
C9orf72-G4C2 Intermediate Repeats and Parkinson’s Disease; A Data-Driven Hypothesis


Abstract
:1. Introduction
2. Methods
2.1. Population
2.2. Determining the G4C2 Hexanucleotide Repeat Length in the C9orf72 Gene
2.3. Assembly of the Risk-Haplotype within the C9orf72 Locus
2.4. Statistical Analyses of C9orf72 G4C2 Hexanucleotide Repeats
3. Results
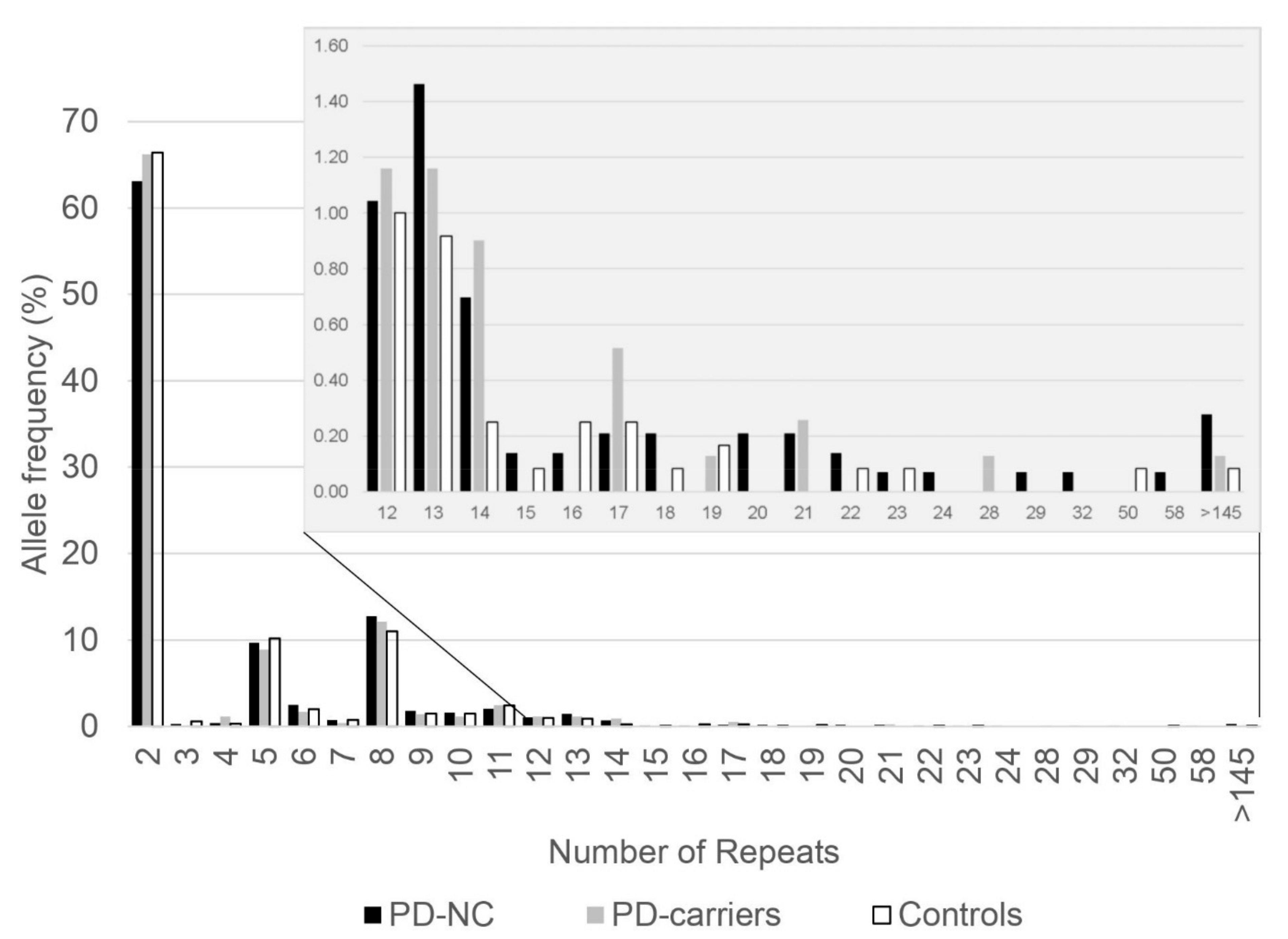
3.1. Allele Frequencies of C9orf72 G4C2 Hexanucleotide Repeats in Ashkenazi PD Patients
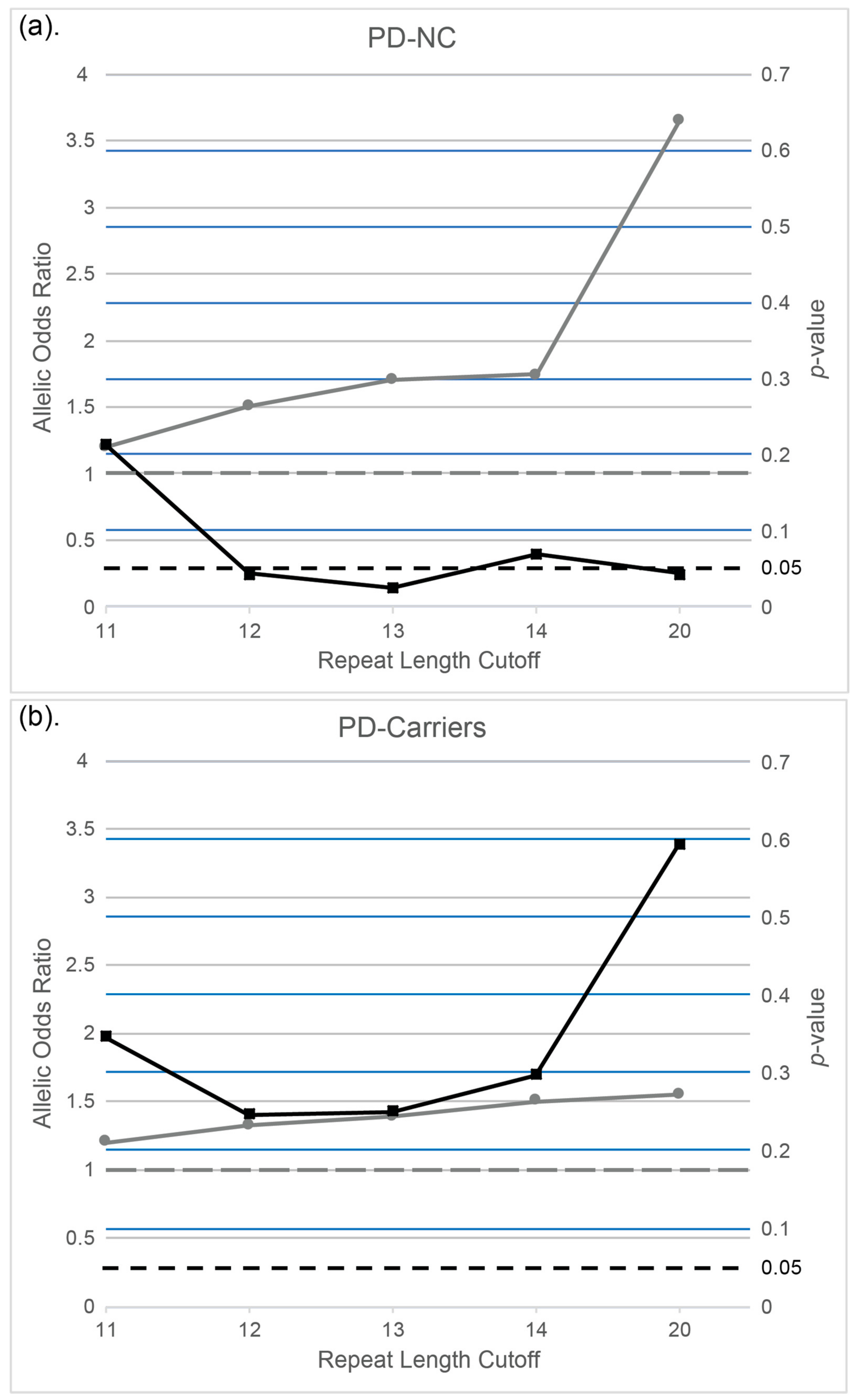
3.2. The Association of C9orf72 G4C2 Hexanucleotide Intermediate Repeat Lengths with PD in Ashkenazi Patients
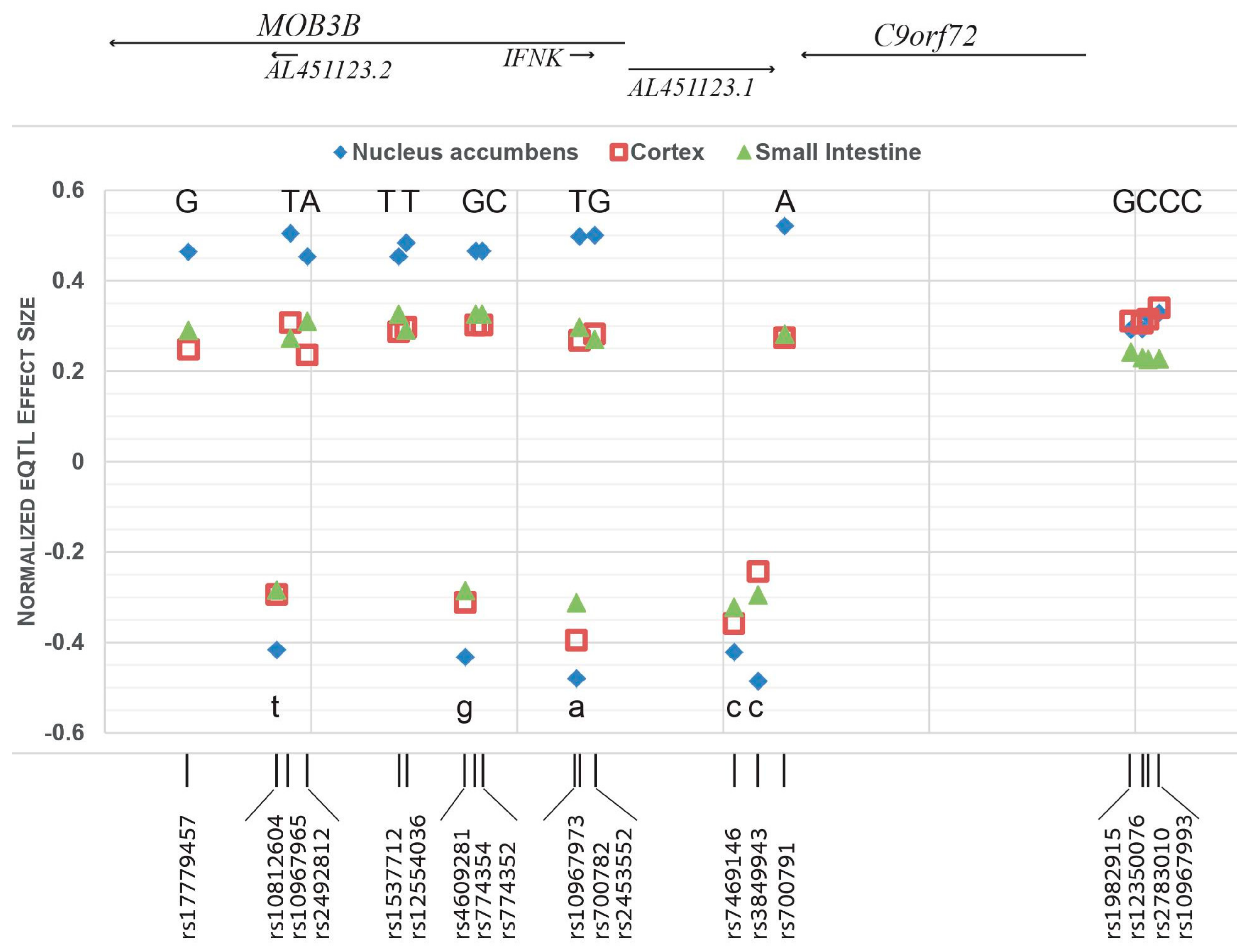
3.3. The C9orf72 Risk-Haplotype Is Associated with Higher RNA Expression Levels and with PD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Farrer, M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet. 2006, 7, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Gasser, T.; Hardy, J.; Mizuno, Y. Milestones in PD genetics. Mov. Disord. 2011, 26, 1042–1048. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Escott-Price, V.; Gibbs, J.R.; Nalls, M.A.; Bras, J.; Price, T.R.; Nicolas, A.; Jansen, I.E.; Mok, K.Y.; Pittman, A.M.; et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: Evidence for oligogenic inheritance. Hum. Mol. Genet. 2016, 25, 5483–5489. [Google Scholar] [CrossRef] [Green Version]
- Van Heesbeen, H.J.; Smidt, M.P. Entanglement of Genetics and Epigenetics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, J.T.; Zong, Y.; Zhou, J.; Tan, L. C9ORF72 mutations in neurodegenerative diseases. Mol. Neurobiol. 