell surface upon neutrophil activation [356]. Consistently, neutrophil-related genes and neutrophils were significantly altered by Pik3ca expression in UCEC [357].
From a clinical point of view, targeting the immune microenvironment is a promising area, and several therapies are currently used in clinical practice, such as antibodies targeting immune-checkpoint molecules (e.g., programmed cell death 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)) [328,358]. However, there are tumors that do not respond to immunotherapy, as is the case for pancreatic cancer [359], and NSCLC patients do not respond to anti-CTLA4 therapy [360]. Thus, there is an urgent need to understand why some patients do not benefit from immunotherapy. The success of the immune-checkpoint blockade depends on the immunogenicity of the tumor, so it is critical to understand the molecular mechanisms that dictate tumor cell PD-L1 expression. K-Ras mutations in lung cancer have been associated with increased PD-L1 expression causing an improved clinical response to anti-PD1 therapy [328], although the mechanism by which oncogenic K-Ras augment PD-L1 expression is controversial. Some studies indicate that PD-L1 is up-regulated by K-Ras mutation through p-ERK but not p-AKT signaling [361,362]. However, Coelho and colleagues showed that oncogenic Ras signaling regulates PD-L1 expression through both MEK and PI3K pathways, with MEK pathway activation stabilizing PD-L1 mRNA through the inhibition of tristetetrapolin (TTP) and the PI3K pathway only affecting protein expression [363]. In agreement with these data, K-Ras-dependent activation of PI3K/AKT/mTOR in human lung adenocarcinma and squamous cell carcinomas tightly regulate PD-L1 expression both in vitro and in vivo. This is further supported by studies carried out on patient samples, suggesting that oncogenic K-Ras can cause an immune escape by AKT/mTOR pathway via PD-L1 [364]. Importantly, PI3K has been shown to regulate PD-L1 immunosuppressive function even in the absence of oncogenic Ras. MDSCs activate the PI3K/AKT/NF-κB pathway in B-cells via the PD-1/PD-L1 axis and drive immunosuppressive effects in breast cancer [365]. K-Ras activation in pancreatic cancer is also associated with increased PD-1 expression, although immunotherapy has limited clinical success in this indication [328,359]. The abundance and complexity of the TME in pancreatic cancer may explain the lack of effectiveness of conventional immunotherapy treatments. Thus, combinatorial strategies targeting the immune system (e.g., PD-L1) and the TME complexity (e.g., with inhibitors of colony stimulating factor receptor 1(CSFR1) or chemokine C-X-C receptor 4 (CXCR4)) are in clinical trials, and it is expected that the reprograming of the TME will improve the benefit of classical immunotherapy treatment [328,359].
6.3. Ras–PI3K in CAFs and ECM Remodeling
CAFs are one of the most abundant stromal cell types in the tumor microenvironment and contribute to several aspects of the malignant phenotype [366]. The relevance of Ras and PI3K signaling in CAFs is, by far, less understood than in cancer cells, but different studies have validated the role played by PI3K in the protumoral functions of CAFs. For example, CAF-derived TGF-α promotes the metastatic colonization of ovarian cancer cells via activation of AKT, epidermal growth factor receptor (EGFR), and extracellular signal-regulated kinase (ERK)-1/2 signaling pathways [367]. Activation of PI3K due to loss of PTEN in stromal fibroblasts accelerated initiation, progression, and malignant transformation of mammary epithelial tumors due to the induction of genes involved in ECM remodeling and the recruitment of macrophages, including Mmp9 and Ccl3 [368]. PI3Kγ has been shown to regulate TNF-mediated secretion of matrix metalloproteinases (MMPs) from fibroblasts, which is crucial for the migration of tumor cells [369]. Ziqian Li et al. reported that in colorectal carcinoma and melanoma, CXCL5 derived from the CAFs activated PI3K/AKT signaling pathway and promoted the expression of PD-L1 in tumor cells [370].
CAFs constitute a heterogeneous population of cells, and their origin is still under debate. The most widely accepted view is that CAFs are early developmental precursors of cells with different origins that respond to signals derived from cancer cells [371]. Among these signals, some are Ras–PI3K-mediated, such as TGF-β, HGF, EGF, and FGF2 [372]. Thus, it is plausible that Ras–PI3K signaling has a prominent role in CAFs expansion and function and that its inhibition may influence tumor growth and progression, although additional studies addressing the relevance of this signaling pathway in CAF functionality are required.
Modulation of cancer-associated fibroblasts by K-Ras mutational changes through Hedgehog signaling (Hh) has been suggested as an important mediator of CAF activation in PDAC models [373,374,375,376,377]. SHh originated from cancer cells was responsible for changes on the fibroblast proteome, promoting the synthesis of ECM components, such as collagen, and matrix metalloproteinases (MMPs) [376]. Cancer-derived Hh also promoted the expression of growth factors such as IGF1 and GAS6 by fibroblasts, which reflected back on cancer cells. Additionally, fibroblast-derived signals significantly alter the phosphoproteome of PDAC cells, not only promoting an increase in the usual cell-autonomous alterations but also activating pathways that are not autonomously triggered. Recently, it was shown that the conditioned medium of K-RasV12-overexpressing colorectal cells strongly increased the migration of intestinal fibroblasts without affecting their proliferation rate and their differentiation status [378]. Together, these studies suggest that oncogenic Ras in cancer cells can alter the behavior of fibroblasts, which in turn affects the microenvironment through ECM changes and growth factor signaling, contributing to tumor progression.
The ECM is mainly secreted by CAFs in the TME and provides structural scaffolding for the surrounding cells as well as biochemical and biomechanical cues for cell differentiation, proliferation, and migration. Within the tumor stroma, not only the cancer cells but also the resident fibroblasts, which differentiate into CAFs, modify the ECM. Growth factors and chemokines, which are tethered to and released from the ECM, as well as metabolic changes of the cells within the tumor bulk, add to the tumor-supporting tumor microenvironment. CAF-mediated ECM remodeling is a highly responsive process of receiving, processing, and responding to the cellular, molecular, and mechanical signals in the TME. The lysyl oxidase (LOX) family and MMPs represent two major types of remodeling enzymes produced by CAFs with a high relevance in tumor progression. As a highly adaptive and mechanically responsive stromal cell type, CAFs sense and respond to the ECM stiffness in a LOX/MMP-dependent manner and further fine-tune the CAF-ECM interactions [379]. LOX is a secreted amine oxidase that modifies the primary tumor microenvironment by crosslinking collagen and elastin in the ECM [380,381] and has been linked to metastasis of CRC cells such as SW480 and its patient-matched metastatic clone, SW620 [382,383]. LOX expression is enhanced by co-expression of Ras G12V through the activation of PI3K/AKT and concomitant accumulation of the hypoxia-inducible factor (HIF)-1α [384,385]. Treatment of cancer cells carrying activated Ras with Kobe0065 [386] (a small-molecule inhibitor of Ras) or a siRNA targeting Ras downregulates the expression of LOX, which has been implicated in metastasis. Moreover, Kobe0065 effectively inhibits not only migration and invasion of cancer cells carrying the activated ras genes but also lung metastasis SW620 carrying the K-Ras G12V mutation. Similarly, in a murine model of pancreatic cancer driven by oncogenic Ras (KPC mice), the targeting of LOX leads to a remarkable increase in survival, especially when combined with gemcitabine. LOX inhibition was associated with increased tumor vascularization, immune cell infiltration, and drug efficacy [387].
Plasticity of the ECM is exploited by tumors during the multi-step process, leading to the acquisition of more aggressive and invasive features [388]. ECM degradation promotes tissue invasion by removing the physical barriers that contain the tumor and prevents its migration towards vessels. The MMP family of proteases degrades ECM proteins and is often deregulated in cancer. Increasing evidence links PI3K/AKT signaling to the control of MMP production in normal and cancer cells [332,369,389,390,391,392,393,394], which is not entirely surprising considering that factors known to be regulated by Ras–PI3K, such as growth factors (TGF-β, EGF, PDGF and FGF-2) and inflammatory cytokines (IL-1, IL-6 and TNF-α), are regulators of MMP levels [395]. Most of our current knowledge on PI3K regulation of CAFs comes from tumor cells, but there is a concerning lack of understanding of how PI3K activation in CAFs influenced by tumor cells regulates MMPs to drive ECM remodeling and tumor cell spread.
A direct role of mutant K-Ras in the modulation of ECM properties has also been described. In K-Ras G12V-driven lung cancer, epithelial cells secreted higher levels of activated MMP-9 [396], and in pancreatic cancer mouse models, increased expression of MMP3 cooperates with K-Ras activation to shape the stromal microenvironment, not only by stimulating immune cell influx but also as a primary proteolytic activator of MMP-9 [397]. Additionally, in pancreatic cancer cells, mutational activation of K-Ras induced the expression of the eukaryotic translation initiation factor 5A (eIF5A) and consequent stimulation of ROCK1 and ROCK2 [398]. ROCK activation and signaling ultimately results in ECM remodeling and collagen degradation by MMPs, thereby enabling invasive tumor growth through elimination of physical restraints [399].
It is now evident that Ras signaling not only exerts its activity to drive cell growth and sustain survival in cancer cells, but also modulates microenvironmental changes. In stromal cells, Ras signaling contributes to directing the response to a multitude of cues that cancer cells produce to modify their microenvironment. It is, thus, an important area to explore, not only from basic knowledge, but also from a therapeutic point of view.
