
Mitochondrial Genetic Heterogeneity in Leber’s Hereditary Optic Neuropathy: Original Study with Meta-Analysis

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Declaration and Patient Evaluation
2.2. DNA Isolation and Sequencing of Complete mtDNA
2.3. Data Analysis
3. Meta-Analysis of LHON-Associated Variants
3.1. Meta-Analysis of Primary LHON Variants
3.2. Meta-Analysis of Two LHON-Associated Variants (m.4216T>C and m.13708G>A)
3.2.1. Search and Selection of Relevant Studies
3.2.2. Data Extraction
3.2.3. Statistical Analysis
4. Results
4.1. Study Samples
4.2. Whole Mitochondrial Genome Screening
4.3. Haplogroup Analysis of Patients with Primary LHON Variants
4.4. Secondary LHON-Associated Variants in Patients Lacking Primary LHON Variants
4.5. Haplogroup Analysis of Patients with Secondary LHON-Associated Variants
4.6. Other Disease-Associated mtDNA Variants
4.7. Meta-Analysis
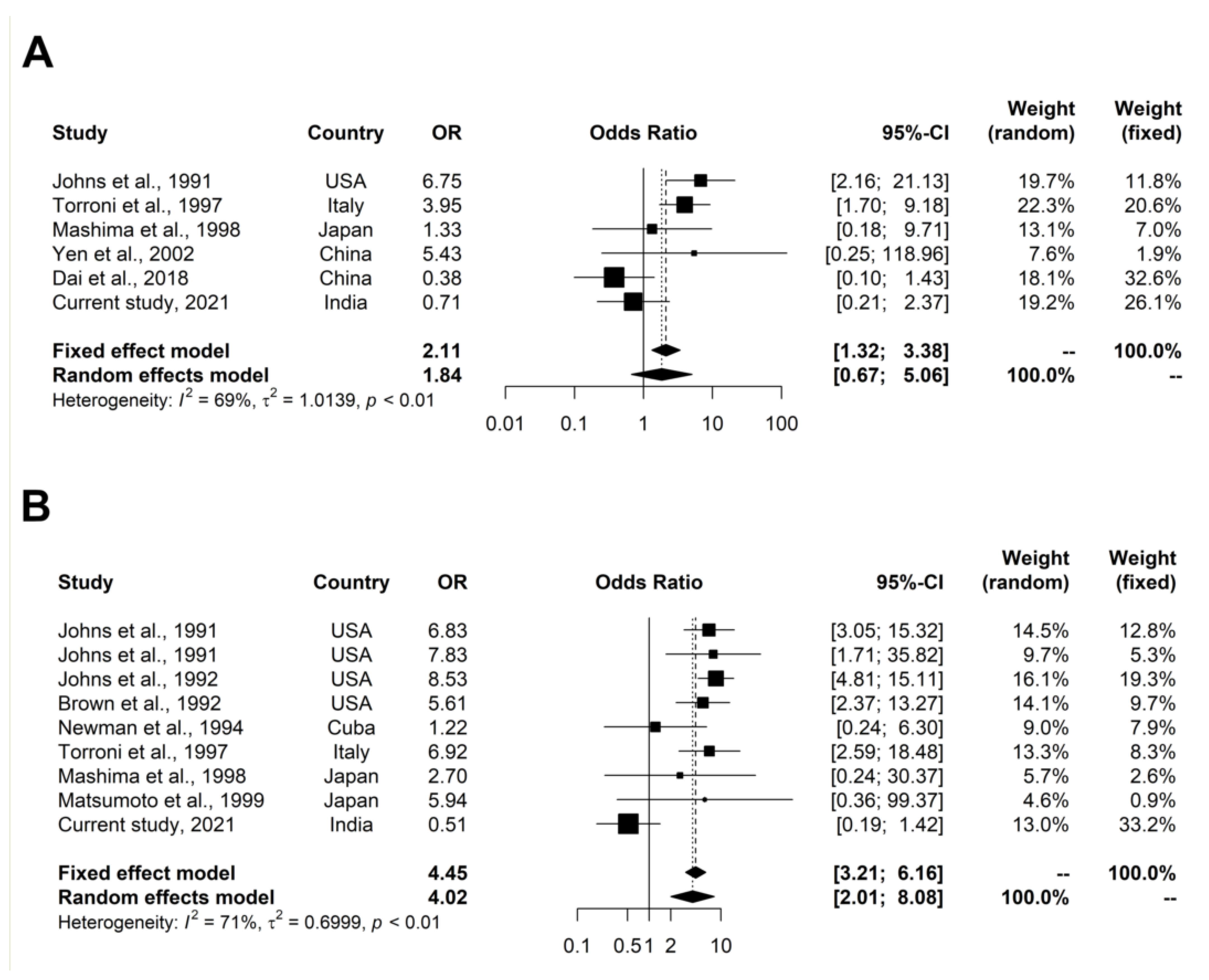
4.7.1. Meta-Analysis of Primary LHON Variants
4.7.2. Meta-Analysis of LHON-Associated Variants
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Wang, H.W.; Jia, X.; Ji, Y.; Kong, Q.P.; Zhang, Q.; Yao, Y.G.; Zhang, Y.P. Strikingly different penetrance of LHON in two Chinese families with primary mutation G11778A is independent of mtDNA haplogroup background and secondary mutation G13708A. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2008, 643, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Man, P.Y.W.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of leber hereditary optic neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy Topic collections Leber hereditary optic neuropathy. J. Med. Genet. 2002, 3939, 162–169. [Google Scholar] [CrossRef]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.Y.; Wang, A.G.; Wei, Y.H. Leber’s hereditary optic neuropathy: A multifactorial disease. Prog. Retin. Eye Res. 2006, 25, 381–396. [Google Scholar] [CrossRef]
- Zhang, A.M.; Jia, X.; Bi, R.; Salas, A.; Li, S.; Xiao, X.; Wang, P.; Guo, X.; Kong, Q.P.; Zhang, Q.; et al. Mitochondrial DNA haplogroup background affects LHON, but not suspected LHON, in Chinese patients. PLoS ONE 2011, 6, e27750. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.A.; Govindaraj, P.; Soumittra, N.; Srilekha, S.; Ambika, S.; Vanniarajan, A.; Meena, A.K.; Uppin, M.S.; Sundaram, C.; Taly, A.B.; et al. Haplogroup Heterogeneity of LHON Patients Carrying the m.14484T>C Mutation in India. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3999–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A.; Govindaraj, P.; Soumittra, N.; Sharma, S.; Srilekha, S.; Ambika, S.; Vanniarajan, A.; Meena, A.K.; Uppin, M.S.; Sundaram, C.; et al. Leber’s hereditary optic neuropathy–specific mutation m.11778G>A exists on diverse mitochondrial haplogroups in India. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3923–3930. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Devi, S.; Saxena, R.; Gupta, N.; Kabra, M.; Chowdhury, M. Frequency of primary mutations of Leber’s hereditary optic neuropathy patients in North Indian population. Indian J. Ophthalmol. 2017, 65, 1156–1160. [Google Scholar]
- Sundaresan, P.; Kumar, S.M.; Thompson, S.; Fingert, J.H. Reduced frequency of known mutations in a cohort of LHON patients from India. Ophthalmic Genet. 2010, 31, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Carelli, V.; Spruijt, L.; Gerards, M.; Mowbray, C.; Achilli, A.; Pyle, A.; Elson, J.; Howell, N.; La Morgia, C.; et al. Clinical expression of leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am. J. Hum. Genet. 2007, 81, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Zhang, A.M.; Jia, X.; Zhang, Y.P.; Xiao, X.; Li, S.; Guo, X.; Bandelt, H.J.; Zhang, Q.; Yao, Y.G. Mitochondrial DNA Haplogroups M7b1′2 and M8a Affect Clinical Expression of Leber Hereditary Optic Neuropathy in Chinese Families with the m.11778G→A Mutation. Am. J. Hum. Genet. 2008, 83, 760–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikia, B.B.; Dubey, S.K.; Shanmugam, M.K.; Sundaresan, P. Whole mitochondrial genome analysis in South Indian patients with Leber’s hereditary optic neuropathy. Mitochondrion 2017, 36, 21–28. [Google Scholar] [CrossRef]
