
Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Editorial Policies and Ethical Considerations
2.2. Variant Identification of SYNE2
2.3. Genetic Study
2.4. Structural Modelling and Visualisation
2.5. Lymphoblastoid (LCL) Cell Generation
2.6. Cell Culture
2.7. Immunofluorescence (IF)
2.8. Western Blotting (WB) Analysis
2.9. Antibodies
2.10. Statistical Analysis
2.11. mRNA Isolation and qPCR
3. Results
3.1. Clinical Features of the Patient
3.2. Genetic Results
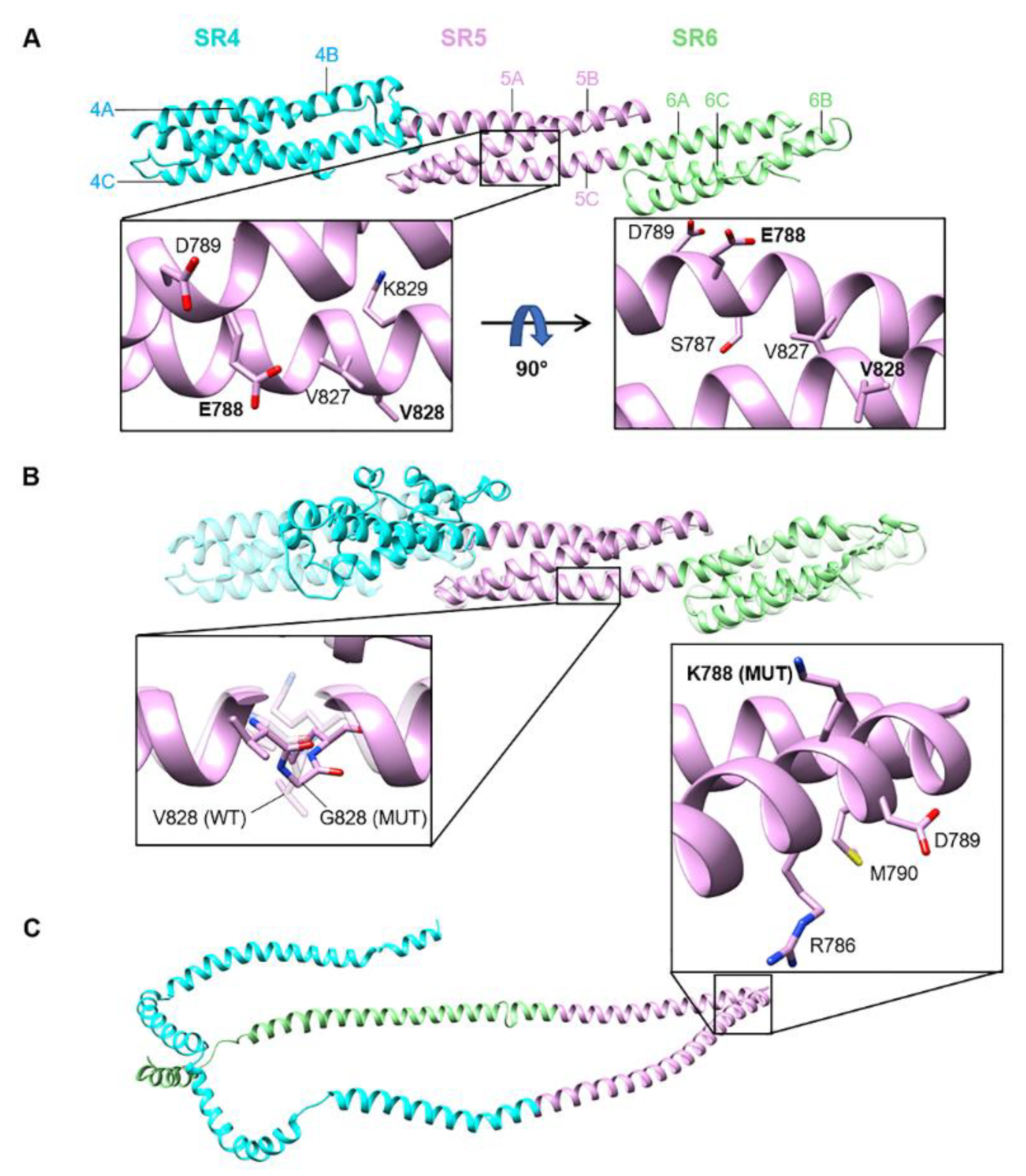
3.3. Position of SYNE2 Mutations within Nesprin-2 giant and Predicted Consequences
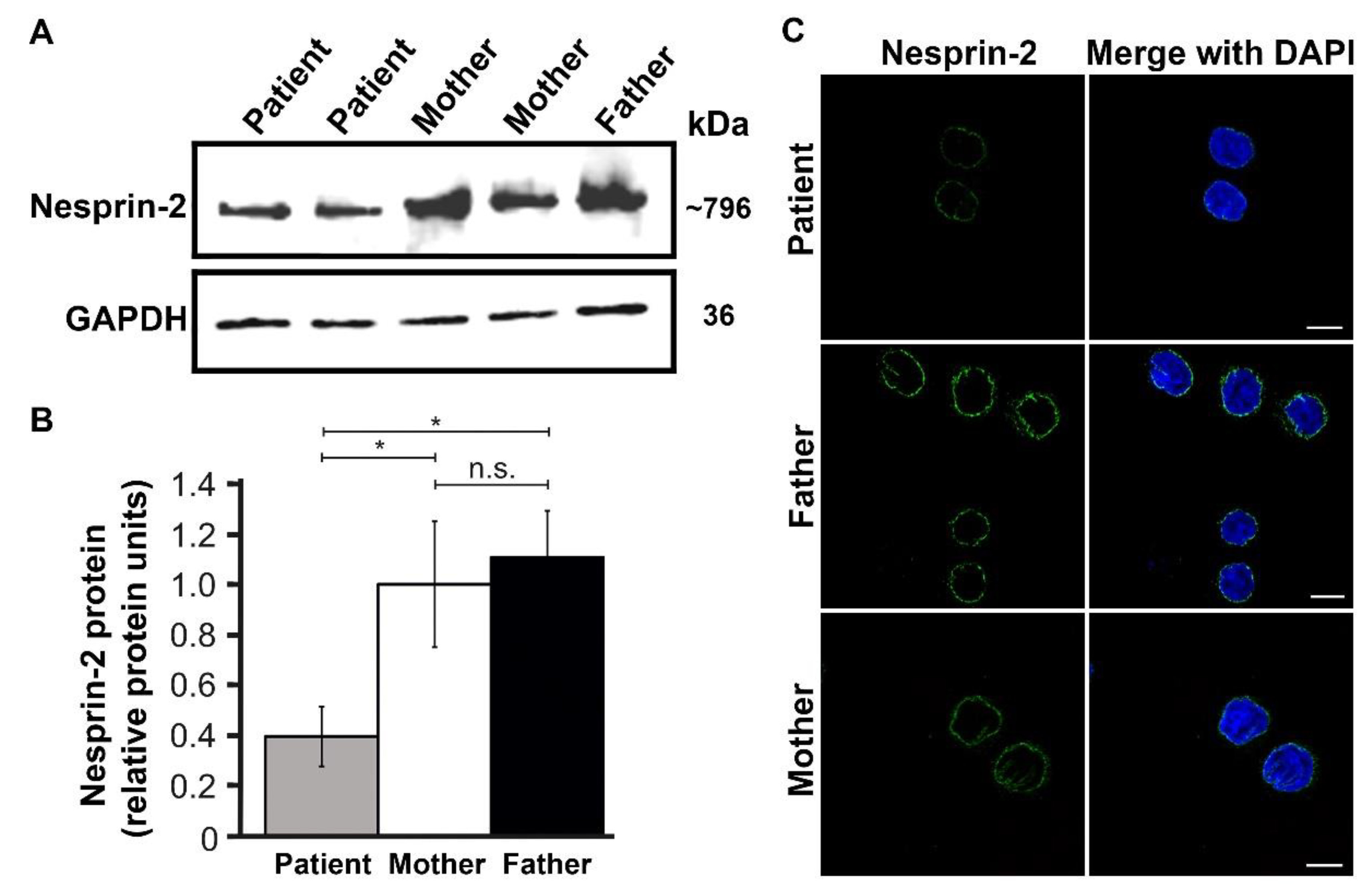
3.4. Expression and Functional Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padmakumar, V.C.; Libotte, T.; Lu, W.; Zaim, H.; Abraham, S.; Noegel, A.A.; Gotzmann, J.; Foisner, R.; Karakesisoglou, I. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J. Cell Sci. 2005, 118, 3419–3430. [Google Scholar] [CrossRef] [Green Version]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, B.A.; Rothballer, A.; Kutay, U.; Schwartz, T.U. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 2012, 149, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M. Nesprins: A novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J. Cell Sci. 2001, 114, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.Y.; Libotte, T.; Munck, M.; Noegel, A.A.; Korenbaum, E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J. Cell Sci. 2002, 115, 3207–3222. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, V.C.; Abraham, S.; Braune, S.; Noegel, A.A.; Tunggal, B.; Karakesisoglou, I.; Korenbaum, E. Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp. Cell Res. 2004, 295, 330–339. [Google Scholar] [CrossRef]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural requirements for the assembly of LINC complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, E.D.; Lewis, R.M.; Grady, R.M.; Sanes, J.R. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J. Biol. Chem. 2000, 275, 31986–31995. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Roux, K.J.; Crisp, M.L.; Liu, Q.; Kim, D.; Kozlov, S.; Stewart, C.L.; Burke, B. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. USA 2009, 106, 2194–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajgor, D.; Shanahan, C.M. Nesprins: From the nuclear envelope and beyond. Expert Rev. Mol. Med. 2013, 15, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartwright, S.; Karakesisoglou, I. Nesprins in health and disease. Semin. Cell Dev. Biol. 2014, 29, 169–179. [Google Scholar] [CrossRef]
- Libotte, T.; Zaim, H.; Abraham, S.; Padmakumar, V.C.; Schneider, M.; Lu, W.S.; Munck, M.; Hutchison, C.; Wehnert, M.; Fahrenkrog, B.; et al. Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol. Biol. Cell. 2005, 16, 3411–3424. [Google Scholar] [CrossRef]