7. Targeting Ras and PI3K in Cancer
K-Ras is a major clinical target, as it is by far the most significant form of RAS in terms of cancer incidence. Regardless of the tremendous attempts in the past decades to develop inhibitors, Ras was considered undruggable for decades [400]. Recently, several small molecules (AMG510, MRTX849, JNJ-74699157, and LY3499446) have been developed to specifically target K-Ras G12C, culminating with the FDA approval of Sotorasiv (AMG510) [401]. Further discussion of the different approaches used over the past decades to inhibit oncogenic Ras is provided by a recent review from Molina-Arcas et al. [402].
Hyperactivation of the PI3K pathway in cancer, and its vital function in cell survival and proliferation, have made it an ideal target for treatment. Furthermore, the inhibition of Ras-effector pathways has for years been considered the most effective approach to target Ras-driven tumors [402,403]. Results from clinical trials with PI3K inhibitors in solid tumors have been, however, largely disappointing [404]. Reasons for this failure include drug resistance, aberrant activation of parallel signaling pathways that in turn stimulate the PI3K pathway, and intolerable toxicity due to lack of specificity, especially with pan-PI3K inhibitors [74,405], while isoform-specific inhibitors seem to have better therapeutic efficacy and an improved toxicity profile [406]. So far, only five PI3K inhibitors have been approved to use in clinical practice. One of them is Alpelisib, a PI3Kα inhibitor approved for breast cancer treatment and investigated in combination with a range of targeted therapies and chemotherapies in multiple clinical trials [407]. The other PI3K inhibitors are used in the treatment of hematological cancers: idealisib, a PI3Kδ inhibitors [404], copanslisib, the only pan-PI3K inhibitor approved [408], duvelisib, a dual inhibitor of the PI3Kδ and PI3Kγ isoforms [409], and umbralisib, a dual inhibitor of PI3Kδ/casein kinase-1εr (www.fda.gov, (accessed on 20 May 2021) [410]).
Considerable efforts have been made to improve the clinical efficacy of PI3K inhibition and to reduce adverse effects. The generation of isoform-selective PI3K inhibitors may reduce the intrinsic toxicity associated with pan-PI3K inhibition, allowing the exploration of combination therapies. In line with the evidence for isoform-specific PI3K targeting in preclinical models, pivotal clinical trials indicate new opportunities for p110α-selective inhibition in selected patient populations. Results from clinical trials with taselisib show that this p110α specific inhibitor can more effectively suppress the PI3K signaling pathway, resulting in greater anti-tumor activity and an improved therapeutic index [411,412]. Since different PI3K isoforms have different expression patterns and activation mechanisms, inhibition of specific PI3K isoforms involved in specific tumors appears to be a way to circumvent toxicity issues. An example of success is the PI3Kδ-specific inhibitor idelalisib, approved for the treatment of B-cell lymphoid malignancies [413]. These promising effects are likely due to the concomitant action of the drug in both tumor and TME.
The emerging roles of Ras and PI3K in shaping the tumor microenvironment provide a new awareness of how kinase inhibitors may contribute to enhancing antitumor immunity and their application in combination therapy to improve the outcome of immunotherapy. For example, PI3Kα or PI3Kβ inhibitors impair tumor cell growth and induce changes in the tumor microvasculature, which correlates with antitumor activity and, more importantly, potentiates the anticancer activity of current cytotoxic therapy [319,321,414,415]. Furthermore, PI3Kβ inhibition could improve the efficacy of immunotherapy by increasing T-cell infiltration in melanoma with PTEN loss [416]. Additionally, in leukemic B-cells, PI3Kδ blockade impairs pro-survival signaling and induces tumor cell death. On the other hand, the inhibition of PI3Kδ in monocyte-derived cells of the lymph node stroma blocks the secretion of pro-survival chemokines sensed by tumor cells, potentiating the pro-apoptotic effect of the drug [417].
The same principle could be applied to PI3Kγ and PI3Kδ specific inhibitors used for the treatment of solid tumors. PI3Kδ inactivation protects against the development of a broad range of cancers, which is mainly associated with the disruption of the function of Tregs and possibly of MDSCs [351]. The anticancer activity of PI3Kγ seems to be associated with its effects on tumor macrophages, which translates into the stimulation of T-cell recruitment and reduced tumor growth [335]. Important results with PI3Kδ and PI3Kγ inhibitors, used alone or in combination with standard anticancer therapy, have been obtained in murine models of pancreatic cancer, a tumor that is highly resistant to chemotherapy and radiation therapy and does not respond to immune checkpoint inhibitors [98,335,351,418,419]. For example, the deletion of PI3Kγ improved survival and responsiveness to standard-of-care chemotherapy in animal models of PDAC [335], and the dual inhibition of PI3Kγ and CSF-1/CSF-1R reduced the tumor infiltration of MDSCs and inhibited tumor growth [420]. Importantly, PI3Kγ inhibition can synergize with anti-PD-1 to reduce tumor growth, extending survival [421,422], and pan-PI3K inhibition can be used to overcome resistance to immune checkpoint inhibitors [423].
As we expand our understanding of the role of PI3K inhibitors in anticancer and procancer immunity, concerns are raised over the implication of isoform-unspecific PI3K inhibitors in anticancer immune response. Although most PI3K inhibitors show a boosting effect on anticancer immunity, it is anticipated that selective PI3K inhibitors can maximally enhance host immunity against cancer and minimize deleterious effects on normal tissues.
8. Conclusions
Signaling networks that are triggered by oncogenic Ras within the cell are complex and highly dynamic. Enormous efforts have been made to elucidate the role of different effector pathways in Ras-controlled functions. In the last few years, our capacity to study the in vivo ramifications of the expression of oncogenic Ras has been constantly improving due to the development of sophisticated genetically engineered mouse models that feature activating mutations of Ras. By achieving tissue- and cell-specific expression in a time-controlled and reversible manner, these models often recapitulate the genetic and biological evolution of human cancers, increasing our understanding of the crucial mediators of RAS-driven oncogenesis and also being instrumental to testing and developing novel targeting strategies directed at Ras.
We now know that activation of K-Ras on cancer cells reshapes the tumor microenvironment, both by oncogenic signaling coming from tumor cells affecting other cells of the stroma, but also by normal signaling within stromal cells. This effect will, in turn, affect tumor cells’ behavior. A complete characterization of the paracrine effects of K-Ras-mutant cancer cells in models with mutant K-Ras would be valuable to obtain an overall view of these effects, identify tumor specificities, and point to possible combination therapies with stromal modulatory approaches. Since metastatic outgrowth relies on the recruitment of non-cancer cells, such as myeloid cells, endothelial cells, fibroblasts, and ECM remodeling, a better understanding of the paracrine effect of Ras signaling would help address the specific role played by mutant K-Ras cancer cells in the regulation of these stromal components and how this would impact on the establishment of metastatic lesions. Additionally, because different K-Ras mutations have distinct transforming potential and preferentially activate different effector pathways, it would be relevant to understand whether they impact the interaction between cancer cells and the microenvironment differently and which signaling pathways have a prevalent role in each process. This would be relevant for the development of mutation-specific therapeutic approaches.
Considering that cancer cell communication with the microenvironment is dynamic and not a unidirectional process and that mutual regulation of cancer cells and ECM exists, understanding the role of Ras in the integration of external signaling and the subsequent cancer cell response will be essential to identifying key molecules that mediate this cross-talk and will undoubtedly impact the design of new combinatorial therapeutic strategies aimed at targeting tumor cells:microenvironment dependencies.