- Khan, N.A.; Govindaraj, P.; Vuskamalla, J.; Meena, A.K.; Thangaraj, K. Co-occurrence of m.1555A>G and m.11778G>A mitochondrial DNA mutations in two Indian families with strikingly different clinical penetrance of leber hereditary optic neuropathy. Mol. Vis. 2013, 19, 1282–1289. [Google Scholar]
- Thangaraj, K.; Joshi, M.B.; Reddy, A.G.; Gupta, N.J.; Chakravarty, B.; Singh, L. CAG repeat expansion in the androgen receptor gene is not associated with male infertility in Indian populations. J. Androl. 2002, 23, 815–818. [Google Scholar] [PubMed]
- Rieder, M.J.; Taylor, S.L.; Tobe, V.O.; Nickerson, D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: Analysis of the human mitochondrial genome. Nucleic Acids Res. 1998, 26, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Kloss-Brandstätter, A.; Pacher, D.; Schönherr, S.; Weissensteiner, H.; Binna, R.; Specht, G.; Kronenberg, F. HaploGrep: A fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum. Mutat. 2011, 32, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Francis, A.; Pooja, S.; Rajender, S.; Govindaraj, P.; Tipirisetti, N.R.; Surekha, D.; Rao, D.R.; Rao, L.; Ramachandra, L.; Vishnupriya, S.; et al. A mitochondrial DNA variant 10398G>A in breast cancer among South Indians: An original study with meta-analysis. Mitochondrion 2013, 13, 559–565. [Google Scholar] [CrossRef]
- Petitti, D.B. Approaches to heterogeneity in meta-analysis. Stat. Med. 2001, 20, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Huedo-Medina, T.B.; Sánchez-Meca, J.; Marín-Martínez, F.; Botella, J. Assessing heterogeneity in meta-analysis: Q statistic or I 2 Index? Psychol. Methods 2006, 11, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Egger, M.; Smith, G.D.; Schneider, M.; Minder, C. Bias in meta-analysis detected by a simple, graphical test. Br. Med. J. 1997, 315, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.Y.; Wang, A.G.; Chang, W.L.; Hsu, W.M.; Liu, J.H.; Wei, Y.H. Leber’s hereditary optic neuropathy-The spectrum of mitochondrial DNA mutations in Chinese patients. Jpn. J. Ophthalmol. 2002, 46, 45–51. [Google Scholar] [CrossRef]
- Dogulu, C.F.; Kansu, T.; Seyrantepe, V.; Ozguc, M.; Topaloglu, H.; Johns, D.R. Mitochondrial DNA analysis in the Turkish Leber’s hereditary optic neuropathy population. Eye 2001, 15, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Verma, I.C.; Bijarnia, S.; Saxena, R.; Kohli, S.; Ratna, D.; Thomas, E.; Chowdhary, D.; Jha, S.N.; Grover, A.K. Leber’s Hereditary Optic Neuropathy with Molecular Characterisation in Two Indian Families. Indian J. Ophthalmol. 2005, 53, 167–171. [Google Scholar] [CrossRef]
- Chan, C.; Mackey, D.A.; Byrne, E. Sporadic Leber hereditary optic neuropathy in Australia and New Zealand. Aust. N. Z. J. Ophthalmol. 1996, 24, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Dawod, P.G.A.; Jancic, J.; Marjanovic, A.; Brankovic, M.; Jankovic, M.; Samardzic, J.; Potkonjak, D.; Djuric, V.; Mesaros, S.; Novakovic, I.; et al. Whole Mitochondrial Genome Analysis in Serbian Cases of Leber’s Hereditary Optic Neuropathy. Genes 2020, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic Mitochondrial DNA Mutations Are Common in the General Population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.X.; Zhou, Y.G.; Zhang, J.P.; Zhang, Q.B.; Liu, W.L.; Ren, C.F.; Li, X.Y. Study on three common mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Chin. J. Med. Genet. 2012, 29, 519–523. [Google Scholar]
- Yum, H.R.; Chae, H.; Shin, S.Y.; Kim, Y.; Kim, M.; Park, S.H. Pathogenic mitochondrial DNA mutations and associated clinical features in Korean patients with Leber’s hereditary optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8095–8101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jančić, J.; Dejanović, I.; Samardžić, J.; Radovanović, S.; Pepić, A.; Kosanović-Jaković, N.; Ćetković, M.; Kostić, V. Leber hereditary optic neuropathy in the population of Serbia. Eur. J. Paediatr. Neurol. 2014, 18, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Fernández, V.; Slabaugh, M.; Seleme, N.; Reyes, N.; Gallardo, P.; Herrera, L.; Peña, L.; Pezo, P.; Moraga, M. Pan-American mDNA haplogroups in Chilean patients with Leber’s hereditary optic neuropathy. Mol. Vis. 2014, 20, 334–340. [Google Scholar]