- Luke, Y.; Zaim, H.; Karakesisoglou, I.; Jaeger, V.M.; Sellin, L.; Lu, W.; Schneider, M.; Neumann, S.; Beijer, A.; Munck, M.; et al. Nesprin-2 Giant (NUANCE) maintains nuclear envelope architecture and composition in skin. J. Cell Sci. 2008, 121, 1887–1898. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.; Lu, W.; Neumann, S.; Brachner, A.; Gotzmann, J.; Noegel, A.A.; Karakesisoglou, I. Molecular mechanisms of centrosome and cytoskeleton anchorage at the nuclear envelope. Cell Mol. Life Sci. 2011, 68, 1593–1610. [Google Scholar] [CrossRef]
- Rashmi, R.; Eckes, B.; Glöckner, G.; Groth, M.; Neumann, S.; Gloy, J.; Sellin, L.; Walz, G.; Schneider, M.; Karakesisoglou, I. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus 2012, 3, 172–186. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bethmann, C.; Worth, N.F.; Davies, J.D.; Wasner, C.; Feuer, A.; Ragnauth, C.D.; Yi, Q.; Mellad, J.A.; Warren, D.T.; et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum. Mol. Genet. 2007, 16, 2816–2833. [Google Scholar] [CrossRef]
- Kandert, S.; Luke, Y.; Kleinhenz, T.; Neumann, S.; Lu, W.; Jaeger, V.M.; Munck, M.; Wehnert, M.; Muller, C.R.; Zhou, Z.; et al. Nesprin-2 giant safeguards nuclear envelope architecture in LMNA S143F progeria cells. Hum. Mol. Genet. 2007, 16, 2944–2959. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chancellor, T.; Lee, J.; Thodeti, C.K.; Lele, T. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys. J. 2010, 99, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Luxton, G.G.; Gomes, E.R.; Folker, E.S.; Vintinner, E.; Gundersen, G.G. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science 2010, 329, 956–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatau, S.B.; Bloom, R.J.; Bajpai, S.; Razafsky, D.; Zang, S.; Giri, A.; Wu, P.-H.; Marchand, J.; Celedon, A.; Hale, C.M. The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Sci. Rep. 2012, 2, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayo, A.; Malboubi, M.; Antoku, S.; Chang, W.; Ortiz-Zapater, E.; Groen, C.; Pfisterer, K.; Tootle, T.; Charras, G.; Gundersen, G.G. Fascin regulates nuclear movement and deformation in migrating cells. Dev. Cell 2016, 38, 371–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, D.T.; Tajsic, T.; Mellad, J.A.; Searles, R.; Zhang, Q.; Shanahan, C.M. Novel nuclear nesprin-2 variants tether active extracellular signal-regulated MAPK1 and MAPK2 at promyelocytic leukemia protein nuclear bodies and act to regulate smooth muscle cell proliferation. J. Biol. Chem. 2010, 285, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, S.; Schneider, M.; Daugherty, R.L.; Gottardi, C.J.; Eming, S.A.; Beijer, A.; Noegel, A.A.; Karakesisoglou, I. Nesprin-2 interacts with α-catenin and regulates Wnt signaling at the nuclear envelope. J. Biol. Chem. 2010, 285, 34932–34938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawe, H.R.; Adams, M.; Wheway, G.; Szymanska, K.; Logan, C.V.; Noegel, A.A.; Gull, K.; Johnson, C.A. Nesprin-2 interacts with meckelin and mediates ciliogenesis via remodelling of the actin cytoskeleton. J. Cell Sci. 2009, 122, 2716–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, N.; Kessler, K.; Schramm, S.