Author Contributions
C.C. and C.A.-A.: writing—original draft preparation. E.C.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Spanish Ministry of Science and Innovation (RTI2018-099161-A-I00), and by Programa de Apoyo a Planes Estratégicos de Investigación de Estructuras de Investigación de Excelencia of the Ministry of Education of the Castilla–León Government (CLC–2017–01).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Claudio Santos for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Martin, M.A.; Arenas, J.; Lucia, A.; Zugaza, J.L. Small GTPases of the Ras superfamily and glycogen phosphorylase regulation in T cells. Small GTPases 2021, 12, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: It’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krygowska, A.A.; Castellano, E. PI3K: A Crucial Piece in the RAS Signaling Puzzle. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.E.; Rubio, I.; Roose, J.P. Regulation of ras exchange factors and cellular localization of ras activation by lipid messengers in T cells. Front. Immunol. 2013, 4, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Mattos, C. The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb. Perspect. Med. 2018, 8, a031427. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; Wink, G.; Mattos, C. Transformation efficiency of Q61 Ras mutants linked to structural features of the switch regions. Structure 2007, 15, 1618–1629. [Google Scholar] [CrossRef] [Green Version]
- Henis, Y.I.; Hancock, J.F.; Prior, I.A. Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 2009, 26, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the und ruggable drugged? Nat. Rev. Drug. Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, A.; Czernilofsky, A.P.; Keri, G.; Smigelskaite, J.; Sill, H.; Troppmair, J. Signaling Through RAS-RAF-MEK-ERK: From Basics to Bedside. Curr. Med. Chem. 2007, 14, 601–623. [Google Scholar] [CrossRef]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenonos, K.; Kyprianou, K. RAS signaling pathways, mutations and their role in colorectal cancer. World J. Int.Gastrointest. Oncol. 2013, 5, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fray, M.J.; Waterfield, M.D.; Donward, J. Phospatidylinositositol-3-OH kinase as a target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal Structure and Functional Analysisof Ras Binding to Its EffectorPhosphoinositide 3-Kinase. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Rebhun, J.F.; Chen, H.; Quilliam, L.A. Identification and Characterization of a New Family of Guanine Nucleotide Exchange Factors for the Ras-related GTPase Ral. J. Biol. Chem. 2000, 275, 13406–13410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent proteinkinase which phosphorylates and activates protein kinase Ba. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Zinatizadeh, M.R.; Miri, S.R.; Zarandi, P.K.; Chalbatani, G.M.; Raposo, C.; Mirzaei, H.R.; Akbari, M.E.; Mahmoodzadeh, H. The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in cancer metastasis. Genes. Dis. 2021, 8, 48–60. [Google Scholar] [CrossRef]
- Kiel, C.; Foglierini, M.; Kuemmerer, N.; Beltrao, P.; Serrano, L. A genome-wide Ras-effector interaction network. J. Mol. Biol. 2007, 370, 1020–1032. [Google Scholar] [CrossRef]
- Ibanez Gaspar, V.; Catozzi, S.; Ternet, C.; Luthert, P.J.; Kiel, C. Analysis of Ras-effector interaction competition in large intestine and colorectal cancer context. Small GTPases 2021, 12, 209–225. [Google Scholar] [CrossRef] [Green Version]
- Erijman, A.; Shifman, J.M. RAS/Effector Interactions from Structural and Biophysical Perspective. Mini. Rev. Med. Chem. 2016, 16, 370–375. [Google Scholar] [CrossRef]
- Catozzi, S.; Halasz, M.; Kiel, C. Predicted ‘wiring landscape’ of Ras-effector interactions in 29 human tissues. NPJ. Syst. Biol. Appl. 2021, 7, 10. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Foster, F.M.; Traer, C.J.; Abraham, S.M.; Fry, M.J. The phosphoinositide (PI) 3-kinase family. J. Cell Sci. 2003, 116, 3037–3040. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.A.; Dornan, G.L.; Hessenberger, M.; Haucke, V. The molecular mechanisms mediating class II PI 3-kinase function in cell physiology. FEBS J. 2021. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhou, W.; Chen, J.; Wei, W. Upstream regulators of phosphoinositide 3-kinase and their role in diseases. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R. The mechanism of PI3Kalpha activation at the atomic level. Chem. Sci. 2019, 10, 3671–3680. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segaliny, A.I.; Tellez-Gabriel, M.; Heymann, M.F.; Heymann, D. Receptor tyrosine kinases: Characterisation, mechanism of action and therapeutic interests for bone cancers. J. Bone Oncol. 2015, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Domchek, S.M.; Auger, K.R.; Chatterjee, S.; Terrence, R.; Burke, J.; Shoelson, S.E. Inhibition of SH2 Domain/Phosphoprotein Association by a Nonhydrolyzable Phosphonopeptide. Bio. Chem. 1992, 31, 9865–9870. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in Signal Transduction: From Phosphotyrosine-SH2 Domain Interactions to Complex Cellular Systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinaseby fibroblast growth factor receptors ismediated by coordinated recruitmentof multiple docking proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, C.L.; Auger, K.R.; Chanudhuri, M.; Yoakim, M.; Schaffhausen, B.; Shoelson, S.; Cantley, L.C. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 1993, 268, 9478–9483. [Google Scholar] [CrossRef]
- Brock, C.; Schaefer, M.; Reusch, H.P.; Czupalla, C.; Michalke, M.; Spicher, K.; Schultz, G.; Nurnberg, B. Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J. Cell Bio Biol. 2003, 160, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes. Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, C.W.; Morgan, M.A.; Bergmann, L. Targeting the Ras signaling pathway: A rational, mechanism-based treatment for hematologic malignancies? Blood 2000, 96, 1655–1669. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulationof Akt/PKB by theRictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Haar, E.V.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples SurvivalSignals to the Cell-Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- She, Q.B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruta, F.; Masuyama, N.; Gotoh, Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 2002, 277, 14040–14047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinaseyAkt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [Green Version]
- Madhunapantula, S.V.; Mosca, P.J.; Robertson, G.P. The Akt signaling pathway: An emerging therapeutic target in malignant melanoma. Cancer Biol. Ther. 2011, 12, 1032–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, G.S.; Lin, C.H.; Yang, Y.L.; Wu, M.S.; Chen, B.C. Ghrelin induces colon cancer cell proliferation through the GHS-R, Ras, PI3K, Akt, and mTOR signaling pathways. Eur. J. Pharm. 2016, 776, 124–131. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Middendorp, S.; Dingjan, G.M.; Hendriks, R.W. Impaired precursor B cell differentiation in Bruton’s tyrosine kinase-deficient mice. J. Immunol. 2002, 168, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Middendorp, S.; Dingjan, G.M.; Maas, A.; Dahlenborg, K.; Hendriks, R.W. Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol. 2003, 171, 5988–5996. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.T.; Berg, L.J. New insights into the regulation and functions of Tec family tyrosine kinases in the immune system. Curr. Opin. Immunol. 2002, 14, 331–340. [Google Scholar] [CrossRef]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.C.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarstrom, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Ann. Annu. Rev. Immunol. 2004, 22, 563–598. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug. Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Herman, S.E.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef] [PubMed]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Ciraolo, E.; Ghigo, A.; Germena, G.; Hirsch, E. Crossroads of PI3K and Rac pathways. Small GTPases 2015, 6, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J.; et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Viernes, D.R.; Choi, L.B.; Kerr, W.G.; Chisholm, J.D. Discovery and development of small molecule SHIP phosphatase modulators. Med. Res. Rev. 2014, 34, 795–824. [Google Scholar] [CrossRef] [PubMed]
- Miletic, A.V.; Anzelon-Mills, A.N.; Mills, D.M.; Omori, S.A.; Pedersen, I.M.; Shin, D.M.; Ravetch, J.V.; Bolland, S.; Morse, H.C., 3rd; Rickert, R.C. Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J. Exp. Med. 2010, 207, 2407–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the "undruggable". Bio. Chim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denley, A.; Kang, S.; Karst, U.; Vogt, P.K. Oncogenic signaling of class I PI3K isoforms. Oncogene 2008, 27, 2561–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1231. [Google Scholar] [CrossRef]
- Buckles, T.C.; Ziemba, B.P.; Masson, G.R.; Williams, R.L.; Falke, J.J. Single-Molecule Study Reveals How Receptor and Ras Synergistically Activate PI3Kalpha and PIP3 Signaling. Biophys. J. 2017, 113, 2396–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate.p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Geering, B.; Cutillas, P.R.; Vanhaesebroeck, B. Regulation of class IA PI3Ks: Is there a role for monomeric PI3K subunits? Biochem. Soc. Trans. 2007, 35, 199–203. [Google Scholar] [CrossRef]
- Burke, J.E.; Perisic, O.; Masson, G.R.; Vadas, O.; Williams, R.L. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc. Natl. Acad. Sci. USA 2012, 109, 15259–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85p110 Phosphatidylinositol 3′-Kinase Stabilization and Inhibition of the p110α Catalytic Subunit. Mol. Cell Biol. 1998, 18, 1379–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.; Schaffhausen, B.; Liu, Y.; Günther, U.L. NMR Structure of the N-SH2 of the p85 Subunit of Phosphoinositide 3-Kinase Complexed to a Doubly Phosphorylated Peptide Reveals a Second Phosphotyrosine Binding Site †. Biochemical 2000, 39, 15860–15869. [Google Scholar] [CrossRef]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kalpha by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.-L.; Wang, J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3085–3096. [Google Scholar]