- Riordan-Eva, P.; Sanders, M.D.; Govan, G.G.; Sweeney, M.G.; Costa, J.D.; Harding, A.E. The clinical features of leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain 1995, 118, 319–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudoyo, H.; Suryadi, H.; Lertrit, P.; Pramoonjago, P.; Lyrawati, D.; Marzuki, S. Asian-specific mtDNA backgrounds associated with the primary G11778A mutation of Leber’s hereditary optic neuropathy. J. Hum. Genet. 2002, 47, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, A.; Bisceglia, L.; Trerotoli, P.; Russo, L.; D’Agruma, L.; Guerriero, S.; Petruzzella, V. Leber’s hereditary optic neuropathy (LHON) in an Apulian cohort of subjects. Acta Myol. 2017, 36, 163–177. [Google Scholar] [PubMed]
- Kumar, M.; Kaur, P.; Kumar, M.; Saxena, R.; Sharma, P.; Dada, R. Clinical characterization and mitochondrial DNA sequence variations in Leber hereditary optic neuropathy. Mol. Vis. 2012, 18, 2687–2699. [Google Scholar]
- Martins, F.T.A.; do Amor Divino Miranda, P.M.; Amaral Fernandes, M.S.; Maciel-Guerra, A.T.; Sartorato, E.L. Optimization of a genotyping screening based on hydrolysis probes to detect the main mutations related to leber hereditary optic neuropathy (LHON). Mol. Vis. 2017, 23, 495–503. [Google Scholar]
- Oostra, R.J.; Bolhuis, P.A.; Wijburg, F.A.; Zorn-Ende, G.; Bleeker-Wagemakers, E.M. Leber’s hereditary optic neuropathy: Correlations between mitochondrial genotype and visual outcome. J. Med. Genet. 1994, 31, 280–286. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Tanwar, M.; Saxena, R.; Sharma, P.; Dada, R. Identification of novel mitochondrial mutations in Leber’s hereditary optic neuropathy. Mol. Vis. 2010, 16, 782–792. [Google Scholar]
- Kapila, A.; Sachdeva, J.; Lal, V.; Wilson, V. Mutation analysis in a cohort of patients with Leber’s hereditary optic neuropathy from India. J. Neurol. Sci. 2017, 381, 841. [Google Scholar] [CrossRef]
- Miranda, P.M.D.A.D.; Da Silva-Costa, S.M.; Balieiro, J.C.; Amaral Fernandes, M.S.; Alves, R.M.; Maciel Guerra, A.T.; Marcondes, A.M.; Sartorato, E.L. Multiplex MALDI-TOF MS detection of mitochondrial variants in Brazilian patients with hereditary optic neuropathy. Mol. Vis. 2016, 22, 1024–1035. [Google Scholar] [PubMed]
- Rosenberg, T.; Nørby, S.; Schwartz, M.; Saillard, J.; Magalhães, P.J.; Leroy, D.; Kann, E.C.; Duno, M. Prevalence and genetics of leber hereditary optic neuropathy in the Danish population. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1370–1375. [Google Scholar] [CrossRef]
- Puomila, A.; Hämäläinen, P.; Kivioja, S.; Savontaus, M.-L.L.; Koivumäki, S.; Huoponen, K.; Nikoskelainen, E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur. J. Hum. Genet. 2007, 15, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Li, S.; Xiao, X.; Guo, X.; Zhang, Q. Molecular epidemiology of mtDNA mutations in 903 Chinese families suspected with Leber hereditary optic neuropathy. J. Hum. Genet. 2006, 51, 851–856. [Google Scholar] [CrossRef]
- Marotta, R.; Chin, J.; Quigley, A.; Katsabanis, S.; Kapsa, R.; Byrne, E.; Collins, S. Diagnostic screening of mitochondrial DNA mutations in Australian adults 1990–2001. Intern. Med. J. 2004, 34, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R.; Neufeld, M.J.; Park, R.D. An ND-6 mitochondrial DNA mutation associated with leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 1551–1557. [Google Scholar] [CrossRef]
- Starikovskaya, E.; Shalaurova, S.; Dryomov, S.; Nazhmidenova, A.; Volodko, N.; Bychkov, I.; Mazunin, I.; Sukernik, R. Mitochondrial DNA Variation of Leber’s Hereditary Optic Neuropathy in Western Siberia. Cells 2019, 8, 1574. [Google Scholar] [CrossRef] [Green Version]