2014, 49, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Sun, Q.; Zhang, Y.; Xu, Q.; Guo, J.; Yan, X.; Tang, B. The Association between C9orf72 Repeats and Risk of Alzheimer’s Disease and Amyotrophic Lateral Sclerosis: A Meta-Analysis. Parkinsons Dis. 2016, 2016, 5731734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dols-Icardo, O.; Garcia-Redondo, A.; Rojas-Garcia, R.; Sanchez-Valle, R.; Noguera, A.; Gomez-Tortosa, E.; Pastor, P.; Hernandez, I.; Esteban-Perez, J.; Suarez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain J. Neurol. 2012, 135, 765–783. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Frolov, A.; Highley, J.R.; Charlesworth, G.; Kirby, J.; Milano, A.; Hartley, J.; Ince, P.G.; McDermott, C.J.; Lashley, T.; et al. C9ORF72 expansions, parkinsonism, and Parkinson disease: A clinicopathologic study. Neurology 2013, 81, 808–811. [Google Scholar] [CrossRef] [Green Version]
- Nuytemans, K.; Bademci, G.; Kohli, M.M.; Beecham, G.W.; Wang, L.; Young, J.I.; Nahab, F.; Martin, E.R.; Gilbert, J.R.; Benatar, M.; et al. C9ORF72 intermediate repeat copies are a significant risk factor for Parkinson disease. Ann. Hum. Genet. 2013, 77, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Majounie, E.; Abramzon, Y.; Renton, A.E.; Keller, M.F.; Traynor, B.J.; Singleton, A.B. Large C9orf72 repeat expansions are not a common cause of Parkinson’s disease. Neurobiol. Aging 2012, 33, 2527.e1–2527.e2. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.; Zinman, L.; Grinberg, Y.; Moreno, D.; Sato, C.; Bilbao, J.M.; Ghani, M.; Hernandez, I.; Ruiz, A.; Boada, M.; et al. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch. Neurol. 2012, 69, 1583–1590. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Le Ber, I.; Condroyer, C.; Broussolle, E.; Gabelle, A.; Thobois, S.; Pasquier, F.; Mondon, K.; Dion, P.A.; Rochefort, D.; et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain J. Neurol. 2013, 136, 385–391. [Google Scholar] [CrossRef]