- Rodriguez-Viciana, P.; Warne, P.H.; Khwaja, A.; Marte, B.M.; Pappin, D.; Das, P.; Waterfield, M.D.; Ridley, A.; Downward, J. Role of Phosphoinositide 3-OH Kinase in Cell Transformation and Control of the Actin Cytoskeleton by Ras. Cell 1997, 87, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Suire, S.; Condliffe, A.M.; Ferguson, G.J.; Ellson, C.D.; Guillou, H.; Davidson, K.; Welch, H.; Coadwell, J.; Turner, M.; Chilvers, E.R.; et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat. Cell Biol. 2006, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Sjolander, A.; Yamamoto, K.; Huber, B.E.; Lapetina, E.G. Association of p21ras with phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 7908–7912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, H.; Shao, J.; DuBois, R.N. Akt/PKB Activity Is Required for Ha-Ras-mediated Transformation of Intestinal Epithelial Cells. J. Biol. Chem. 2001, 276, 14498–14504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.A.; Park, M.T.; Kang, C.M.; Um, H.D.; Bae, S.; Lee, K.H.; Kim, T.H.; Kim, J.H.; Cho, C.K.; Lee, Y.S.; et al. Opposite effects of Ha-Ras and Ki-Ras on radiation-induced apoptosis via differential activation of PI3K/Akt and Rac/p38 mitogen-activated protein kinase signaling pathways. Oncogene 2004, 23, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritsman, K.; Yuzugullu, H.; Von, T.; Yan, H.; Clayton, L.; Fritsch, C.; Maira, S.M.; Hollingworth, G.; Choi, C.; Khandan, T.; et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J. Clin. Investig. 2014, 124, 1794–1809. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Iwanaga, K.; Raso, M.G.; Wislez, M.; Hanna, A.E.; Wieder, E.D.; Molldrem, J.J.; Wistuba, I.I.; Powis, G.; Demayo, F.J.; et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS ONE 2008, 3, e2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Han, Y.K.; Kim, J.Y.; Lee, E.G.; Lee, H.W.; Lee, G.M. Effect of constitutively active Ras overexpression on cell growth in recombinant Chinese hamster ovary cells. Biotechnol. Prog. 2011, 27, 577–580. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P.; Gilbert, E.U.C.; Coffert, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef]
- Orme, M.H.; Alrubaie, S.; Bradley, G.L.; Walker, C.D.; Leevers, S.J. Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat. Cell Biol. 2006, 8, 1298–1302. [Google Scholar] [CrossRef]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes. Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Chun-Jen Lin, C.; Stern, M. Ras-dependent and Ras-independent effects of PI3K in Drosophila motor neurons. Genes. Brain Behav. 2012, 11, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Murillo, M.M.; Rana, S.; Spencer-Dene, B.; Nye, E.; Stamp, G.; Downward, J. Disruption of the Interaction of RAS with PI 3-Kinase Induces Regression of EGFR-Mutant-Driven Lung Cancer. Cell Rep. 2018, 25, 3545–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, A.; Becattini, B.; Mazzoli, A.; Bleve, A.; Radici, L.; Maxvall, I.; Sopasakis, V.R.; Molinaro, A.; Backhed, F.; Solinas, G. Insulin-Driven PI3K-AKT Signaling in the Hepatocyte Is Mediated by Redundant PI3Kalpha and PI3Kbeta Activities and Is Promoted by RAS. Cell Metab. 2019, 29, 1400–1409.e1405. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Klinghoffer, R.; Prestwich, G.D.; Toker, A.; Kazlauskas, A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Curr. Biol. 1999, 9, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Whiteford, C.C.; Best, C.; Kazlauskas, A.; Ulug, E.T. D-3 phosphoinositide metabolism in cells treated with platelet-derived growth factor. Bio. Chem. J. 1996, 319, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auger, K.R.; Serunian, L.A.; Soltoff, S.P.; Libby, P.; Cantley, L.C. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 1989, 57, 167–175. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Gire, V.; Marshall, C.; Wynford-Thomas, D. PI-3-kinase is an essential anti-apoptotic effector in the proliferative response of primary human epithelial cells to mutant RAS. Oncogene 2000, 19, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Sethi, T.; Ginsberg, M.H.; Downward, J.; Hughes, P.E. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol. Biol. Cell 1999, 10, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Ruoslahti, E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 2005, 11, 1346–1350. [Google Scholar] [CrossRef]
- Li, F.; Sawada, J.; Komatsu, M. R-Ras-Akt axis induces endothelial lumenogenesis and regulates the patency of regenerating vasculature. Nat. Commun. 2017, 8, 1720. [Google Scholar] [CrossRef]
- Sawada, J.; Urakami, T.; Li, F.; Urakami, A.; Zhu, W.; Fukuda, M.; Li, D.Y.; Ruoslahti, E.; Komatsu, M. Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 2012, 22, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, G.S.; Grundl, M.; Allen, J.S., 3rd; Matter, M.L. R-Ras interacts with filamin a to maintain endothelial barrier function. J. Cell Physiol. 2011, 226, 2287–2296. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.I.; Sato, T.; Nagamine, K.; Hori, T. The inhibition of Bax activation-induced apoptosis by RasGRP2 via R-Ras-PI3K-Akt signaling pathway in the endothelial cells. Sci. Rep. 2019, 9, 16717. [Google Scholar] [CrossRef] [Green Version]
- Nakagami, H.; Morishita, R.; Yamamoto, K.; Yoshimura, S.I.; Taniyama, Y.; Aoki, M.; Matsubara, H.; Kim, S.; Kaneda, Y.; Ogihara, T. Phosphorylation of p38 mitogen-activated protein kinase downstream of bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes 2001, 50, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Jiang, Y.; Fu, T.; Hao, Y.; Zhu, X.; Lu, Y. Naringin inhibits lipopolysaccharide-induced damage in human umbilical vein endothelial cells via attenuation of inflammation, apoptosis and MAPK pathways. Cytotechnology 2016, 68, 1473–1487. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.A.; Rawlings, D.; Reynolds, H.; Vigorito, E.; et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Bio. Biophys. Acta. Mol. Cell Biol. Lipids 2015, 1851, 882–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banham-Hall, E.; Clatworthy, M.R.; Okkenhaug, K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. Open Rheumatol. J. 2012, 6, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Perez de Castro, I.; Diaz, R.; Malumbres, M.; Hernandez, M.I.; Jagirdar, J.; Jimenez, M.; Ahn, D.; Pellicer, A. Mice deficient for N-ras: Impaired antiviral immune response and T-cell function. Cancer Res. 2003, 63, 1615–1622. [Google Scholar]
- Kurig, B.; Shymanets, A.; Bohnacker, T.; Prajwal; Brock, C.; Ahmadian, M.R.; Schaefer, M.; Gohla, A.; Harteneck, C.; Wymann, M.P.; et al. Ras is an indispensable coregulator of the class IB phosphoinositide 3-kinase p87/p110gamma. Proc. Natl. Acad. Sci. USA 2009, 106, 20312–20317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, P.; Dorner, M.B.; Schaefer, M. Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006, 281, 9977–9986. [Google Scholar] [CrossRef] [Green Version]
- Nürnberg, B.; Beer-Hammer, S. Function, Regulation and Biological Roles of PI3Kγ Variants. Biol. Mol. 2019, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Shymanets, A.; Prajwal; Bucher, K.; Beer-Hammer, S.; Harteneck, C.; Nurnberg, B. p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J. Biol. Chem. 2013, 288, 31059–31068. [Google Scholar] [CrossRef] [Green Version]
- Rynkiewicz, N.K.; Anderson, K.E.; Suire, S.; Collins, D.M.; Karanasios, E.; Vadas, O.; Williams, R.; Oxley, D.; Clark, J.; Stephens, L.R.; et al. Gbetagamma is a direct regulator of endogenous p101/p110gamma and p84/p110gamma PI3Kgamma complexes in mouse neutrophils. Sci. Signal 2020, 13, 1289. [Google Scholar] [CrossRef] [PubMed]
- Deladeriere, A.; Gambardella, L.; Pan, D.; Anderson, K.E.; Hawkins, P.T.; Stephens, L.R. The regulatory subunits of PI3Kgamma control distinct neutrophil responses. Sci. Signal 2015, 8, ra8. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.R.; Gogvadze, E.; Xavier, A.R.; Bohnacker, T.; Voelzmann, J.; Wymann, M.P. PI3Kgamma Regulatory Protein p84 Determines Mast Cell Sensitivity to Ras Inhibition-Moving Towards Cell Specific PI3K Targeting? Front. Immunol. 2020, 11, 585070. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a New Gβγ-Activated Regulatory Subunit of the Type IB Phosphoinositide 3-Kinase p110γ. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Suire, S.; Baltanas, F.C.; Segonds-Pichon, A.; Davidson, K.; Santos, E.; Hawkins, P.T.; Stephens, L.R. Frontline Science: TNF-alpha and GM-CSF1 priming augments the role of SOS1/2 in driving activation of Ras, PI3K-gamma, and neutrophil proinflammatory responses. J. Leukoc. Biol. 2019, 106, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suire, S.; Lecureuil, C.; Anderson, K.E.; Damoulakis, G.; Niewczas, I.; Davidson, K.; Guillou, H.; Pan, D.; Jonathan, C.; Phillip, T.H.; et al. GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J. 2012, 31, 3118–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffargue, M.; Calvez, R.; Finan, P.; Trifilieff, A.; Barbier, M.; Altruda, F.; Hirsch, E.; Wymann, M.P. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 2002, 16, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, A.; Ayesh, H.S.K.; Halawani, H. PIK3CA Gene Mutations in Solid Malignancies: Association with Clinicopathological Parameters and Prognosis. Cancers 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.E.; Williams, R.L. Synergy in activating class I PI3Ks. Trends Biochem. Sci. 2015, 40, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Miled, N.; Yan, Y.; Perisic, W.-C.H.O.; Inbar, M.Z.Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of Two Classes of Cancer Mutations in the Phosphoinositide 3-Kinase Catalytic Subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, A.; Bussaglia, E.; Pallares, J.; Dolcet, X.; Llobet, D.; Encinas, M.; Llecha, N.; Palacios, J.; Prat, J.; Matias-Guiu, X. PIK3CA gene mutations in endometrial carcinoma: Correlation with PTEN and K-RAS alterations. Hum. Pathol. 2006, 37, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lee, J.E.; Lee, S.S.; Kim, C.; Lee, S.J.; Jang, W.S.; Park, S. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int. J. Cancer 2013, 133, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, H.; Pan, Y.; Wang, R.; Li, Y.; Shen, L.; Yu, Y.; Li, H.; Cai, D.; Sun, Y.; et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE 2014, 9, e88291. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef]