- Maciel-Guerra, A.T.; Zanchetta, L.M.; Amaral Fernandes, M.S.; Andrade, P.B.; do Amor Divino Miranda, P.M.; Sartorato, E.S. Leber’s hereditary optic neuropathy: Clinical and molecular profile of a Brazilian sample. Ophthalmic Genet. 2010, 31, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, C.; Nie, Z.; Han, J.; Chen, T.; Zhao, X.; Ai, C.; Ji, Y.; Gao, T.; Jiang, P. Mutation analysis of Leber’s hereditary optic neuropathy using a multi-gene panel. Biomed. Rep. 2018, 8, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Gowri, P.; Kumar, S.M.; Vanniarajan, A.; Bharanidharan, D.; Sundaresan, P. A hospital-based five-year prospective study on the prevalence of Leber’s hereditary optic neuropathy with genetic confirmation. Mol. Vis. 2020, 26, 789–796. [Google Scholar]
- Dimitriadis, K.; Leonhardt, M.; Yu-Wai-Man, P.; Kirkman, M.A.; Korsten, A.; De Coo, I.F.; Chinnery, P.F.; Klopstock, T. Leber’s hereditary optic neuropathy with late disease onset: Clinical and molecular characteristics of 20 patients. Orphanet J. Rare Dis. 2014, 9, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macmillan, C.; Kirkham, T.; Fu, K.; Allison, V.; Andermann, E.; Chitayat, D.; Fortier, D.; Gans, M.; Hare, H.; Quercia, N.; et al. Pedigree analysis of French Canadian families with T14484C Leber’s hereditary optic neuropathy. Neurology 1998, 50, 417–422. [Google Scholar] [CrossRef]
- Gürkan, H.; Özal, S.A.; Esgin, H. Results of mitochondrial DNA sequence analysis in patients with clinically diagnosed Leber’s hereditary optic neuropathy. Balkan Med. J. 2012, 29, 306–309. [Google Scholar] [PubMed]
- Yamada, K. DNA diagnosis of leber’s hereditary optic neuropathy performed at Keio university hospital. J. Jpn. Ophthalmol. Soc. 2001, 105, 608–613. [Google Scholar] [CrossRef]
- Yamada, K.; Oguchi, Y.; Hotta, Y.; Nakamura, M.; Isashiki, Y.; Mashima, Y. Multicenter study on the frequency of three primary mutations of mitochondrial DNA in Japanese pedigrees with Leber’s hereditary optic neuropathy: Comparison with American and British counterparts. Neuro-ophthalmology 1999, 22, 187–193. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Jia, X.; Xiao, X.; Li, S.; Guo, X. Genetic and Clinical Analyses of DOA and LHON in 304 Chinese Patients with Suspected Childhood-Onset Hereditary Optic Neuropathy. PLoS ONE 2017, 12, e0170090. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Morizane, Y.; Shiraga, F.; Shikishima, K.; Ishikawa, H.; Wakakura, M.; Nakamura, M. Nationwide epidemiological survey of Leber hereditary optic neuropathy in Japan. J. Epidemiol. 2017, 27, 447–450. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hayasaka, S.; Kadoi, C.; Hotta, Y.; Fujiki, K.; Fujimaki, T.; Takeda, M.; Ishida, N.; Endo, S.; Kanai, A. Secondary mutations of mitochondrial DNA in Japanese patients with Leber’s hereditary optic neuropathy. Ophthalmic Genet. 1999, 20, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Tasaki, M.; Soemantri, A.; Dyat, M.; Susanto, J.C.; Tamam, M.; Sudarmanto, B.; Ishida, T. Leber’s hereditary optic neuropathy with 14484 mutation in Central Java, Indonesia. J. Hum. Genet. 2003, 48, 385–389. [Google Scholar] [CrossRef]
- Torroni, A.; Petrozzi, M.; D’Urbano, L.; Sellitto, D.; Zeviani, M.; Carrara, F.; Carducci, C.; Leuzzi, V.; Carelli, V.; Barboni, P.; et al. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 1997, 60, 1107–1121. [Google Scholar]
- Mashima, Y.; Yamada, K.; Wakakura, M.; Kigasawa, K.; Kudoh, J.; Shimizu, N.; Oguchi, Y. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber’s hereditary optic neuropathy. Curr. Eye Res. 1998, 17, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R.; Berman, J. Alternative, simultaneous complex I mitochondrial DNA mutations in Leber’s hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1991, 174, 1324–1330. [Google Scholar] [CrossRef]
