1. Introduction
Metagenomics is the study of microorganism genomes in the environment, such as the soil, ocean, and living organisms, achieved by extracting and sequencing DNA. This method can reveal the existence and ratio of microorganisms in a specific clade within an environment, and also provide genomic information of unknown and uncultured microorganisms.
The DNA sequences of most microorganisms in the environment are unknown; thus, metagenomic analysis refers to the genomic information of not only the same species, but also that of closely related organisms. A homology search must be performed against enormous databases to identify sequences that are similar to those obtained by DNA sequencing. Since changes in genomic information occur at the amino acid level, a six-frame translation of the DNA sequence is performed prior to the homology search, focusing only on the coding region. While DNA sequences are represented by four letters (A, T, G, and C), protein sequences are represented by 20 standard amino acid letters. Further, while comparisons between DNA sequences often distinguish only two states of each base (match or mismatch), similarities between protein sequences are based on a substitution matrix that represents the likelihood of each amino acid being substituted with another amino acid [
1]. Therefore, performing a homology search on protein sequences is more challenging than on DNA sequences.
The Smith–Waterman algorithm [
2] is the most rigorous method to obtain optimal sequence alignment by means of dynamic programming during a homology search. This algorithm is implemented in SSEARCH [
3] and other software; however, SSEARCH is too slow to search against a large number of sequences within a realistic execution time. For this reason, programs that perform fast homology searches are now widely used, such as BLAST [
4,
5]. Nevertheless, the amount of data has drastically increased with the advent of high-throughput next-generation DNA sequencers. For example, the Illumina NovaSeq6000 DNA sequencer can output up to 6T bases in a single run of a few days, requiring hundreds of thousands of CPU days for BLAST to process that volume of data. Thus, faster homology search programs such as RAPSearch2 [
6], GHOSTZ [
7], and DIAMOND [
8] have been proposed. While there is a tradeoff between search speed and accuracy, further acceleration while maintaining high accuracy is required for comprehensive metagenomic analyses.
In general, homology search tools identify subsequences (seeds) that have a high degree of similarity between database and query sequences, thereby substantially reducing the number of candidate sequences in the database. This is called a seed search strategy. RAPSearch2, GHOSTZ, and DIAMOND achieve highly efficient and accurate searches by employing reduced amino acid alphabets in their seed search strategies. These reduced amino acid alphabets cluster the 20 standard amino acids according to the scores between them, yielding an alphabet with a smaller number of representative characters. The reduced amino acid alphabets proposed by Murphy et al. [
9] are commonly used in existing methods. Performing a seed search based on a sequence converted to a reduced amino acid alphabet enables treating similar amino acids, such as Asp and Glu, as the same character. As a result, database sequences similar to the query sequence can be quickly detected by exact-match search. Existing methods have used single patterns of reduced alphabets. However, the amino acids that constitute the functional sites of proteins are strongly conserved, whereas the peripheral amino acids that maintain the structure are loosely conserved. Therefore, we hypothesized that significant seed hits could be identified more rapidly by searching for exact matches in the center of the seed using a less compressed amino acid alphabet, while searching at both ends of the seed using a more compressed amino acid alphabet.
In this study, we proposed a new seed search algorithm that employs multiple reduced amino acid alphabets with different numbers of characters to search for similar subsequences in a two-step seed search (TSSS) strategy, with the aim of implementing a faster but accurate homology search program. The implementation is open-sourced at
https://github.com/akiyamalab/tsss (accessed on 17 September 2021) under the MIT license.