- Leone, V.; di Palma, A.; Ricchi, P.; Acquaviva, F.; Giannouli, M.; Di Prisco, A.M.; Iuliano, F.; Acquaviva, A.M. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G673–G681. [Google Scholar] [CrossRef]
- Zhan, H.; Bhattacharya, S.; Cai, H.; Iglesias, P.A.; Huang, C.H.; Devreotes, P.N. An Excitable Ras/PI3K/ERK Signaling Network Controls Migration and Oncogenic Transformation in Epithelial Cells. Dev. Cell 2020, 54, 608–623. [Google Scholar] [CrossRef]
- Jiang, K.; Sun, J.; Cheng, J.; Djeu, J.Y.; Wei, S.; Sebti, S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol. Cell Biol. 2004, 24, 5565–5576. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Sheridan, C.; Thin, M.Z.; Nye, E.; Spencer-Dene, B.; Diefenbacher, M.E.; Moore, C.; Kumar, M.S.; Murillo, M.M.; Gronroos, E.; et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell 2013, 24, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Counter, C.M. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell 2005, 8, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Hancock, D.C.; Sheridan, C.; Kumar, M.S.; Downward, J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013, 3, 548–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebi, H.; Corcoran, R.B.; Singh, A.; Chen, Z.; Song, Y.; Lifshits, E.; Ryan, D.P.; Meyerhardt, J.A.; Benes, C.; Settleman, J.; et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J. Clin. Investig. 2011, 121, 4311–4321. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Cintas, C.; Dufresne, M.; Cassant-Sourdy, S.; Schonhuber, N.; Planque, L.; Lulka, H.; Couderc, B.; Bousquet, C.; Garmy-Susini, B.; et al. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes. Dev. 2014, 28, 2621–2635. [Google Scholar] [CrossRef] [Green Version]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, D.W.; Watson, T.; Kung, H.F.; Curran, T. Microinjection of transforming ras protein induces c-fos expression. Mol. Cell Biol. 1987, 7, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Gutman, A.; Wasylyk, C.; Wasylyk, B. Cell-specific regulation of oncogene-responsive sequences of the c-fos promoter. Mol. Cell Biol. 1991, 11, 5381–5387. [Google Scholar] [CrossRef] [Green Version]
- Urich, M.; Senften, M.; Shaw, P.E.; Ballmer-Hofer, K. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene 1997, 14, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwick, J.K.; Cox, A.D.; Der, C.J.; Cobb, M.H.; Hibi, M.; Karin, M.; Brenner, D.A. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 6030–6034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finco, T.S.; Westwick, J.K.; Norris, J.L.; Beg, A.A.; Der, C.J.; Baldwin, A.S., Jr. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J. Biol. Chem. 1997, 272, 24113–24116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Pellicer, A. RAS pathways to cell cycle control and cell transformation. Front. Biol. Sci. 1998, 3, d887–d912. [Google Scholar] [CrossRef]
- Filmus, J.; Robles, A.I.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar] [PubMed]
- Albanese, C.; Johnson, J.; Watanabe, G.; Eklund, N.; Vu, D.; Arnold, A.; Pestell, R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995, 270, 23589–23597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, J.T.; Coats, S.R.; Wang, Y.Z.; Pledger, W.J. Regulation of the cell cycle machinery by oncogenic ras. Oncogene 1996, 12, 127–134. [Google Scholar]
- Liu, J.J.; Chao, J.R.; Jiang, M.C.; Ng, S.Y.; Yen, J.J.; Yang-Yen, H.F. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell Biol. 1995, 15, 3654–3663. [Google Scholar] [CrossRef] [Green Version]
- Gille, H.; Downward, J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwick, J.K.; Lambert, Q.T.; Clark, G.J.; Symons, M.; Van Aelst, L.; Pestell, R.G.; Der, C.J. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol. Cell Biol. 1997, 17, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol. Cell Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes. Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Ottmann, O.G.; Deininger, M.; Hochhaus, A. Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica 2014, 99, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Janas, M.L.; Turner, M. Interaction of Ras with P110γ Is Required for Thymic β-Selection in the Mouse. J. Immunol. 2011, 187, 4667–4675. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Alam, H.; Tang, M.; Maitituoheti, M.; Dhar, S.S.; Kumar, M.; Han, C.Y.; Ambati, C.R.; Amin, S.B.; Gu, B.; Chen, T.Y.; et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 2020, 37, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Kim, K.H.; Sung, H.; Paraiso, K.H.; Dannenberg, J.H.; Bosenberg, M.; Chin, L.; Kim, M. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 2010, 29, 6222–6232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakimoto, H.; Tanaka, S.; Curry, W.T.; Loebel, F.; Zhao, D.; Tateishi, K.; Chen, J.; Klofas, L.K.; Lelic, N.; Kim, J.C.; et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res. 2014, 20, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Blenis, J. A nexus for cellular homeostasis: The interplay between metabolic and signal transduction pathways. Curr. Opin. Biotechnol. 2015, 34, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.G.; Sanders, A.J.; Katoh, M.; Ungefroren, H.; Gieseler, F.; Prince, M.; Thompson, S.K.; Zollo, M.; Spano, D.; Dhawan, P.; et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35, S244–S275. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Chiu, H.C.; Li, C.J.; Yiang, G.T.; Tsai, A.P.; Wu, M.Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef]
- Qu, T.; Zhao, Y.; Chen, Y.; Jin, S.; Fang, Y.; Jin, X.; Sun, L.; Ma, Y. Down-regulated MAC30 expression inhibits breast cancer cell invasion and EMT by suppressing Wnt/beta-catenin and PI3K/Akt signaling pathways. Int. J. Clin. Exp. Pathol. 2019, 12, 1888–1896. [Google Scholar]
- Xu, E.; Xia, X.; Jiang, C.; Li, Z.; Yang, Z.; Zheng, C.; Wang, X.; Du, S.; Miao, J.; Wang, F.; et al. GPER1 Silencing Suppresses the Proliferation, Migration, and Invasion of Gastric Cancer Cells by Inhibiting PI3K/AKT-Mediated EMT. Front. Cell Dev. Biol. 2020, 8, 591239. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Liang, C.; Li, Z.; An, D.; Ren, S.; Yue, C.; Wu, J. DcR3 induces proliferation, migration, invasion, and EMT in gastric cancer cells via the PI3K/AKT/GSK-3beta/beta-catenin signaling pathway. Onco Targets Ther. 2018, 11, 4177–4187. [Google Scholar] [CrossRef] [Green Version]
- Andreolas, C.; Kalogeropoulou, M.; Voulgari, A.; Pintzas, A. Fra-1 regulates vimentin during Ha-RAS-induced epithelial mesenchymal transition in human colon carcinoma cells. Int. J. Cancer 2008, 122, 1745–1756. [Google Scholar] [CrossRef]
- Li, B.; Xu, W.W.; Lam, A.K.Y.; Wang, Y.; Hu, H.F.; Guan, X.Y.; Qin, Y.R.; Saremi, N.; Tsao, S.W.; He, Q.Y.; et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 2017, 8, 38755–38766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.N.; Herrera, B.; Mikula, M.; Proell, V.; Fuchs, E.; Gotzmann, J.; Schulte-Hermann, R.; Beug, H.; Mikulits, W. Integration of Ras subeffector signaling in TGF-beta mediated late stage hepatocarcinogenesis. Carcinogenesis 2005, 26, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre-Perona, A.; Riesco-Eizaguirre, G.; Zaballos, M.A.; Santisteban, P. beta-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget 2016, 7, 49435–49449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annibaldi, A.; Dousse, A.; Martin, S.; Tazi, J.; Widmann, C. Revisiting G3BP1 as a RasGAP binding protein: Sensitization of tumor cells to chemotherapy by the RasGAP 317-326 sequence does not involve G3BP1. PLoS ONE 2011, 6, e29024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitard, E.; Parker, F.; Millon, R.; Abecassis, J.; Tocque, B. G3BP is overexpressed in human tumors and promotes S phase entry. Cancer Lett. 2001, 162, 213–221. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.H.; He, H.W.; Zhang, C.X.; Yu, D.K.; Shao, R.G. Downregulation of G3BPs inhibits the growth, migration and invasion of human lung carcinoma H1299 cells by suppressing the Src/FAK-associated signaling pathway. Cancer Gene Ther. 2013, 20, 622–629. [Google Scholar] [CrossRef]
- Zhang, L.N.; Zhao, L.; Yan, X.L.; Huang, Y.H. Loss of G3BP1 suppresses proliferation, migration, and invasion of esophageal cancer cells via Wnt/beta-catenin and PI3K/AKT signaling pathways. J. Cell Physiol. 2019, 234, 20469–20484. [Google Scholar] [CrossRef]
- Janda, E.; Litos, G.; Grunert, S.; Downward, J.; Beug, H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene 2002, 21, 5148–5159. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Giehl, K. Oncogenic Ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef]
- Grunert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; Borel Rinkes, I.H.; Voest, E.E.; Kranenburg, O. Control of colorectal metastasis formation by K-Ras. Bio.Chim. Biophys. Acta 2005, 1756, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005, 65, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- McCormick, B.; Chu, J.Y.; Vermeren, S. Cross-talk between Rho GTPases and PI3K in the neutrophil. Small GTPases 2019, 10, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Soriano, O.; Alcón-Pérez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819. [Google Scholar] [CrossRef]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [Green Version]