- Jiao, B.; Guo, J.F.; Wang, Y.Q.; Yan, X.X.; Zhou, L.; Liu, X.Y.; Zhang, F.F.; Zhou, Y.F.; Xia, K.; Tang, B.S.; et al. C9orf72 mutation is rare in Alzheimer’s disease, Parkinson’s disease, and essential tremor in China. Front. Cell. Neurosci. 2013, 7, 164. [Google Scholar] [CrossRef] [Green Version]
- Theuns, J.; Verstraeten, A.; Sleegers, K.; Wauters, E.; Gijselinck, I.; Smolders, S.; Crosiers, D.; Corsmit, E.; Elinck, E.; Sharma, M.; et al. Global investigation and meta-analysis of the C9orf72 (G4C2)n repeat in Parkinson disease. Neurology 2014, 83, 1906–1913. [Google Scholar] [CrossRef] [Green Version]
- Bourinaris, T.; Houlden, H. C9orf72 and its Relevance in Parkinsonism and Movement Disorders: A Comprehensive Review of the Literature. Mov. Disord. Clin. Pract. 2018, 5, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Orr-Urtreger, A.; Shifrin, C.; Rozovski, U.; Rosner, S.; Bercovich, D.; Gurevich, T.; Yagev-More, H.; Bar-Shira, A.; Giladi, N. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: Is there a gender effect? Neurology 2007, 69, 1595–1602. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, O.; Gana-Weisz, M.; Nefussy, B.; Vainer, B.; Nayshool, O.; Bar-Shira, A.; Traynor, B.J.; Drory, V.E.; Orr-Urtreger, A. High frequency of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis patients from two founder populations sharing the same risk haplotype. Neurobiol. Aging 2017. [Google Scholar] [CrossRef]
- Goldstein, O.; Nayshool, O.; Nefussy, B.; Traynor, B.J.; Renton, A.E.; Gana-Weisz, M.; Drory, V.E.; Orr-Urtreger, A. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 2016, 86, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, O.; Gana-Weisz, M.; Cohen-Avinoam, D.; Shiner, T.; Thaler, A.; Cedarbaum, J.M.; John, S.; Lalioti, M.; Gurevich, T.; Bar-Shira, A.; et al. Revisiting the non-Gaucher-GBA-E326K carrier state: Is it sufficient to increase Parkinson’s disease risk? Mol. Genet. Metab. 