2. Methods
2.1. Reduced Amino Acid Alphabets
A reduced amino acid character represents a cluster of standard amino acids, as shown in
Figure 1. Treating a match of amino acids represented by the same reduced amino acid alphabet as a perfect match provides a fast relaxed search that allows the substitution of an amino acid with a similar amino acid. In this study, we used the reduced amino acid alphabets generated using the method proposed by Murphy et al., based on the BLOSUM62 substitution matrix, which describes the similarity score between each of the 20 standard amino acids. First, correlation coefficients were calculated for all pairs of amino acids based on the BLOSUM62, and then the amino acids were grouped by hierarchical clustering. The size of the reduced amino acid alphabet was user-defined, ranging from 4 to 18 characters in the current study. The maximum score obtained by a perfect match of any standard amino acid in the group was used to define the match score for a matching reduced amino acid alphabet character in an identical group, as an example is shown in the bottom part of
Figure 1.
2.2. Two-Step Seed Search (TSSS)
In TSSS, the seed search was divided into two steps employing different reduced amino acid alphabets, as shown in
Figure 2. In the first step, the database and query sequences were converted into the first reduced amino acid alphabet, A
1. Subsequences starting from all possible positions of the query sequence were enumerated (referred to as
seed1) with a residue length of L
1. Then, matching portions were searched against the database where
seed1 matched, with a Hamming distance ≤ H
1.