- Cain, R.J.; Ridley, A.J. Phosphoinositide 3-kinases in cell migration. Biol. Cell 2009, 101, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.E.; van Delft, S.; ten Klooster, J.P.; Reid, T.; van der Kammen, R.A.; Michiels, F.; Collard, J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 1998, 143, 1385–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liliental, J.; Moon, S.Y.; Lesche, R.; Mamillapalli, R.; Li, D.; Zheng, Y.; Sun, H.; Wu, H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 2000, 10, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaduwal, S.; Jeong, W.J.; Park, J.C.; Lee, K.H.; Lee, Y.M.; Jeon, S.H.; Lim, Y.B.; Min do, S.; Choi, K.Y. Sur8/Shoc2 promotes cell motility and metastasis through activation of Ras-PI3K signaling. Oncotarget 2015, 6, 33091–33105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, S.; Abdellatef, S.; Haddad, M.A.; Fakhoury, I.; El-Sibai, M. Hypoxia and EGF Stimulation Regulate VEGF Expression in Human Glioblastoma Multiforme (GBM) Cells by Differential Regulation of the PI3K/Rho-GTPase and MAPK Pathways. Cells 2019, 8, 1397. [Google Scholar] [CrossRef] [Green Version]
- Jebri, I.; Tsujita, K.; Fujita, Y.; Itoh, T. Non-cell-autonomous migration of RasV12-transformed cells towards the basal side of surrounding normal cells. Biochem. Biophys. Res. Commun. 2021, 543, 15–22. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Hung, M.C. Induction of Akt activity by chemotherapy confers acquired resistance. J. Formos. Med. Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Iida, M.; Harari, P.M.; Wheeler, D.L.; Toulany, M. Targeting AKT/PKB to improve treatment outcomes for solid tumors. Mutat. Res. 2020, 819–820, 111690. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Bakanauskas, V.J.; Cerniglia, G.J.; Cheng, Y.; Bernhard, E.J.; Muschel, R.J.; McKenna, W.G. The Ras radiation resistance pathway. Cancer Res. 2001, 61, 4278–4282. [Google Scholar] [PubMed]
- Toulany, M.; Dittmann, K.; Baumann, M.; Rodemann, H.P. Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS. Radiother Oncol. 2005, 74, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; Bae, S.S.; Fernandes, A.; Wu, J.; Muschel, R.J.; McKenna, W.G.; Birnbaum, M.J.; Bernhard, E.J. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 2005, 65, 7902–7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.Y.; Hu, S.L.; Xu, X.Y.; Wang, R.Z.; Yu, H.Y.; Xu, J.Y.; Chen, L.; Dong, G.L. Nimotuzumab enhances the radiosensitivity of cancer cells in vitro by inhibiting radiation-induced DNA damage repair. PLoS ONE 2013, 8, e70727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; McKenna, W.G.; Weber, C.N.; Feldman, M.D.; Goldsmith, J.D.; Mick, R.; Machtay, M.; Rosenthal, D.I.; Bakanauskas, V.J.; Cerniglia, G.J.; et al. Local recurrence in head and neck cancer: Relationship to radiation resistance and signal transduction. Clin. Cancer Res. 2002, 8, 885–892. [Google Scholar]
- Kim, T.J.; Lee, J.W.; Song, S.Y.; Choi, J.J.; Choi, C.H.; Kim, B.G.; Lee, J.H.; Bae, D.S. Increased expression of pAKT is associated with radiation resistance in cervical cancer. Br. J. Cancer 2006, 94, 1678–1682. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [Green Version]
- Minjgee, M.; Toulany, M.; Kehlbach, R.; Giehl, K.; Rodemann, H.P. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int. J. Radiatoncol. Biol. 2011, 81, 1506–1514. [Google Scholar] [CrossRef]
- Oeck, S.; Al-Refae, K.; Riffkin, H.; Wiel, G.; Handrick, R.; Klein, D.; Iliakis, G.; Jendrossek, V. Activating Akt1 mutations alter DNA double strand break repair and radiosensitivity. Sci. Rep. 2017, 7, 42700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, F.M.; Prior, I.A.; MacEwan, D.J. The importance of Ras in drug resistance in cancer. Br. J. Pharm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.J.; Ryu, Y.K.; Kim, S.Y.; Wu, H.G.; Kim, J.S.; Kim, I.H.; Kim, I.A. Targeting epidermal growth factor receptor-associated signaling pathways in non-small cell lung cancer cells: Implication in radiation response. Mol. Cancer Res. 2010, 8, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef] [PubMed]
- Weykamp, F.; Seidensaal, K.; Rieken, S.; Green, K.; Mende, S.; Zaoui, K.; Freier, K.; Adeberg, S.; Debus, J.; Welte, S.E. Age-dependent hemato- and nephrotoxicity in patients with head and neck cancer receiving chemoradiotherapy with weekly cisplatin. Strahlenther. Onkol. 2020, 196, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Caiola, E.; Salles, D.; Frapolli, R.; Lupi, M.; Rotella, G.; Ronchi, A.; Garassino, M.C.; Mattschas, N.; Colavecchio, S.; Broggini, M.; et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget 2015, 6, 30072–30087. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 2014, 20, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Garassino, M.C.; Marabese, M.; Rusconi, P.; Rulli, E.; Martelli, O.; Farina, G.; Scanni, A.; Broggini, M. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann. Oncol. 2011, 22, 235–237. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Luo, H.; Luo, D.; Yang, H.; Zhou, X. Src homology phosphotyrosyl phosphatase 2 mediates cisplatin-related drug resistance by inhibiting apoptosis and activating the Ras/PI3K/Akt1/survivin pathway in lung cancer cells. Oncol. Rep. 2018, 39, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Overcoming resistance to BRAF inhibitors. Ann. Transl. Med. 2017, 5, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, Y.N.; Deng, W.; Woodman, S.E.; Komurov, K.; Ram, P.; Smith, P.D.; Davies, M.A. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010, 70, 8736–8747. [Google Scholar] [CrossRef] [Green Version]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, P.P.; Cardone, C.; Martini, G.; Ciardiello, D.; Belli, V.; Matrone, N.; Barra, G.; Napolitano, S.; Della Corte, C.; Turano, M.; et al. Receptor tyrosine kinase-dependent PI3K activation is an escape mechanism to vertical suppression of the EGFR/RAS/MAPK pathway in KRAS-mutated human colorectal cancer cell lines. J. Exp. Clin. Cancer Res. 2019, 38, 41. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Won, J.K.; Yang, H.W.; Shin, S.Y.; Lee, J.H.; Heo, W.D.; Cho, K.H. The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J. Mol. Cell Biol. 2012, 4, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Amgen overcomes historically undruggable target, with FDA nod for first KRAS inhibitor. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Hata, A.N.; Shaw, A.T. Resistance looms for KRAS(G12C) inhibitors. Nat. Med. 2020, 26, 169–170. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRAS (G12C) Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.B.; Fece de la Cruz, F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS (G12C) Inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Massarelli, E.; Varella-Garcia, M.; Tang, X.; Xavier, A.C.; Ozburn, N.C.; Liu, D.D.; Bekele, B.N.; Herbst, R.S.; Wistuba, I.I. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin. Cancer Res. 2007, 13, 2890–2896. [Google Scholar] [CrossRef] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef]
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixido, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzon, M.; Codony-Servat, C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [Green Version]
- Riess, C.; Shokraie, F.; Classen, C.F.; Kreikemeyer, B.; Fiedler, T.; Junghanss, C.; Maletzki, C. Arginine-Depleting Enzymes—An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol. Biochem. 2018, 51, 854–870. [Google Scholar] [CrossRef]
- Tsai, W.B.; Aiba, I.; Long, Y.; Lin, H.K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Gebbink, M.F.; Voest, E.E. Stimulation of angiogenesis by Ras proteins. Biochim. Biophys. Acta. 2004, 1654, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Okada, F.; Rak, J.W.; Croix, B.S.; Lieubeau, B.; Kaya, M.; Roncari, L.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Impact of oncogenes in tumor angiogenesis: Mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3609–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar] [PubMed]
- Zhang, X.; Gaspard, J.P.; Chung, D.C. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001, 61, 6050–6054. [Google Scholar] [PubMed]
- Jung, F.; Haendeler, J.; Hoffmann, J.; Reissner, A.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Hypoxic induction of the hypoxia-inducible factor is mediated via the adaptor protein Shc in endothelial cells. Circ. Res. 2002, 91, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Blancher, C.; Moore, J.W.; Robertson, N.; Harris, A.L. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1alpha, HIF-2alpha, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3’-kinase/Akt signaling pathway. Cancer Res. 2001, 61, 7349–7355. [Google Scholar] [PubMed]
- Lee, E.; Yim, S.; Lee, S.K.; Park, H. Two transactivation domains of hypoxia-inducible factor-1alpha regulated by the MEK-1/p42/p44 MAPK pathway. Mol. Cells 2002, 14, 9–15. [Google Scholar]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Jo, W.S.; Duerr, E.M.; Gala, M.; Li, J.; Zhang, X.; Zimmer, M.A.; Iliopoulos, O.; Zukerberg, L.R.; Kohgo, Y.; et al. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 2005, 11, 992–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Pathak, P.S.; Fukumura, D. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol 3’-kinase signaling pathways contributes to expression of interleukin 8 in human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunaga, N.; Imai, H.; Shimizu, K.; Shames, D.S.; Kakegawa, S.; Girard, L.; Sato, M.; Kaira, K.; Ishizuka, T.; Gazdar, A.F.; et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int. J. Cancer 2012, 130, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Ancrile, B.B.; O’Hayer, K.M.; Counter, C.M. Oncogenic ras-induced expression of cytokines: A new target of anti-cancer therapeutics. Mol. Interv. 2008, 8, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Rak, J.; Mitsuhashi, Y.; Sheehan, C.; Tamir, A.; Viloria-Petit, A.; Filmus, J.; Mansour, S.J.; Ahn, N.G.; Kerbel, R.S. Oncogenes and tumor angiogenesis: Differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000, 60, 490–498. [Google Scholar] [PubMed]