2019, 128, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Ozelius, L.J.; Clark, L.N.; Bar-Shira, A.; Gana-Weisz, M.; Gurevich, T.; Gusev, A.; Kedmi, M.; Kenny, E.E.; Liu, X.; et al. Genome-wide mapping of IBD segments in an Ashkenazi PD cohort identifies associated haplotypes. Hum. Mol. Genet. 2014, 23, 4693–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Lee, T.; Li, Y.R.; Ingre, C.; Weber, M.; Grehl, T.; Gredal, O.; de Carvalho, M.; Meyer, T.; Tysnes, O.B.; Auburger, G.; et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum. Mol. Genet. 2011, 20, 1697–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattante, S.; Pomponi, M.G.; Conte, A.; Marangi, G.; Bisogni, G.; Patanella, A.K.; Meleo, E.; Lunetta, C.; Riva, N.; Mosca, L.; et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol. Aging 2017. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.; Zhang, M.; Bruni, A.C.; Maletta, R.G.; Colao, R.; Fratta, P.; Polke, J.M.; Sweeney, M.G.; Mudanohwo, E.; Nacmias, B.; et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015, 129, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Waite, A.J.; Baumer, D.; East, S.; Neal, J.; Morris, H.R.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779.e5–1779.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, C.P.; Patino, M.; Tai, Y.K.; Ho, W.Y.; McLean, C.A.; Morris, C.M.; Seeley, W.W.; Miller, B.L.; Gaig, C.; Vonsattel, J.P.G.; et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 2019, 138, 795–811. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Non-Carrier PD Patients a | PD Patients Carriers of LRRK2, GBA, or SMPD1 Mutations | |
|---|---|---|
| N | 718 | 388 c |
| Women, N (%) | 266 (37.0) | 171 (44.1) |
| AAE, mean (SD), y | 68.5 (10.2) d | 65.4 (10.1) |
| AAO, mean (SD), y | 61.4 (11.5) | 58.4 (10.6) |
| Family history of PD b, N (%) | 144 (20) | 120 (30.9) |