In the second step, the seed1 hits obtained in the first step were extended in both directions with a residue length of L2. Each extended area at both ends was named seed2. The seed2 subsequences were converted into the second reduced amino acid alphabet, A2, and Hamming distances between the query and database sequences were calculated individually for both seed2 areas. If the Hamming distances of both seed2 areas were less than or equal to threshold H2, the whole region (seed1 plus both seed2 areas) was treated as a final seed hit for the following alignment calculation.
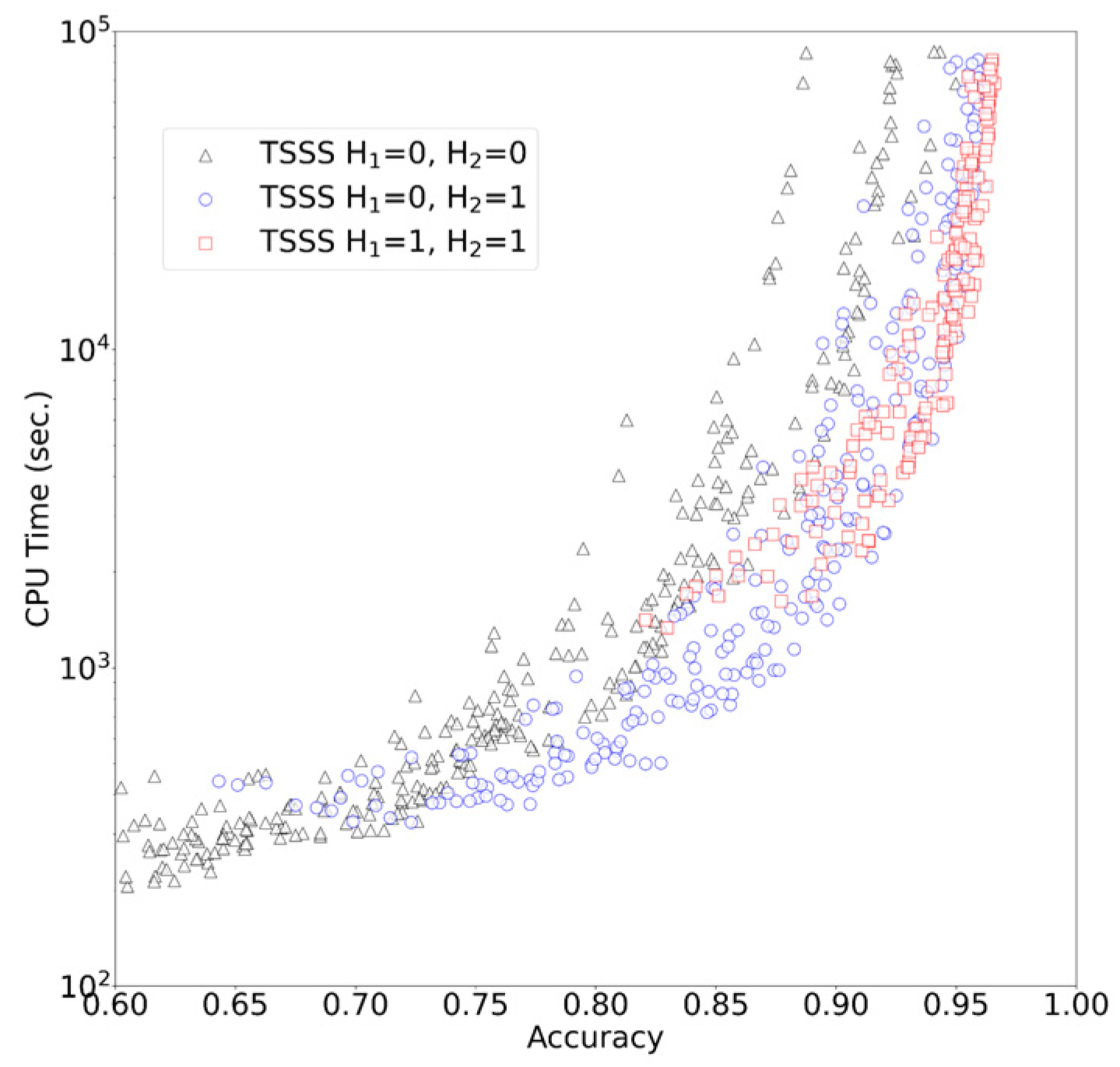
In the case of H1 > 0 (or H2 > 0), we generated all words within the designated Hamming distance for the query seed and then searched database positions that exactly matched with one of the words.
Larger values of A1, A2, L1, or L2 corresponded with smaller numbers of obtained seed hits for the alignment step, which resulted in faster processing but degraded sensitivity. Similarly, smaller values of H1 or H2 resulted in faster and less sensitive processing. Therefore, the combination of values greatly affected the specifications of TSSS, necessitating optimization of these parameters.
There is a possible alternative searching strategy where the seed1 plus both seed2 areas are at first searched with a small (loose) reduced amino acid alphabet, and then candidates are narrowed down by searching the seed1 area with a large (tight) reduced amino acid alphabet. Nevertheless, this approach might not be so efficient with the current TSSS procedure because enumerating all patterns within a Hamming distance requires high computational cost in the first step for a longer seed.
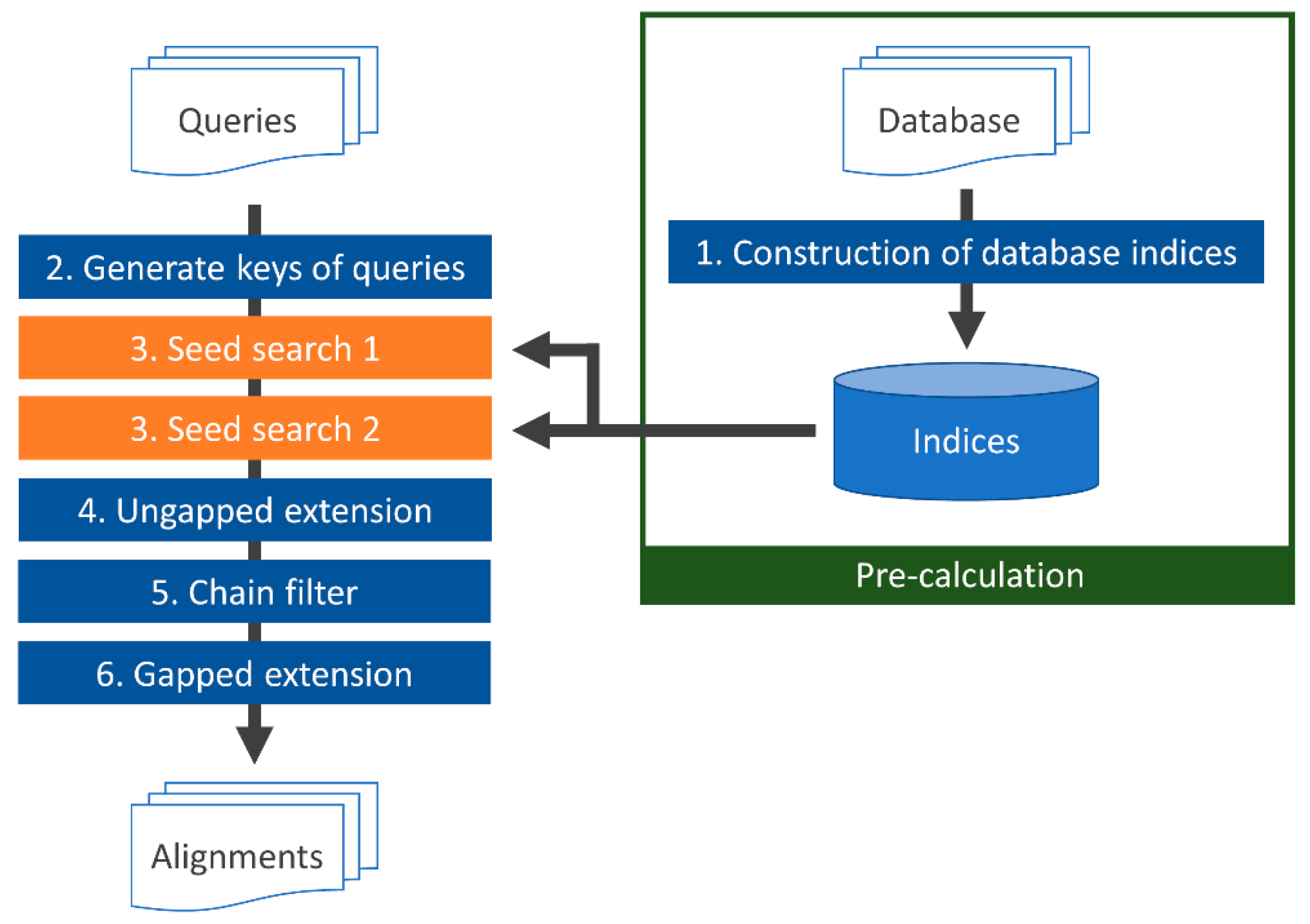
2.3. TSSS Procedure
Figure 3 displays a flowchart of TSSS. First, (1) indices of the
seed1 and
seed2 subsequences in the database were constructed with the corresponding reduced amino acid alphabets A
1 and A