- Watnick, R.S.; Cheng, Y.N.; Rangarajan, A.; Ince, T.A.; Weinberg, R.A. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell 2003, 3, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Mazure, N.M.; Chen, E.Y.; Laderoute, K.R.; Giaccia, A.J. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997, 90, 3322–3331. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Meng, Q.; Cao, Z.; Shi, X.; Jiang, B.H. Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J. Cell Physiol. 2006, 209, 56–66. [Google Scholar] [CrossRef]
- Borrello, M.G.; Degl’Innocenti, D.; Pierotti, M.A. Inflammation and cancer: The oncogene-driven connection. Cancer Lett. 2008, 267, 262–270. [Google Scholar] [CrossRef]
- Feng, Y.; Santoriello, C.; Mione, M.; Hurlstone, A.; Martin, P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: Parallels between tumor initiation and wound inflammation. PLoS Biol. 2010, 8, e1000562. [Google Scholar] [CrossRef] [Green Version]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Sawaoka, H.; Hori, M.; DuBois, R.N. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 93, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Dormond, O.; Foletti, A.; Paroz, C.; Ruegg, C. NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat. Med. 2001, 7, 1041–1047. [Google Scholar] [CrossRef]
- Sui, W.; Zhang, Y.; Wang, Z.; Wang, Z.; Jia, Q.; Wu, L.; Zhang, W. Antitumor effect of a selective COX-2 inhibitor, celecoxib, may be attributed to angiogenesis inhibition through modulating the PTEN/PI3K/Akt/HIF-1 pathway in an H(2)(2) murine hepatocarcinoma model. Oncol. Rep. 2014, 31, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Jia, Q.; Song, H.Y.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J. Biol. Chem. 1995, 270, 6729–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, H.P.; McMurtrey, A.; Kowalski, J.; Yan, M.; Keyt, B.A.; Dixit, V.; Ferrara, N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998, 273, 30336–30343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, T.L.; Choi, H.S.; Matsui, A.; Benes, C.; Lifshits, E.; Luo, J.; Frangioni, J.V.; Cantley, L.C. Class 1A PI3K regulates vessel integrity during development and tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 9739–9744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo, M.M.; Zelenay, S.; Nye, E.; Castellano, E.; Lassailly, F.; Stamp, G.; Downward, J. RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J. Clin. Investig. 2014, 124, 3601–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, A.; Serra, H.; Pearce, W.; Angulo, A.; Guillermet-Guibert, J.; Friedman, L.S.; Vinals, F.; Gerhardt, H.; Casanovas, O.; Graupera, M.; et al. Inhibition of the p110alpha isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J. Exp. Med. 2013, 210, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, V.; Toh, H.C.; Abastado, J.P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Houghton, A.M.; Mariani, T.J.; Perera, S.; Kim, C.B.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.A.; Hezel, A.; et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Deng, S.; Clowers, M.J.; Velasco, W.V.; Ramos-Castaneda, M.; Moghaddam, S.J. Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 2019, 9, 1556. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [Green Version]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes. Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Mummidi, S.; Cortez, D.M.; Prabhu, S.D.; Valente, A.J.; Chandrasekar, B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J. Physiol. Heart Circ. Physiol. 2008, 294, H2078–H2087. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Baker, J.R.; Vuppusetty, C.; Fenwick, P.; Donnelly, L.E.; Ito, K.; Barnes, P.J. Decreased phosphatase PTEN amplifies PI3K signaling and enhances proinflammatory cytokine release in COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L230–L239. [Google Scholar] [CrossRef] [Green Version]
- Hutti, J.E.; Pfefferle, A.D.; Russell, S.C.; Sircar, M.; Perou, C.M.; Baldwin, A.S. Oncogenic PI3K mutations lead to NF-kappaB-dependent cytokine expression following growth factor deprivation. Cancer Res. 2012, 72, 3260–3269. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulielmaki, E.; Bermudez-Brito, M.; Andreou, M.; Tzenaki, N.; Tzardi, M.; de Bree, E.; Tsentelierou, E.; Makrigiannakis, A.; Papakonstanti, E.A. Pharmacological inactivation of the PI3K p110delta prevents breast tumour progression by targeting cancer cells and macrophages. Cell Death Dis. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar]
- Weijzen, S.; Velders, M.P.; Kast, W.M. Modulation of the immune response and tumor growth by activated Ras. Leukemia 1999, 13, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, S.; Sasaki, M.; Scharer, C.D.; Kissick, H.T.; Patterson, D.G.; Magliocca, K.R.; Seykora, J.T.; Sapkota, B.; Gutman, D.A.; Cooper, L.A.; et al. Phosphoinositide 3-Kinase Signaling Can Modulate MHC Class I and II Expression. Mol. Cancer Res. 2019, 17, 2395–2409. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNgamma receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- El-Jawhari, J.J.; El-Sherbiny, Y.M.; Scott, G.B.; Morgan, R.S.; Prestwich, R.; Bowles, P.A.; Blair, G.E.; Tanaka, T.; Rabbitts, T.H.; Meade, J.L.; et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol. Immunol. 2014, 58, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, S.; Wollscheid, U.; Huber, C.; Seliger, B. Multiple levels of MHC class I down-regulation by ras oncogenes. Scand. J. Immunol. 1996, 43, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Bacchetta, R.; Schulz, U.; Roncarolo, M.G. The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef] [Green Version]
- Granville, C.A.; Memmott, R.M.; Balogh, A.; Mariotti, J.; Kawabata, S.; Han, W.; Lopiccolo, J.; Foley, J.; Liewehr, D.J.; Steinberg, S.M.; et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS ONE 2009, 4, e5061. [Google Scholar] [CrossRef] [PubMed]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant K-Ras Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Bilate, A.M.; Lafaille, J.J. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 2012, 30, 733–758. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.D.; Bork, J.P.; Krueger, B.; Patsenker, E.; Schulze-Krebs, A.; Hahn, E.G.; Schuppan, D. VEGF induces proliferation, migration, and TGF-beta1 expression in mouse glomerular endothelial cells via mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 2005, 334, 1049–1060. [Google Scholar] [CrossRef]
- Lee, S.J.; Yang, E.K.; Kim, S.G. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: Role for Zf9 dephosphorylation. Mol. Pharm. 2006, 70, 415–425. [Google Scholar] [CrossRef]
- Lee, K.S.; Park, S.J.; Kim, S.R.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Lee, Y.R.; Kim, J.S.; Hong, S.J.; et al. Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur. Respir. J. 2008, 31, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018, 103, 1065–1076. [Google Scholar] [CrossRef]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Lim, E.L.; Bouabe, H.; Scudamore, C.L.; Hancox, T.; Maecker, H.; et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Carnevalli, L.S.; Sinclair, C.; Taylor, M.A.; Gutierrez, P.M.; Langdon, S.; Coenen-Stass, A.M.L.; Mooney, L.; Hughes, A.; Jarvis, L.; Staniszewska, A.; et al. PI3Kalpha/delta inhibition promotes anti-tumor immunity through direct enhancement of effector CD8(+) T-cell activity. J. Immunother Cancer 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Sarabi, M.; Pereira Abrantes, M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Jia, L.P.; Liu, Y.; Han, Y.; Deng, Q. Alteration of tumor associated neutrophils by PIK3CA expression in endometrial carcinoma from TCGA data. J. Ovarian. Res. 2019, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef] [Green Version]
- Torphy, R.J.; Zhu, Y.; Schulick, R.D. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann. Gastroenterol. Surg. 2018, 2, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Scilla, K.; Caglevic, C.; Miller, K.; Oliveira, J.; Rolfo, C. Immunotherapy in Lung Cancer: A New Age in Cancer Treatment. Adv. Exp. Med. Biol. 2018, 995, 65–95. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.; de Carne Trecesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wei, F.; Wang, J.; Yu, W.; Shen, M.; Liu, T.; Zhang, D.; Wang, Y.; Ren, X.; Sun, Q. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1(-)PD-L1(+) Bregs through PD-L1/PI3K/AKT/NF-kappaB axis in breast cancer. Cell Death Dis. 2021, 12, 465. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, T.S.; Chan, L.K.; Wong, E.C.; Hui, C.W.; Sneddon, K.; Cheung, T.H.; Yim, S.F.; Lee, J.H.; Yeung, C.S.; Chung, T.K.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFalpha-TGFalpha-EGFR. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, A.E.; Kandalam, V.; Chakrabarti, S.; Wang, X.; Penninger, J.M.; Davidge, S.T.; Oudit, G.Y.; Kassiri, Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am. J. Physiol. Cell Physiol. 2010, 298, C679–C692. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Bio. Sci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasca di Magliano, M.; Sekine, S.; Ermilov, A.; Ferris, J.; Dlugosz, A.A.; Hebrok, M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes. Dev. 2006, 20, 3161–3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic K-Ras Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; Pasca di Magliano, M.; Fernandez-Zapico, M.E. Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating K-Ras oncogene-induced transformation. J. Biol. Chem. 2013, 288, 11786–11794. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Saotome, T.; Usui, T.; Ohama, T.; Sato, K. Regulation of intestinal myofibroblasts by KRas-mutated colorectal cancer cells through heparin-binding epidermal growth factor-like growth factor. Oncol. Rep. 2017, 37, 3128–3136. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erler, J.T.; Weaver, V.M. Three-dimensional context regulation of metastasis. Clin. Exp. Metastasis 2009, 26, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Cox, T.R.; Bird, D.; Lang, G.; Murray, G.I.; Sun, X.F.; Southall, S.M.; Wilson, J.R.; Erler, J.T. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J. Natl. Cancer Inst. 2011, 103, 407–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Erler, J.T. Lysyl oxidase in colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G659–G666. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Takano, O.; Kato, I.; Takahashi, Y.; Shima, F.; Kataoka, T. Ras inhibitors display an anti-metastatic effect by downregulation of lysyl oxidase through inhibition of the Ras-PI3K-Akt-HIF-1alpha pathway. Cancer Lett. 2017, 410, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Wolfman, J.C.; Wolfman, A. K-ras regulates the steady-state expression of matrix metalloproteinase 2 in fibroblasts. J. Biol. Chem. 2003, 278, 31871–31878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Wang, Q.; Fu, X.H.; Huang, X.H.; Chen, X.L.; Cao, L.Q.; Chen, L.Z.; Tan, H.X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 2009, 39, 177–186. [Google Scholar] [CrossRef]