-F.; Boldt, K.; Becirovic, E.; Michalakis, S.; Regus-Leidig, H.; Noegel, A.A.; Ueffing, M.; Thiel, C.T. Functional analyses of Pericentrin and Syne-2 interaction in ciliogenesis. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.R.; You, L.R.; Wang, W.J.; Huang, W.S.; Chu, C.T.; Chi, Y.H.; Chen, H.C. Lamin A-mediated nuclear lamina integrity is required for proper ciliogenesis. EMBO Rep. 2020, 21, e49680. [Google Scholar] [CrossRef]
- Brosig, M.; Ferralli, J.; Gelman, L.; Chiquet, M.; Chiquet-Ehrismann, R. Interfering with the connection between the nucleus and the cytoskeleton affects nuclear rotation, mechanotransduction and myogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 1717–1728. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambliss, A.B.; Khatau, S.B.; Erdenberger, N.; Robinson, D.K.; Hodzic, D.; Longmore, G.D.; Wirtz, D. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci. Rep. 2013, 3, 1087. [Google Scholar] [CrossRef] [Green Version]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Arsenovic, P.T.; Ramachandran, I.; Bathula, K.; Zhu, R.; Narang, J.D.; Noll, N.A.; Lemmon, C.A.; Gundersen, G.G.; Conway, D.E. Nesprin-2G, a component of the nuclear LINC complex, is subject to myosin-dependent tension. Biophys. J. 2016, 110, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Balikov, D.A.; Brady, S.K.; Ko, U.H.; Shin, J.H.; de Pereda, J.M.; Sonnenberg, A.; Sung, H.-J.; Lang, M.J. The nesprin-cytoskeleton interface probed directly on single nuclei is a mechanically rich system. Nucleus 2017, 8, 534–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter-Featherstone, E.; Young, N.; Chamberlain, K.; Cubillas, P.; Hulette, B.; Wei, X.; Tiesman, J.P.; Bascom, C.C.; Benham, A.M.; Goldberg, M.W. Culturing Keratinocytes on Biomimetic Substrates Facilitates Improved Epidermal Assembly In Vitro. Cells 2021, 10, 1177. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Rao, L.; Warren, D.T.; Shanahan, C.M.; Zhang, Q. Mouse models of nesprin-related diseases. Biochem. Soc. Trans. 2018, 46, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros-Louis, F.; Dupré, N.; Dion, P.; Fox, M.A.; Laurent, S.; Verreault, S.; Sanes, J.R.; Bouchard, J.-P.; Rouleau, G.A. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat. Genet. 2007, 39, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Smets, K.; Mallaret, M.; Di Bella, D.; Gallenmüller, C.; Baets, J.; Schulze, M.; Magri, S.; Sarto, E.; Mustafa, M. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: A large multi-centre study. Brain 2016, 139, 1378–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, Y.; Miyamoto, R.; Morino, H.; Yoshizawa, A.; Nishinaka, K.; Udaka, F.; Kameyama, M.; Maruyama, H.; Kawakami, H. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology 2013, 80, 600–601. [Google Scholar] [CrossRef]