| Cohort | Non-Carrier PD Patients a | PD Patients Carriers of LRRK2, GBA, or SMPD1 Mutations | Controls b |
|---|---|---|---|
| 2–19 repeats, N (%) | 701 (98.2) | 384 (99.2) | 596 (99.5) |
| 20–60 repeats, N (%) | 13 (1.8) | 3 (0.8) | 3 (0.5) |
| Odds Ratio (95% CI) | 3.684 (1.045–12.990) | 1.552 (0.312–7.729) | |
| p-value c | 0.041 | 0.684 |
| Location, chr9 (hg38) | rs ID | Gene | Risk Allele | Ref > Alt a | Highest eQTL for C9orf72, NES (Tissue) | eQTL for C9orf72 in Cerebellum, NES | Highest sQTL for C9orf72, NES (Tissue) | GnomAD v3.1- AF for the Risk Allele (in Non-Neuro Cases) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| AJ | European (Non-Finnish) | All Populations | ||||||||
| 27488094 | rs17779457 | MOB3B | G | T > G | 0.464 (N.A.) | 0.341 | −0.88 (Cer) | 0.2097 | 0.2428 | 0.2490 |
| 27496663 | rs10812604 | MOB3B | T | T > G | −0.416 (N.A.) | −0.343 | 0.70 (Cer) | 0.2252 | 0.2861 | 0.3555 |
| 27497990 | rs10967965 | MOB3B | T | A > T | 0.505 (N.A.) | 0.360 | −0.96 (C.H.) | 0.1742 | 0.2185 | 0.1547 |
| 27499629 | rs2492812 | MOB3B | A | C > A | 0.454 (N.A.) | 0.334 | −0.88 (C.H.) | 0.2091 | 0.2419 | 0.2486 |
| 27508491 | rs1537712 | MOB3B | T | C > T | 0.454 (N.A.) | 0.332 | −0.75 (Cer) | 0.2323 | 0.2753 | 0.2915 |
| 27509213 | rs12554036 | MOB3B | T | G > T | 0.484 (N.A.) | 0.345 | −1.0 (C.H.) | 0.2054 | 0.2335 | 0.1796 |
| 27514964 | rs4609281 | MOB3B | G | G > T | −0.432 (N.A.) | −0.364 | 0.71 (Cer) | 0.2417 | 0.2865 | 0.3509 |
| 27515969 | rs774354 | MOB3B | G | A > G | 0.466 (N.A.) | 0.345 | −0.80 (C.H.) | 0.2354 | 0.2754 | 0.2909 |
| 27516592 | rs774352 | MOB3B | C | T > C | 0.466 (N.A.) | 0.345 | −0.80 (C.H.) | 0.2354 | 0.2755 | 0.2910 |
| 27525753 | rs10967973 | IFNK | A | A > G | −0.602 (Cer) | −0.602 | 0.66 (Cer) | 0.3570 | 0.4640 | 0.5599 |
| 27526049 | rs700782 | IFNK | T | C > T | 0.498 (N.A.) | 0.370 | −0.98 (C.H.) | 0.2109 | 0.2440 | 0.2461 |