- Newman, N.J.; Torroni, A.; Brown, M.D.; Lott, M.T.; Fernandez, M.M.; Wallace, D.C.; Philen, R.M.; Malilay, J.; Flanders, W.D.; Olson, D.; et al. Epidemic neuropathy in Cuba not associated with mitochondrial DNA mutations found in Leber’s hereditary optic neuropathy patients. Am. J. Ophthalmol. 1994, 118, 158–168. [Google Scholar] [CrossRef]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; Torroni, A.; Yang, C.-C.; Wallace, D.C. Mitochondrial DNA Complex I and I11 Mutations Associated With Leber’s Hereditary Optic Neuropathy. Genetics 1992, 130, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R.; Neufeld, M.J. Cytochrome b mutations in Leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun. 1991, 181, 1358–1364. [Google Scholar] [CrossRef]
- Reich, D.; Thangaraj, K.; Patterson, N.; Price, A.L.; Singh, L. Reconstructing Indian population history. Nature 2009, 461, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuka, N.; Moorjani, P.; Rai, N.; Sarkar, B.; Tandon, A.; Patterson, N.; Bhavani, G.S.; Girisha, K.M.; Mustak, M.S.; Srinivasan, S.; et al. The promise of discovering population-specific disease-associated genes in South Asia. Nat. Genet. 2017, 49, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Catarino, C.B.; Ahting, U.; Gusic, M.; Iuso, A.; Repp, B.; Peters, K.; Biskup, S.; von Livonius, B.; Prokisch, H.; Klopstock, T. Characterization of a Leber’s hereditary optic neuropathy (LHON) family harboring two primary LHON mutations m.11778G > A and m.14484T > C of the mitochondrial DNA. Mitochondrion 2017, 36, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, A.; Kumar, S.; Sreenath, J.; Sarkar, B.N.; Urade, B.P.; Mallick, S.; Bandopadhyay, S.S.; Barua, P.; Barik, S.S.; Basu, D.; et al. Updating Phylogeny of Mitochondrial DNA Macrohaplogroup M in India: Dispersal of Modern Human in South Asian Corridor. PLoS ONE 2009, 4, e7447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauser, S.; Luberichs, J.; Besch, D.; Leo-Kottler, B. Sequence analysis of the complete mitochondrial genome in patients with Leber’s hereditary optic neuropathy lacking the three most common pathogenic DNA mutations. Biochem. Biophys. Res. Commun. 2002, 295, 342–347. [Google Scholar] [CrossRef]
- Palanichamy, M.G.; Mitra, B.; Zhang, C.L.; Debnath, M.; Li, G.M.; Wang, H.W.; Agrawal, S.; Chaudhuri, T.K.; Zhang, Y.P. West Eurasian mtDNA lineages in India: An insight into the spread of the Dravidian language and the origins of the caste system. Hum. Genet. 2015, 134, 637–647. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Chinnery, P.F.; Turnbull, D.M.; Howell, N.; Turnbuu, D.M. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet. 1997, 13, 450–455. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age | Age of Onset | Sex | Visual Acuity | Fundus Findings Disc Pallor | Colour Vision Affected | Visual Fields | Optic Atrophy | Family History | |
|---|---|---|---|---|---|---|---|---|---|---|
| Logmar (RE) | Logmar (LE) | |||||||||
| P1 | 17 | 14 | M | 0.795 | ND | Yes | Yes | BE-large CS | Yes | Yes |
| P2 | 37 | 10 | M | 0.795 | 0.795 | TP | Yes | RE-few CD | No | No |
| P3 | 18 | 15 | M | 1.204 | 1.000 | Yes | Yes | BE-large CS | Yes | Yes |
| P4 | 44 | 42 | M | 0.301 | 0.698 | Yes | Yes | ND | Yes (LE) | Yes |
| P5 | 13 | 13 | M | 1.204 | 1.204 | Yes | Yes | BE-CS | No | No |
| P6 | 35 | 35 | M | 1.800 | 1.800 | Yes | Yes | ND | No | No |
| P7 | 40 | 40 | M | ND | ND | ND | ND | ND | No | ND |
| P8 | 16 | 16 | M | 0.300 | 0.602 | Yes | Yes | LE-CS; RE-few central depressed spots | No | No |
| P9 | 17 | 17 | M | 1.000 | 1.000 | HD | Yes | BE-cs | No | Yes |
| P10 | 28 | 28 | M | 2.000 | 2.000 | Yes | Yes | BE-cs | No | No |
| P11 | 15 | 15 | M | 1.800 | 1.204 | Yes | Yes | RE-cs; LE-CS | No | ND |
| P12 | 43 | 38 | M | 0 | 0 | RE-SP; LE-PD | Yes | BE-IFD | Yes | No |
| P13 | 16 | 16 | M | 1.301 | 0.795 | Yes | Only RE | BE-CS | No | Yes |
| P14 | 47 | 45 | M | 0.795 | 0.698 | Yes | Yes | RE-AFL; LE-CS | No | Yes |
| P15 | 8 | 8 | M | 0.477 | 1.301 | TP | Only LE | BE-cs | No | No |
| P16 | 31 | 31 | M | 1.602 | 1.800 | TP | Yes | ND | No | No |
| P17 | 30 | 29 | M | 1.800 | 2.000 | DP | Yes | ND | No | No |
