
Genome-Wide Survey for Microdeletions or -Duplications in 155 Patients with Lower Urinary Tract Obstructions (LUTO)

 , , , , , , , , , , ,
, , , , , , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and DNA Isolation
2.2. Array Genomic Hybridization Analysis
2.3. CNV Analysis
2.3.1. Quality Check
2.3.2. Filter Algorithms
2.4. CNV Validation Using Quantitative PCR (qPCR)
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LUTO | Lower Urinary Tract Obstruction |
| PUV | Posterior Urethral Valves |
| CNV | Copy Number Variation |
| ESRD | End Stage Renal Disease |
| CAKUT | Congenital Anomalies of the Kidneys and Urinary Tract |
| DGV | Database of Genomic Variants |
| eGFR | estimated Glomerular Filtration Rate |
| CKD | Chronic Kidney Disease |
| VCUG | Voiding Cystourethrography |
| Kbp | kilo base pairs |
| qPCR | quantitative PCR |
References
- Robertson, W.B.; Hayes, J.A. Congenital diaphragmatic obstruction of the male posterior urethra. Br. J. Urol. 1969, 41, 592–598. [Google Scholar] [CrossRef]
- Cheung, K.W.; Morris, R.K. Congenityl urinary tract obstrucion. Best Pract. Res. Clin. Obstet. Gynaecol. 2019, 58, 78–92. [Google Scholar] [CrossRef]
- Morris, R.K.; Kilby, M.D. Long-term renal and neurodevelopmental outcome in infants with LUTO, with and without fetal intervention. Early Hum. Dev. 2011, 87, 607–610. [Google Scholar] [CrossRef]
- Malin, G.; Tonks, A.M.; Morris, R.K.; Gardosi, J.; Kilby, M.D. Congenital lower urinary tract obstruction: A population-based epidemiological study. BJOG Int. J. Obstet. Gynaecol. 2012, 119, 1455–1464. [Google Scholar] [CrossRef]
- Bildau, J.; Enzensberger, C.; Degenhardt, J.; Kawecki, A.; Tenzer, A.; Kohl, T.; Stressig, R.; Ritgen, J.; Utsch, B.; Axt-Fliedner, R. Lower urinary tract obstruction (LUTO)—Clinical picture, prenatal diagnostics and therapeutic options. Z. Geburtshilfe Neonatol. 2014, 218, 18–26. [Google Scholar] [PubMed]
- Coquillette, M.; Lee, R.S.; Pagni, S.E.; Cataltepe, S.; Stein, D.R. Renal outcomes of neonates with early presentation of posterior urethral valves: A 10-year single center experience. J. Perinatol. Off. J. Calif. Perinat Assoc. 2020, 40, 112–117. [Google Scholar] [CrossRef]
- Kolvenbach, C.M.; Dworschak, G.C.; Frese, S.; Japp, A.S.; Schuster, P.; Wenzlitschke, N.; Yilmaz, Ö.; Lopes, F.M.; Pryalukhin, A.; Schierbaum, L.; et al. Rare Variants in BNC2 Are Implicated in Autosomal-Dominant Congenital Lower Urinary-Tract Obstruction. Am. J. Hum. Genet. 2019, 104, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, S.; Habbig, S.; Weber, L.T.; Liebau, M.C. Molecular causes of congenital anomalies of the kidney and urinary tract (CAKUT). Mol. Cell Pediatr. 2021, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Boghossian, N.S.; Sicko, R.J.; Kay, D.; Rigler, S.L.; Caggana, M.; Tsai, M.; Yeung, E.; Pankratz, N.; Cole, B.R.; Druschel, C.M.; et al. Rare copy number variants implicated in posterior urethral valves. Am. J. Med. Genet. A 2016, 170, 622–633. [Google Scholar] [CrossRef]
- Verbitsky, M.; Westland, R.; Perez, A.; Kiryluk, K.; Liu, Q.; Krithivasan, P.; Mitrotti, A.; Fasel, D.A.; Batourina, E.; Sampson, M.G.; et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat. Genet. 2019, 51, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinforma Oxf. Engl. 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Rice, A.M.; McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 2017, 8, 14366. [Google Scholar] [CrossRef]