- Nadaf, S.N.; Chakor, R.T.; Kothari, K.V.; Mannan, A.U. Synaptic nuclear envelope Protein 1 (SYNE 1) ataxia with amyotrophic lateral sclerosis-like presentation: A novel synaptic nuclear envelope Protein 1 (SYNE 1) gene deletion mutation from India. Ann. Indian Acad. Neurol. 2020, 23, 539. [Google Scholar] [CrossRef]
- Attali, R.; Warwar, N.; Israel, A.; Gurt, I.; McNally, E.; Puckelwartz, M.; Glick, B.; Nevo, Y.; Ben-Neriah, Z.; Melki, J. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum. Mol. Genet. 2009, 18, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Steichen-Gersdorf, E.; Krabichler, B.; Petersen, B.-S.; Weber, U.; Schmidt, W.M.; Zschocke, J.; Müller, T.; Bittner, R.E.; Janecke, A.R. Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: A genotype–phenotype correlation. Eur. J. Hum. Genet. 2017, 25, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandra, M.; Maria Pia, L.; Stefano, C.; Pietro, P.; Crociani, P.; Aldo, R.; Giuseppe, D.S.; Massimo, C. Emery-Dreifuss muscular dystrophy type 4: A new SYNE1 mutation associated with hypertrophic cardiomyopathy masked by a perinatal distress-related spastic diplegia. Clin. Case Rep. 2019, 7, 1078–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puckelwartz, M.J.; Kessler, E.J.; Kim, G.; DeWitt, M.M.; Zhang, Y.; Earley, J.U.; Depreux, F.F.; Holaska, J.; Mewborn, S.K.; Pytel, P. Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Li, C.; Zhou, B.; Sun, H.; Koullourou, V.; Holt, I.; Puckelwartz, M.J.; Warren, D.T.; Hayward, R.; Lin, Z. Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum. Mol. Genet. 2017, 26, 2258–2276. [Google Scholar] [CrossRef]
- Schuurs-Hoeijmakers, J.H.; Vulto-van Silfhout, A.T.; Vissers, L.E.; Van De Vondervoort, I.I.; Van Bon, B.W.; De Ligt, J.; Gilissen, C.; Hehir-Kwa, J.Y.; Neveling, K.; Del Rosario, M. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 2013, 50, 802–811. [Google Scholar] [CrossRef]
- Özoğuz, A.; Uyan, Ö.; Birdal, G.; Iskender, C.; Kartal, E.; Lahut, S.; Ömür, Ö.; Agim, Z.S.; Eken, A.G.; Sen, N.E. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol. Aging 2015, 36, 1764.e9–1764.e18. [Google Scholar] [CrossRef]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Toda, T.; Tsuji, S. Juvenile amyotrophic lateral sclerosis with complex phenotypes associated with novel SYNE1 mutations. Amyotroph. Lateral Scler. Front. Degener. 2020. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Timothy, W.Y.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013, 77, 259–273. [Google Scholar]
- Sklar, P.; Ripke, S.; Scott, L.J.; Andreassen, O.A.; Cichon, S.; Craddock, N.; Edenberg, H.J.; Nurnberger, J.I., Jr.; Rietschel, M.; Blackwood, D. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977. [Google Scholar]
- Green, E.K.; Grozeva, D.; Forty, L.; Gordon-Smith, K.; Russell, E.; Farmer, A.; Hamshere, M.; Jones, I.R.; Jones, L.; McGuffin, P. Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol. Psychiatry 2013, 18, 614–617. [Google Scholar] [CrossRef]