| P18 | 19 | 19 | M | 2.300 | 2.300 | Yes | Yes | ND | No | Yes |
| P19 | 18 | 16 | M | 1.800 | 1.800 | Yes | Yes | ND | No | No |
| P20 | 23 | 23 | M | 1.000 | 1.800 | Yes | Yes | RE-cs; LE-ND | No | No |
| P21 | 31 | 24 | M | 0.795 | 0.795 | TP | Yes | BE-GFL | No | No |
| P22 | 21 | 21 | M | 1.000 | 1.778 | Yes | Yes | BE-AFL | No | Yes |
| P23 | 18 | 18 | M | 1.204 | 1.477 | RE-mild blurring of disc margin; LE-mild pallor setting in | Yes | ND | No | No |
| P24 | 35 | 35 | M | 1.800 | 1.800 | Yes | Yes | BE-GFL | No | Yes |
| P25 | 21 | 21 | M | 0.602 | 0.903 | BE-Mild RNFL edema | Yes | BE- enlargement of blind spot with cs | No | Yes |
| P26 | 17 | 17 | M | 1.800 | 1.800 | Yes | Yes | BE-DF | No | No |
| P27 | 18 | 18 | M | 2.000 | 2.000 | DP | Yes | ND | No | Yes |
| P28 | 17 | 17 | M | 1.778 | 2.000 | Trace pallor | Yes | ND | No | No |
| P29 | 17 | 17 | M | 0 | 0 | Sectoral DP | Yes | BE-CS | No | No |
| P30 | 17 | 17 | M | 0.795 | 1.204 | TP | Yes | BE-large cs | No | No |
| P31 | 18 | 16 | M | 0.698 | 1.000 | Minimal TP | Yes | BE-CS | No | No |
| P32 | 38 | 38 | M | 0.477 | 1.100 | N | ND | BE-cs | No | No |
| P33 | 17 | 16 | M | 1.000 | 0.795 | RNFL opacification | Yes | ND | No | No |
| Sample ID | Patient Haplogroup | Primary LHON Variants | ||
|---|---|---|---|---|
| m.3460G>A (p.Ala52Thr) | m.11778G>A (p.Arg340His) | m.14484T>C (p.Met64Val) | ||
| P1, P3 | U2a1 | - | + | - |
| P2, P6 | M5a | - | + | - |
| P4 | M65a1 | - | + | - |
| P5, P9 | M3a1 | - | + | - |
| P7 | H | - | + | - |
| P8 | M18a | - | + | - |
| P10 | M34a | - | + | - |
| P11 | M30c | - | + | - |
| P12 | M3a2 | - | + | - |
| P13 | M35a1 | - | + | - |
| P14 | R8b1 | - | + | - |
| P15 | M36 | - | + | - |
| P16 | M65a | - | + | - |
| P17 | W4 | - | + | - |
| P18 | M5a2a | - | + | - |
| P19 | R | - | + | - |
| P20 | R8a1a1b | - | + | - |
| P21 | M2a3 | - | + | - |
| P22 | R5a2 | - | + | - |
| P23 | R2 | - | + | - |
| P24 | D4 | - | + | - |
| P25 | M2a1c | - | + | - |
| P26 | M5a1b | - | + | - |
| P27 | M | - | + | - |
| P28 | W | - | + | - |
| P29 | I1 | - | - | + |
| P30 | M34A | - | - | + |
| P31 | U7 | - | - | + |
| P32 | M33a3 | - | - | + |
| P33 | M42b2 | - | - | + |
| Sample ID | Nucleotide Change | Amino Acid Change | Gene | Conservation (%) | Haplogroup Specific Variant | Patient Haplogroup | Disease Association |
|---|---|---|---|---|---|---|---|
| P2 * | m.13708G>A | p.A458T | MT-ND5 | 33.33 | Yes | M5a | LHON, increased MS risk, higher frequency in PD-ADS |
| m.15927G>A | - | MT-TT | 35.56 | No | LHON, MS, deaf1555 increased penetrance, CHD | ||
| P4 * | m.15924A>G | - | MT-TT | 71.11 | Yes | M65a1 | LIMM |
| P6 * | m.1391T>C | - | MT-RNR1 | 24.44 | No | M5a | HCM |
| m.11084A>G | p.T109A | MT-ND4 | 86.67 | No | AD, PD, MELAS | ||
| m.12477T>C | p.S47S | MT-ND5 | 97.78 | No | HCM | ||
| P16 * | m.13708G>A | p.A458T | MT-ND5 | 33.33 | Yes | M65a | LHON, increased MS risk, higher frequency in PD-ADS |
| P18 * | m.4454T>C | - | MT-TM | 55.56 | No | M5a2a | Possible contributor to mitochondrial dysfunction, hypertension |
| P19 * | m.1116A>G | - | MT-RNR1 | 51.11 | No | R | Deafness |
| P20 * | m.2755A>G | - | MT-RNR2 | 55.56 | No | R8a1a1b | LVNC |
| P23 * | m.4216T>C | p.Y304H | MT-ND1 | 24.44 | Yes | R2 | LHON, insulin resistance, possible adaptive high-altitude variant, miscarriage |
| P24 * | m.3010G>A | - | MT-RNR2 | 20 | Yes | D4 | Cyclic vomiting syndrome with migraine, high altitude adaptation |
| m.5178C>A | p.L237M | MT-ND2 | 22.22 | Yes | Longevity, extraversion, diabetes, AMS protection, blood iron metabolism, correlation with myocardial infarction, atherosclerosis | ||
| m.8414C>T | p.L17F | MT-ATP8 | 31.11 | Yes | Increased risk of T2DM and HAPC in haplogroup D4, longevity | ||
| m.11253T>C | p.I165T | MT-ND4 | 42.22 | No | LHON, PD | ||
| m.14668C>T | p.M2M | MT-ND6 | 24.44 | Yes | Depressive disorder associated | ||
| P26 * | m.15287T>C | p.F181L | MT-CYB | 82.22 | No | M5a1b | Deaf helper mutation |
| P27 * | m.5460G>A | p.A331T | MT-ND2 | 4.44 | Yes | M | AD, PD, LHON |