- Chen, X.C.; Wei, X.T.; Guan, J.H.; Shu, H.; Chen, D. EGF stimulates glioblastoma metastasis by induction of matrix metalloproteinase-9 in an EGFR-dependent mechanism. Oncotarget 2017, 8, 65969–65982. [Google Scholar] [CrossRef] [Green Version]
- Ding, P.; Zhang, X.; Jin, S.; Duan, B.; Chu, P.; Zhang, Y.; Chen, Z.N.; Xia, B.; Song, F. CD147 functions as the signaling receptor for extracellular divalent copper in hepatocellular carcinoma cells. Oncotarget 2017, 8, 51151–51163. [Google Scholar] [CrossRef] [Green Version]
- Kuang, W.; Deng, Q.; Deng, C.; Li, W.; Shu, S.; Zhou, M. Hepatocyte growth factor induces breast cancer cell invasion via the PI3K/Akt and p38 MAPK signaling pathways to up-regulate the expression of COX2. Am. J. Transl. Res. 2017, 9, 3816–3826. [Google Scholar]
- Zhou, R.; Xu, L.; Ye, M.; Liao, M.; Du, H.; Chen, H. Formononetin inhibits migration and invasion of MDA-MB-231 and 4T1 breast cancer cells by suppressing MMP-2 and MMP-9 through PI3K/AKT signaling pathways. Horm Metab. Res. 2014, 46, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Robert, L. Tumors perturbing extracellular matrix biosynthesis. The case of von Recklinghausen’s disease. Pathol. Biol. 2014, 62, 118–122. [Google Scholar] [CrossRef]
- Mehner, C.; Miller, E.; Khauv, D.; Nassar, A.; Oberg, A.L.; Bamlet, W.R.; Zhang, L.; Waldmann, J.; Radisky, E.S.; Crawford, H.C.; et al. Tumor cell-derived MMP3 orchestrates Rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol. Cancer Res. 2014, 12, 1430–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, K.; Choi, S.; Wyse, M.; Strnadel, J.; Wright, T.; Klemke, R. Eukaryotic Translation Initiation Factor 5A (EIF5A) Regulates Pancreatic Cancer Metastasis by Modulating RhoA and Rho-associated Kinase (ROCK) Protein Expression Levels. J. Biol. Chem. 2015, 290, 29907–29919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta. Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA approves first-in-class KRASinhibitor. Nat. Rev. Drug. Discov. 2021, 20, 496. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef] [PubMed]
- Stalnecker, C.A.; Der, C.J. RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci. Signal 2020, 13, 624. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Krause, G.; Hassenruck, F.; Hallek, M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug. Des. Devel. Ther. 2018, 12, 2577–2590. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Samaniego, F.; Jurczak, W.; Ghosh, N.; Derenzini, E.; Reeves, J.A.; Knopińska-Posłuszny, W.; Cheah, C.Y.; Phillips, T.; Lech-Maranda, E.; et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Saura, C.; Richards, D.A.; Krop, I.E.; Cervantes, A.; Bedard, P.L.; Patel, M.R.; Pusztai, L.; Oliveira, M.; Cardenas, A.K.; et al. Phase II Study of Taselisib (GDC-0032) in Combination with Fulvestrant in Patients with HER2-Negative, Hormone Receptor-Positive Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 4380–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Sanabria Bohorquez, S.M.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.J.; Furman, M.; Winer, E.S. CAL-101: A phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Exp. Opin. Investig. Drug. 2012, 21, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Qayum, N.; Im, J.; Stratford, M.R.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin. Cancer Res. 2012, 18, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Azad, A.K.; Zhabyeyev, P.; Vanhaesebroeck, B.; Eitzen, G.; Oudit, G.Y.; Moore, R.B.; Murray, A.G. Inactivation of endothelial cell phosphoinositide 3-kinase beta inhibits tumor angiogenesis and tumor growth. Oncogene 2020, 39, 6480–6492. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release 2020, 321, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, J.; Owens, P.; Novitskiy, S.V.; Hawkins, O.E.; Vilgelm, A.E.; Yang, J.; Sobolik, T.; Lavender, N.; Johnson, A.C.; McClain, C.; et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clin. Cancer Res. 2017, 23, 3371–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K pathway increases immune infiltrate in muscle-invasive bladder cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef]
Figure 1.
Ras effector pathways. GTP binding to Ras induces changes in protein conformation that increases Ras affinity for downstream effectors, thus activating many different signal transduction pathways. Among them, Raf/MEK/ERK, PI3K/AKT, and Ral are the best understood. Further, Ras crosslinks with other signaling pathways such as proteins from the Rho family and YAP. Through the regulation of the different downstream pathways, Ras proteins play a key role in the control of proliferation, survival, metastasis, apoptosis, migration, angiogenesis, and endocytosis.
Figure 1.
Ras effector pathways. GTP binding to Ras induces changes in protein conformation that increases Ras affinity for downstream effectors, thus activating many different signal transduction pathways. Among them, Raf/MEK/ERK, PI3K/AKT, and Ral are the best understood. Further, Ras crosslinks with other signaling pathways such as proteins from the Rho family and YAP. Through the regulation of the different downstream pathways, Ras proteins play a key role in the control of proliferation, survival, metastasis, apoptosis, migration, angiogenesis, and endocytosis.

Figure 2.
Mechanisms of activation of PI3K and downstream effectors. GCPRs and RTKs are upstream signals that control PI3K activation through direct interaction with the regulatory subunit of PI3K. Further, RTK can activate PI3K indirectly through Ras activation that in turn activates PI3K in a p110-dependent manner. Once activated, PI3K generates PIP3 that promotes AKT phosphorylation, which subsequently phosphorylates a large number of downstream targets to control cell survival, proliferation and apoptosis. Other PI3K effectors are TEC family tyrosine kinase, such as BTK, and GTPases of the Rho/Rac/cdc42 family. Activation of PI3K-AKT pathway is an important mechanism in the development of resistance to chemotherapy (through protection of drug-induced apoptosis [79]) and radiotherapy (through repair of radiation-induced DSBs [80,81]).
Figure 2.
Mechanisms of activation of PI3K and downstream effectors. GCPRs and RTKs are upstream signals that control PI3K activation through direct interaction with the regulatory subunit of PI3K. Further, RTK can activate PI3K indirectly through Ras activation that in turn activates PI3K in a p110-dependent manner. Once activated, PI3K generates PIP3 that promotes AKT phosphorylation, which subsequently phosphorylates a large number of downstream targets to control cell survival, proliferation and apoptosis. Other PI3K effectors are TEC family tyrosine kinase, such as BTK, and GTPases of the Rho/Rac/cdc42 family. Activation of PI3K-AKT pathway is an important mechanism in the development of resistance to chemotherapy (through protection of drug-induced apoptosis [79]) and radiotherapy (through repair of radiation-induced DSBs [80,81]).

Figure 3.
PI3K activation. Under unstimulated conditions, interactions between the regulatory and the catalytic subunit keep PI3Ks in the inactive state. The nSH2 and iSH2 (coiled portion) domains of p85 are the minimal fragment of the regulatory subunit required for full inhibition of p110α lipid kinase activity. In PI3K activation by RTKs, binding of p85 to phosphorylated RTKs disrupts the inhibitory contact between the regulatory and the catalytic subunit, which generates conformational changes in p110α for substrate catalysis. In PI3K activation by Ras, the interaction of Ras with p110 displaces nSH2 and iSH2 in p85 from the p110α subunit, facilitating the full activation of PI3Kα.
Figure 3.
PI3K activation. Under unstimulated conditions, interactions between the regulatory and the catalytic subunit keep PI3Ks in the inactive state. The nSH2 and iSH2 (coiled portion) domains of p85 are the minimal fragment of the regulatory subunit required for full inhibition of p110α lipid kinase activity. In PI3K activation by RTKs, binding of p85 to phosphorylated RTKs disrupts the inhibitory contact between the regulatory and the catalytic subunit, which generates conformational changes in p110α for substrate catalysis. In PI3K activation by Ras, the interaction of Ras with p110 displaces nSH2 and iSH2 in p85 from the p110α subunit, facilitating the full activation of PI3Kα.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cuesta, C.; Arévalo-Alameda, C.; Castellano, E. The Importance of Being PI3K in the RAS Signaling Network. Genes 2021, 12, 1094. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071094
AMA Style
Cuesta C, Arévalo-Alameda C, Castellano E. The Importance of Being PI3K in the RAS Signaling Network. Genes. 2021; 12(7):1094. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071094
Chicago/Turabian StyleCuesta, Cristina, Cristina Arévalo-Alameda, and Esther Castellano. 2021. "The Importance of Being PI3K in the RAS Signaling Network" Genes 12, no. 7: 1094. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071094
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.