- Hou, L.; Bergen, S.E.; Akula, N.; Song, J.; Hultman, C.M.; Landen, M.; Adli, M.; Alda, M.; Ardau, R.; Arias, B.; et al. Genome-wide association study of 40,000 individuals identifies two novel loci associated with bipolar disorder. Hum. Mol Genet. 2016, 25, 3383–3394. [Google Scholar] [CrossRef]
- Xu, W.; Cohen-Woods, S.; Chen, Q.; Noor, A.; Knight, J.; Hosang, G.; Parikh, S.V.; De Luca, V.; Tozzi, F.; Muglia, P.; et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med. Genet. 2014, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, Y.; Luo, B.; Zhang, Y.; Song, X.; Li, M.; Lv, L. Association of SYNE1 locus with bipolar disorder in Chinese population. Hereditas 2019, 156, 19. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, S.; Choi, E.; Shin, S.; Park, J. A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy. Clin. Chim. Acta 2020, 506, 50–54. [Google Scholar] [CrossRef]
- Mroß, C.; Marko, M.; Munck, M.; Glöckner, G.; Motameny, S.; Altmüller, J.; Noegel, A.A.; Eichinger, L.; Peche, V.S.; Neumann, S. Depletion of Nesprin-2 is associated with an embryonic lethal phenotype in mice. Nucleus 2018, 9, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, N.; Joachimsthaler, A.; Kessler, K.; Lux, U.T.; Noegel, A.A.; Kremers, J.; Brandstätter, J.H.; Gießl, A. Lack of a retinal phenotype in a Syne-2/Nesprin-2 knockout mouse model. Cells 2019, 8, 1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Lei, K.; Zhou, M.; Craft, C.M.; Xu, G.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. KASH protein Syne-2/Nesprin-2 and SUN proteins SUN1/2 mediate nuclear migration during mammalian retinal development. Hum. Mol. Genet. 2011, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddox, D.M.; Collin, G.B.; Ikeda, A.; Pratt, C.H.; Ikeda, S.; Johnson, B.A.; Hurd, R.E.; Shopland, L.S.; Naggert, J.K.; Chang, B. A mutation in Syne2 causes early retinal defects in photoreceptors, secondary neurons, and Müller glia. Investig. Vis. Sci. 2015, 56, 3776–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.-C.; Wu, S.; Hsu, C.-S.; Huang, S.-M.; Hong, J.-S.; Hu, C.-F. Association of SYNE2 variants in accelerating the progress of DYT1 early-onset isolated dystonia. bioRxiv 2019, 807891. [Google Scholar]
- Eilbeck, K.; Quinlan, A.; Yandell, M. Settling the score: Variant prioritization and Mendelian disease. Nat. Rev. Genet. 2017, 18, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Autore, F.; Pfuhl, M.; Quan, X.; Williams, A.; Roberts, R.G.; Shanahan, C.M.; Fraternali, F. Large-scale modelling of the divergent spectrin repeats in nesprins: Giant modular proteins. PLoS ONE 2013, 8, e63633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Philip McCoy, J.P., Jr. Handling, storage, and preparation of human blood cells. Curr. Protoc. Cytom. 1997. [Google Scholar] [CrossRef]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Nurnberg, G.; Farooq, M.; Ahmad, I.; Alef, T.; Hennies, H.C.; Technau, M.; Altmuller, J.; et al. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am. J. Hum. Genet. 2012, 90, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Le Rumeur, E.; Hubert, J.-F.; Winder, S.J. A new twist to coiled coil. FEBS Lett. 2012, 586, 2717–2722. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Machnicka, B.; Czogalla, A.; Hryniewicz-Jankowska, A.; Bogusławska, D.M.; Grochowalska, R.; Heger, E.; Sikorski, A.F. Spectrins: A structural platform for stabilization and activation of membrane channels, receptors and transporters. Biochim. Biophys. Acta (BBA)-Biomembr. 2014, 1838, 620–634. [Google Scholar] [CrossRef] [Green Version]
- Hanspal, M.; Hanspal, J.S.; Sahr, K.E.; Fibach, E.; Nachman, J.; Palek, J. Molecular basis of spectrin deficiency in hereditary pyropoikilocytosis. Blood 1993, 82, 1652–1660. [Google Scholar] [CrossRef]
- Acsadi, G.; Moore, S.A.; Chéron, A.; Delalande, O.; Bennett, L.; Kupsky, W.; El-Baba, M.; Le Rumeur, E.; Hubert, J.-F. Novel mutation in spectrin-like repeat 1 of dystrophin central domain causes protein misfolding and mild Becker muscular dystrophy. J. Biol. Chem. 2012, 287, 18153–18162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.M. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, X.; Golhar, R.; Otieno, F.G.; He, M.; Hou, C.; Kim, C.; Keating, B.; Lyon, G.J.; Wang, K. Whole-genome sequencing in an autism multiplex family. Mol. Autism 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demily, C.; Assouline, M.; Boddaert, N.; Barcia, G.; Besmond, C.; Poisson, A.; Sanlaville, D.; Munnich, A. Apports de la génétique au diagnostic des troubles du spectre autistique. Neuropsychiatr. L’enfance L’Adolescence 2016, 64, 395–401. [Google Scholar] [CrossRef]
- Krumm, N.; O’Roak, B.J.; Shendure, J.; Eichler, E.E. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014, 37, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T. The genetic basis of Mendelian phenotypes: Discoveries, challenges, and opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet. Med. 2017, 19, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanis, J.; Samocha, K.E.; Wiel, L.; Zhang, Z.; Arvai, K.J.; Eberhardt, R.Y.; Gallone, G.; Lelieveld, S.H.; Martin, H.C.; McRae, J.F. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020, 586, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.P.; Basson, M.A. The neuroanatomy of autism–a developmental perspective. J. Anat. 2017, 230, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.; Man, H.-Y. Fundamental elements in autism: From neurogenesis and neurite growth to synaptic plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, G.; Zarbalis, K.S. Prenatal neurogenesis in autism spectrum disorders. Front. Chem. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Schneider, M.; Neumann, S.; Jaeger, V.-M.; Taranum, S.; Munck, M.; Cartwright, S.; Richardson, C.; Carthew, J.; Noh, K. Nesprin interchain associations control nuclear size. Cell Mol. Life Sci. 2012, 69, 3493–3509. [Google Scholar] [CrossRef] [Green Version]