| P32 ** | m.2361G>A | - | MT-RNR2 | 2.22 | Yes | M33a3 | LVNC |
| P33 ** | m.921T>C | - | MT-RNR1 | 66.67 | No | M42b2 | LVNC |
| Sample ID | Age | Sex | Haplogroup | Nucleotide Change | Amino Acid Change | Gene | Conservation (%) | Haplogroup-Specific Variant | Disease Associated |
|---|---|---|---|---|---|---|---|---|---|
| P34 | 24 | M | T2 | m.4216T>C | p.Y304H | MT-ND1 | 24.44 | Yes | LHON, insulin resistance, possible adaptive high-altitude variant, miscarriage |
| m.4917A>G | p.N150D | MT-ND2 | 91.11 | Yes | LHON, insulin resistance, AMD, NRTI-PN | ||||
| m.5556G>A | - | MT-TW | 93.33 | No | Combined OXPHOS defects | ||||
| m.15928G>A | - | MT-TT | 48.89 | Yes | MS, idiopathic repeat miscarriage, AD protection | ||||
| P35 | 29 | M | F1d | m.6962G>A | p.L353L | MT-CO1 | 100 | Yes | Possible helper variant for 15927A |
| P36 | 31 | F | M65b | m.13708G>A | p.A458T | MT-ND5 | 33.33 | Yes | LHON, increased MS risk, higher frequency in PD-ADS |
| P37 | 19 | M | M35a1 | m.4136A>G | p.Y277C | MT-ND1 | 97.78 | No | LHON |
| P38 | 14 | M | M2a1b | m.4216T>C | p.Y304H | MT-ND1 | 24.44 | Yes | LHON, insulin resistance, possible adaptive high-altitude variant, miscarriage |
| P39 | 15 | M | M43b | m.8950G>A | p.V142I | MT-ATP6 | 51.11 | No | LDYT |
| m.11696G>A | p.V313I | MT-ND4 | 6.67 | No | LHON, LDYT, deafness, hypertension helper mutation | ||||
| P40 | 25 | M | U2b1a | m.12372G>A | p.L12L | MT-ND5 | 80 | Yes | Altered brain pH, sCJD patients |
| m.13708G>A | p.A458T | MT-ND5 | 33.33 | No | LHON, increased MS risk, higher frequency in PD-ADS | ||||
| m.15257G>A | p.D171N | MT-CYB | 95.57 | No | LHON | ||||
| P41 | 28 | M | M5a | m.12477T>C | p.S47S | MT-ND5 | 97.78 | Yes | Possible HCM susceptibility |
| m.13708G>A | p.A458T | MT-ND5 | 33.33 | Yes | LHON, increased MS risk, higher frequency in PD-ADS | ||||
| m.15927G>A | - | MT-TT | 35.56 | No | LHON, MS, deaf1555 increased penetrance, CHD | ||||
| P42 | 17 | M | R30a1b | m.3316G>A | p.A4T | MT-ND1 | 4.44 | Yes | Diabetes, LHON, PEO |
| m.9966G>A | p.V254I | MT-CO3 | 82.22 | No | LHON possible helper variant | ||||
| P43 | 39 | M | F1c1a2 | m.6962G>A | p.L353L | MT-CO1 | 100 | Yes | Possible helper variant for 15927A |
| m.10454T>C | - | MT-TR | 35.56 | Yes | Deaf helper mutation | ||||
| P44 | 30 | M | R30a1b | m.3316G>A | p.A4T | MT-ND1 | 4.44 | Yes | Diabetes, LHON, PEO |
| m.9966G>A | p.V254I | MT-CO3 | 82.22 | No | LHON possible helper variant | ||||
| P45 | 15 | M | M42b1 | m.7598G>A | p.A5T | MT-CO2 | 17.78 | Yes | Possible LHON helper variant |
| P46 | 16 | M | R2 | m.4216T>C | p.Y304H | MT-ND1 | 24.44 | Yes | LHON, insulin resistance, possible adaptive high-altitude variant, miscarriage |
| P47 | 20 | M | M6a1b | m.14693A>G | - | MT-TE | 91.11 | Yes | MELAS, LHON, deafness, hypertension helper |
| P48 | 22 | M | D4j1b | m.4883C>T | p.P138P | MT-ND2 | 100 | Yes | Glaucoma |
| m.11696G>A | p.V313I | MT-ND4 | 6.67 | No | LHON, LDYT, deafness, hypertension helper mutation | ||||
| m.14668C>T | p.M2M | MT-ND6 | 24.44 | Yes | Depressive disorder associated | ||||
| P49 | 24 | M | M43b | m.8950G>A | p.V142I | MT-ATP6 | 51.11 | No | LDYT |
| m.11696G>A | p.V313I | MT-ND4 | 6.67 | No | LHON, LDYT, deafness, hypertension helper mutation | ||||
| P50 | 50 | M | M38a | m.9966G>A | p.V254I | MT-CO3 | 82.22 | Yes | LHON possible helper variant |
| P51 | 26 | M | T1 | m.4216T>C | p.Y304H | MT-ND1 | 24.44 | Yes | LHON, insulin resistance, possible adaptive high-altitude variant, miscarriage |
| P52 | 29 | M | R6a2 | m.3700G>A | p.A132T | MT-ND1 | 93.33 | No | LHON |
| P53 | 32 | M | HV1b1b | m.7598G>A | p.A5T | MT-CO2 | 17.78 | No | Possible LHON helper variant |
| m.15927G>A | - | MT-TT | 35.56 | No | LHON, MS, deaf1555 increased penetrance, CHD | ||||
| P54 | 16 | M | M5b | m.6261G>A | p.A120T | MT-CO1 | 97.78 | No | Prostate cancer, LHON |
| m.13708G>A | p.A458T | MT-ND5 | 33.33 | Yes | LHON, increased MS risk, higher frequency in PD-ADS | ||||
| P55 | 14 | M | W3a1b | m.4386T>C | - | MT-TQ | 24.44 | No | Heart disease, myopathy, hypertension |