- Draaken, M.; Giesen, C.A.; Kesselheim, A.L.; Jabs, R.; Aretz, S.; Kugaudo, M.; Chrzanowska, K.H.; Krajewska-Walasek, M.; Ludwig, M. Maternal de novo triple mosaicism for two single OCRL nucleotide substitutions (c.1736A>T, c.1736A>G) in a Lowe syndrome family. Hum. Genet. 2011, 129, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E. Mouse Genome Database Group. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Patel, P.; Verma, M. Biomarkers in prostate cancer epidemiology. Cancers 2011, 3, 3773–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, A.A.; Ameh, E.A.; Abdur-Rahman, L.O.; Adeniran, J.O.; Abraham, M.K. Posterior urethral valve. World J. Pediatr. WJP 2011, 7, 205–216. [Google Scholar] [CrossRef]
- Luo, Z.; Zheng, Y.; Zhang, W. Pleiotropic functions of miR107 in cancer networks. OncoTargets Ther. 2018, 11, 4113–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, R.M.; Todd, J.N.; Sandholm, N.; Cole, J.B.; Chen, W.M.; Andrews, D.; Pezzolesi, M.G.; McKeigue, P.M.; Hiraki, L.T.; Qiu, C.; et al. Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. JASN 2019, 30, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, H.V.; Richards, S.M.; Bevan, P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Giardino, D.; Finelli, P.; Amico, F.P.; Gottardi, G.; Civa, R.; Corona, G.; Nocera, G.; Larizza, L. Unbalanced segregation of a complex four-break 5q23-31 insertion in the 5p13 band in a malformed child. Eur. J. Hum. Genet. EJHG 2004, 12, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Kruepunga, N.; Hikspoors, J.P.; Mekonen, H.K.; Mommen, G.M.; Meemon, K.; Weerachatyanukul, W.; Asuvapongpatana, S.; Eleonore Köhler, S.; Lamers, W.H. The development of the cloaca in the human embryo. J. Anat. 2018, 233, 724–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluth, M.; Galal, R.; Krohn, A.; Weischenfeldt, J.; Tsourlakis, C.; Paustian, L.; Ahrary, R.; Ahmed, M.; Scherzai, S.; Meyer, A.; et al. Prevalence of chromosomal rearrangements involving non-ETS genes in prostate cancer. Int. J. Oncol. 2015, 46, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Rieke, J.M.; Zhang, R.; Braun, D.; Yilmaz, Ö.; Japp, A.S.; Lopes, F.M.; Pleschka, M.; Hilger, A.C.; Schneider, S.; Newman, W.G.; et al. SLC20A1 is involved in Urinary Tract and Urorectal Development. Front. Cell Dev. Biol. 2020, 8, 567. [Google Scholar] [CrossRef] [PubMed]


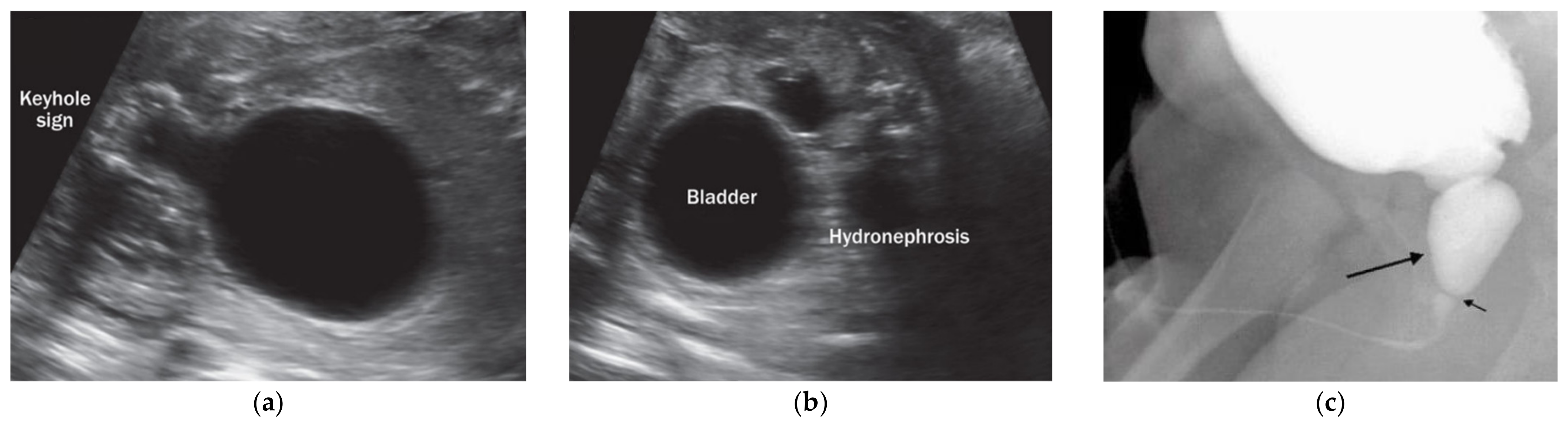{kind=link}
| Patient ID | Phenotype | CNV | Length | Mode of Inheritance | Affected Genes |
|---|---|---|---|---|---|
| 100243 | PUV | dup5q23.2 | 852.7 kbp | de novo | LINC02201/LOC101927357/LOC105379152 /MGC32805/PPIC/PRDM6/ SNCAIP/SNX2/SNX24 |
| 100295 | PUV | dup10q23.31 | 131.3 kbp | no parental DNA | MIR107/PANK1/SLC16A12 |
| 100009 | LUTO | dup1p36.21 | 65.7 kbp | no parental DNA | FBLIM1/PLEKHM2/SLC25A34/ SLC25A34-AS1/TMEM82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schierbaum, L.M.; Schneider, S.; Herms, S.; Sivalingam, S.; Fabian, J.; Reutter, H.; Weber, S.; Merz, W.M.; Tkaczyk, M.; Miklaszewska, M.; et al. Genome-Wide Survey for Microdeletions or -Duplications in 155 Patients with Lower Urinary Tract Obstructions (LUTO). Genes 2021, 12, 1449. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091449
Schierbaum LM, Schneider S, Herms S, Sivalingam S, Fabian J, Reutter H, Weber S, Merz WM, Tkaczyk M, Miklaszewska M, et al. Genome-Wide Survey for Microdeletions or -Duplications in 155 Patients with Lower Urinary Tract Obstructions (LUTO). Genes. 2021; 12(9):1449. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091449
Chicago/Turabian StyleSchierbaum, Luca M., Sophia Schneider, Stefan Herms, Sugirthan Sivalingam, Julia Fabian, Heiko Reutter, Stefanie Weber, Waltraut M. Merz, Marcin Tkaczyk, Monika Miklaszewska, and et al. 2021. "Genome-Wide Survey for Microdeletions or -Duplications in 155 Patients with Lower Urinary Tract Obstructions (LUTO)" Genes 12, no. 9: 1449. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091449