2. Once the homology search was initiated, (2) the DNA query sequence was translated into a protein sequence, followed by conversion of the translated sequence to both the first and second reduced amino acid alphabets A
1 and A
2. Then, seeds of the query were enumerated and translated with the same reduced amino acid alphabets. Next, (3) the seed was searched against the database to find positions (seed hits) with high similarity between the database and query sequences. Finally, (4–6) the identified seeds were extended (ungapped and gapped seed extensions) with alignment, and the score was calculated. The details of each process are described in the following sections.
2.3.1. Building a Database Subsequence Index (1)
First, all protein sequences in the database were concatenated by inserting a predetermined delimiter character such as ‘#’. All subsequences with the lengths of the two seeds, seed1 L1 and seed2 L2, were obtained by repeatedly shifting one character at a time in the concatenated sequence. Subsequences containing the delimiter were discarded. Then, depending on parameters A1, A2, L1, and L2, the seed1 and seed2 areas were enumerated and indices were generated that mapped each subsequence to a position in the database.
2.3.2. Generating Keys for Query (2) and Searching for Seeds (3)
DNA sequence reads obtained by a DNA sequencer were translated into six frames and concatenated using the delimiter. The same operation described in
Section 2.3.1 was performed on the concatenated query to enumerate keys of the subsequence. Keys containing the termination codon were discarded.
In the seed search, parameters H
1 and H
2 (described in
Section 2.2) were used to search for database positions of the seed hits. An identified seed hit was recorded as a candidate for alignment, and the next operation was executed.