- Titlow, J.; Robertson, F.; Järvelin, A.; Ish-Horowicz, D.; Smith, C.; Gratton, E.; Davis, I. Syncrip/hnRNP Q is required for activity-induced Msp300/Nesprin-1 expression and new synapse formation. J. Cell Biol. 2020, 219, e201903135. [Google Scholar] [CrossRef] [Green Version]
- Tulgren, E.D.; Turgeon, S.M.; Opperman, K.J.; Grill, B. The Nesprin family member ANC-1 regulates synapse formation and axon termination by functioning in a pathway with RPM-1 and β-Catenin. PLoS Genet. 2014, 10, e1004481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-h.; Narzisi, G.; Leotta, A. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthew, J.; Karakesisoglou, I. Detection of diverse and high molecular weight Nesprin-1 and Nesprin-2 isoforms using Western blotting. In The Nuclear Envelope; Springer: Berlin/Heidelberg, Germany, 2016; pp. 221–232. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Tool | c.2362Gv>vA; p.v(Glu788Lys) | c.2483Tv>vG; p.v(Val828Gly) |
|---|---|---|
| PANTHER | Neutral (0.128) | Disease causing (0.723) |
| PhD-SNP | Disease causing (0.886) | Disease causing (0.862) |
| SIFT | Disease causing (0.000) | Disease causing (0.000) |
| SNAP | Disease causing (0.595) | Disease causing (0.555) |
| Meta SNP | Disease causing (0.681) | Disease causing (0.681) |
| Provean | Neutral (−2.272) | Disease causing (−3.649) |
| MuPro | Decrease protein stability (delta delta G: −1.14) | Decrease protein stability (delta delta G: −2.32) |
| SNPs and GO | Neutral | Neutral |
| Polyphen-2 | Probably damaging (0.999) | Possibly damaging (0.465) |
| Mutation Taster | Polymorphism (56) | Polymorphism (109) |
| CADD score | 27.2 | 13.2 |
| FATHMM | Tolerated (0.58) | Tolerated (0.72) |
| ACMG Interpretation | Uncertain significance (PM2) | Uncertain significance (PM2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, N.; Asif, M.; Jackson, M.; Fernández-Mayoralas, D.M.; de la Peña, M.J.; Calleja-Pérez, B.; Álvarez, S.; Hunter-Featherstone, E.; Noegel, A.A.; Höhne, W.; et al. Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism. Genes 2021, 12, 1294. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091294
Young N, Asif M, Jackson M, Fernández-Mayoralas DM, de la Peña MJ, Calleja-Pérez B, Álvarez S, Hunter-Featherstone E, Noegel AA, Höhne W, et al. Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism. Genes. 2021; 12(9):1294. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091294
Chicago/Turabian StyleYoung, Natalie, Maria Asif, Matthew Jackson, Daniel Martín Fernández-Mayoralas, Mar Jimenez de la Peña, Beatriz Calleja-Pérez, Sara Álvarez, Eve Hunter-Featherstone, Angelika A. Noegel, Wolfgang Höhne, and et al. 2021. "Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism" Genes 12, no. 9: 1294. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091294