| m.5460G>A | p.A331T | MT-ND2 | 4.44 | Yes | AD, PD, LHON | ||||
| P56 | 29 | M | U2e1b | m.988G>A | - | MT-RNR1 | 77.78 | No | Possible deaf risk factor |
| m.14831G>A | p.A29T | MT-CYB | 42.22 | No | LHON | ||||
| P57 | 8 | M | R30a1b1 | m.3316G>A | p.A4T | MT-ND1 | 4.44 | Yes | Diabetes, LHON, PEO |
| P58 | 11 | M | W3a1 | m.5460G>A | p.A331T | MT-ND2 | 4.44 | Yes | AD, PD, LHON |
| Sample ID | Age | Sex | Haplogroup | Nucleotide Change | Amino Acid Change | Gene | Conservation (%) | Haplogroup Specific Variant | Disease Associated |
|---|---|---|---|---|---|---|---|---|---|
| P59 | 21 | M | M33a2a | m.15908T>C | - | MT-TT | 57.78 | Yes | Deaf helper mutation |
| P60 | 15 | M | M3a1 | m.5556G>A | - | MT-TW | 93.33 | No | Combined OXPHOS defects |
| P61 | 16 | M | R5a1 | m.14864T>C | p.C40R | MT-CYB | 97.78 | No | MELAS |
| P62 | 20 | M | M33a2a | m.15908T>C | - | MT-TT | 57.78 | Yes | Deaf helper mutation |
| P63 | 17 | F | M4a | m.7859G>A | p.D92N | MT-CO2 | 24.44 | Yes | Progressive encephalomyopathy |
| P64 | 23 | M | M35a1 | m.15043G>A | p.G99G | MT-CYB | 97.78 | Yes | MDD-associated, possible role in high-altitude sickness |
| m.15924A>G | - | MT-TT | 71.11 | Yes | LIMM | ||||
| P65 | 17 | M | M | m.15908T>C | - | MT-TT | 57.78 | Yes | Deaf helper mutation |
| P66 | 19 | M | R31b | m.1452T>C | - | MT-RNR1 | 91.11 | No | Deafness |
| P67 | 37 | M | M5a | m.1391T>C | MT-RNR1 | 24.44 | No | HCM | |
| m.12477T>C | p.S47S | MT-ND5 | 97.78 | Yes | Possible HCM susceptibility | ||||
| m.15043G>A | p.G99G | MT-CYB | 97.78 | Yes | MDD-associated, possible role in high altitude sickness | ||||
| P68 | 56 | F | M2b | m.1453A>G | - | MT-RNR1 | 80 | Yes | Possible deaf risk factor |
| P69 | 32 | M | M6a2 | m.12236G>A | - | MT-TS2 | 71.11 | Yes | Deaf |
| P70 | 29 | M | F1d | m.5628T>C | - | MT-TA | 95.56 | No | CPEO, deaf enhancer, gout, tic disorder |
| P71 | 24 | M | M2b1 | m.1453A>G | - | MT-RNR1 | 80 | Yes | Possible deaf risk factor |
| P72 | 26 | M | R31b | m.1452T>C | - | MT-RNR1 | 91.11 | No | Deafness |
| P73 | 38 | M | U2c1 | m.9098T>C | p.I191T | MT-ATP6 | 75.56 | No | Predisposition to anti-retroviral mitochondrial disease |
| P74 | 37 | M | H5a1 | m.4336T>C | - | MT-TQ | 42.22 | Yes | ADPD, hearing loss & migraine, autism spectrum, intellectual disability |
| P75 | 44 | M | M33a2 | m.15908T>C | - | MT-TT | 57.78 | Yes | Deaf helper mutation |
| P76 | 38 | M | M33a2a | m.2361G>A | MT-RNR2 | 2.22 | Yes | Possibly LVNC-associated | |
| m.15908T>C | - | MT-TT | 57.78 | Yes | Deaf helper mutation | ||||
| P77 | 19 | M | M3a2a | m.5783G>A | - | MT-TC | 82.22 | No | Myopathy, deafness, gout, tic disorder |
| P78 | 18 | M | K1a4 | m.9055G>A | p.A177T | MT-ATP6 | 86.67 | Yes | PD protective factor |
| P79 | 15 | M | M5a2a | m.4454T>C | - | MT-TM | 55.56 | No | Possible contributor to mitochondrial dysfunction, hypertension |
| P80 | 24 | M | M5a2a1 | m.4454T>C | - | MT-TM | 55.56 | No | Possible contributor to mitochondrial dysfunction, hypertension |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jha, R.K.; Dawar, C.; Hasan, Q.; Pujar, A.; Gupta, G.; Vishnu, V.Y.; Kekunnaya, R.; Thangaraj, K. Mitochondrial Genetic Heterogeneity in Leber’s Hereditary Optic Neuropathy: Original Study with Meta-Analysis. Genes 2021, 12, 1300. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091300
Jha RK, Dawar C, Hasan Q, Pujar A, Gupta G, Vishnu VY, Kekunnaya R, Thangaraj K. Mitochondrial Genetic Heterogeneity in Leber’s Hereditary Optic Neuropathy: Original Study with Meta-Analysis. Genes. 2021; 12(9):1300. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091300
Chicago/Turabian StyleJha, Rajan Kumar, Chhavi Dawar, Qurratulain Hasan, Akhilesh Pujar, Gaurav Gupta, Venugopalan Y. Vishnu, Ramesh Kekunnaya, and Kumarasamy Thangaraj. 2021. "Mitochondrial Genetic Heterogeneity in Leber’s Hereditary Optic Neuropathy: Original Study with Meta-Analysis" Genes 12, no. 9: 1300. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091300