2.3.3. Ungapped Extension (4) and Chain Filtering (5)
In the extension step, an ungapped extension procedure was performed around the seed hit to further narrow down significant database positions. Although the seed search used reduced amino acid alphabets, the process after the ungapped extension step calculated an alignment score including the seed region based on the 20 standard amino acids. Termination codons were not discarded in the ungapped and gapped extension procedures, and were regarded as a single virtual amino acid. The similarity score between a termination codon and each amino acid was defined by the BLOSUM62 matrix. In the ungapped extension step, as in BLAST, X-dropoff [
4] was used to terminate the extension when the score decrease from the peak value was greater than the value determined by the score matrix and user-defined parameter. Only subsequences with scores exceeding the threshold were recorded as candidates for the next gapped extension step.
Where a long region in the database matched the query, many subsequences had almost the same results in the gapped extension. For this reason, TSSS used chain filtering to eliminate redundant gapped extension trials by combining subsequences located in neighboring positions into one subsequence [
5]. Subsequences were combined into one longer subsequence when subsequences of the ungapped extension overlapped, or when the ungapped extension score between two subsequences was greater than or equal to the threshold.
2.3.4. Gapped Extension (6)
The gapped extension in TSSS used the same method as BLAST. As in the ungapped extension, we employed X-dropoff to terminate the extension when the score decrease from the peak value was greater than the threshold. We also used a Gotoh algorithm [
10] that considered affine gaps, which reduced the penalty for consecutive gaps in alignment.
2.4. Evaluation Procedure
2.4.1. Computing Environment and Comparison Programs
We evaluated the computational speed and accuracy of TSSS using the f_node of the TSUBAME3.0 supercomputer at Tokyo Institute of Technology. This computing environment consisted of two Intel Xeon E5-2680 v4 (14 cores, 2.4 GHz) CPUs and 256 GB of memory. We used GCC (version 4.8.5) as the compiler, with the optimization option -O3.
BLAST [
4,
5] (version 2.7.1), RAPSearch2 [
6] (version 2.22), GHOSTZ [
7] (version 1.0.2), and DIAMOND [
8] (version 0.9.14.115) were used as comparison programs. The following options for all programs were employed: BLOSUM62 for the substitution matrix, no SEG filter to ignore low-complexity regions of sequences, and 10 alignments per query. Each program supported multi-thread operations, but was executed with a single thread unless otherwise noted. The detailed options for each program are presented in
Table 1.
The sizes of the reduced amino acid alphabets used in the comparison programs are listed in
Table 2. Although most alphabets were 10 characters in size, the seed search algorithms of each program differed, making a simple comparison difficult. For example, DIAMOND used multiple patterns of spaced seeds that do not necessarily require an exact match of contiguous subsequences.
2.4.2. Datasets
We employed the KEGG GENES prokaryotes database (acquired in February 2019) [
11,
12] as the protein sequence database. This database consists of approximately 17.7 million protein sequences, with a total residue length of approximately 5.6 billion residues. SRR5788325 [
13], a set of DNA sequences obtained from the NCBI Sequence Read Archive, was used as the query data. The DNA sequences were quality controlled using PRINSEQ-lite [
14] (version 0.20.4) prior to the experiments.
2.4.3. Calculation of Accuracy
To evaluate the search accuracy of each program, the optimal hits with the Smith–Waterman algorithm were needed for each query sequence. We used SSEARCH with the E-value threshold of 10
−5 to obtain them. Reference hits used in the evaluation consisted of up to 10 optimal hits, and thus, the maximum number of reference hits was 10 for each query. Then, the top 10 hits of each homology search program were retained, and the obtained hits resulting in the same sequences as the reference hits were counted as matches. The accuracy of the search program was measured as the ratio of the number of matches to the number of reference hits (≤10). The E-value of the alignment was based on the output value of each program, although a previous study suggests that for a more precise calculation, the E-value should consider frameshift alignment [
15].