| 27527514 | rs2453552 | MOB3B | G | T > G | 0.501 (N.A.) | 0.36 | −1.0 (C.H.) | 0.2110 | 0.2450 | 0.2650 |
| 27541043 | rs7469146 | C9orf72 | C | C > T | −0.542 (Cer) | −0.542 | 0.65 (Cer) | 0.3736 | 0.5119 | 0.5916 |
| 27543384 | rs3849943 | C9orf72 | C | C > T | −0.485 (N.A.) | −0.336 | 0.96 (Cer, C.H.) | 0.2005 | 0.2362 | 0.2178 |
| 27545962 | rs700791 | C9orf72 | A | C > A | 0.521 (N.A.) | 0.388 | −1.1 (C.H.) | 0.1958 | 0.2293 | 0.1979 |
| 27579562 | rs1982915 | Intergenic | G | A > G | 0.407 (Cer) | 0.407 | −0.49 (Cer) | 0.4212 | 0.4982 | 0.4745 |
| 27580676 | rs12350076 | Intergenic | C | A > C | 0.408 (Cer) | 0.408 | −0.51 (Cer) | 0.4262 | 0.5104 | 0.4964 |
| 27581241 | rs2783010 | Intergenic | C | T > C | 0.418 (Cer) | 0.418 | −0.56 (Cer) | 0.4273 | 0.4645 | 0.4808 |
| 27582313 | rs10967993 | Intergenic | C | T > C | 0.424 (Cer) | 0.424 | −0.54 (F.C.) | 0.4025 | 0.5044 | 0.5032 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobo, H.; Goldstein, O.; Gana-Weisz, M.; Bar-Shira, A.; Gurevich, T.; Thaler, A.; Mirelman, A.; Giladi, N.; Orr-Urtreger, A. C9orf72-G4C2 Intermediate Repeats and Parkinson’s Disease; A Data-Driven Hypothesis. Genes 2021, 12, 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081210
Kobo H, Goldstein O, Gana-Weisz M, Bar-Shira A, Gurevich T, Thaler A, Mirelman A, Giladi N, Orr-Urtreger A. C9orf72-G4C2 Intermediate Repeats and Parkinson’s Disease; A Data-Driven Hypothesis. Genes. 2021; 12(8):1210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081210
Chicago/Turabian StyleKobo, Hila, Orly Goldstein, Mali Gana-Weisz, Anat Bar-Shira, Tanya Gurevich, Avner Thaler, Anat Mirelman, Nir Giladi, and Avi Orr-Urtreger. 2021. "C9orf72-G4C2 Intermediate Repeats and Parkinson’s Disease; A Data-Driven Hypothesis" Genes 12, no. 8: 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12081210