4. Conclusions
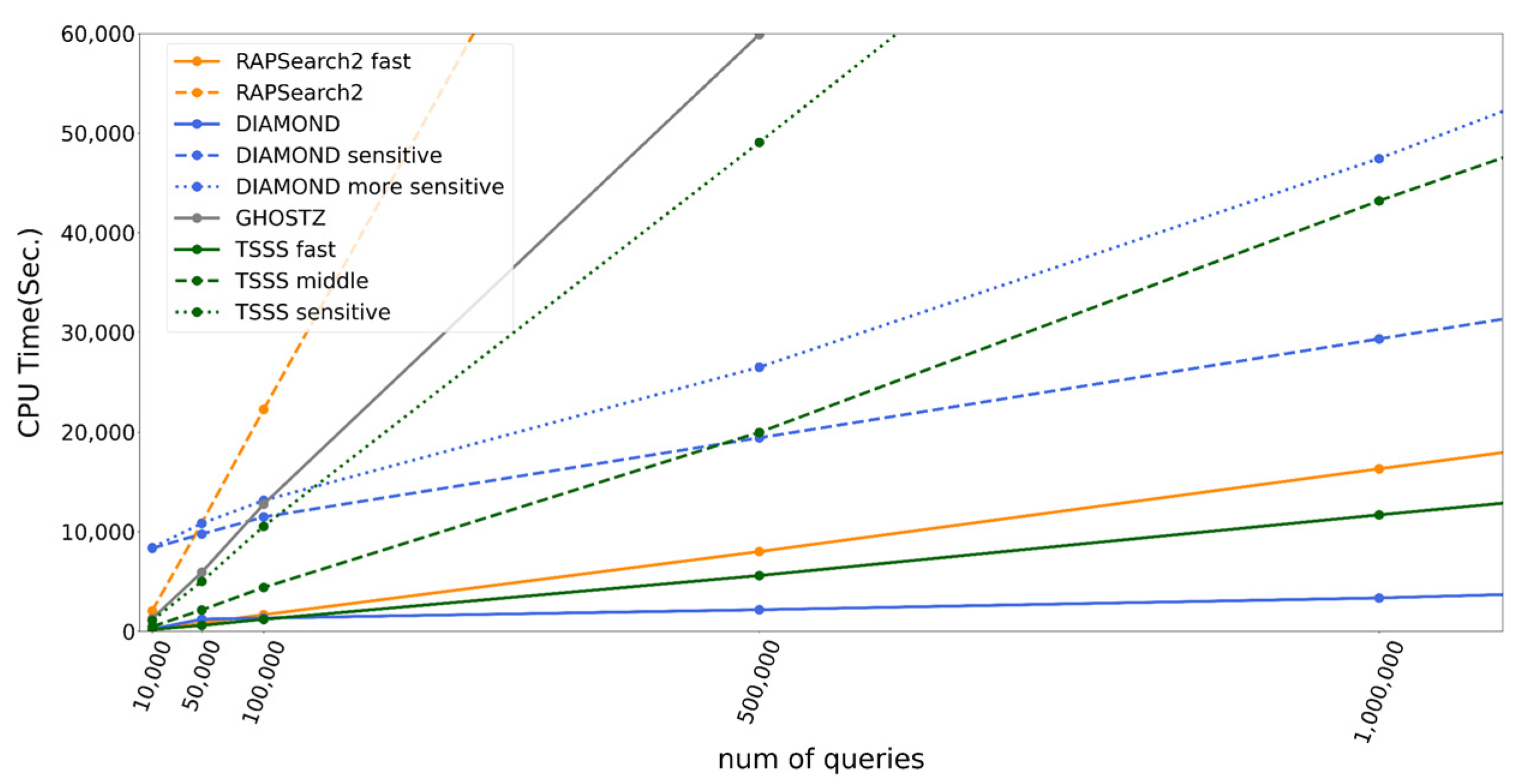
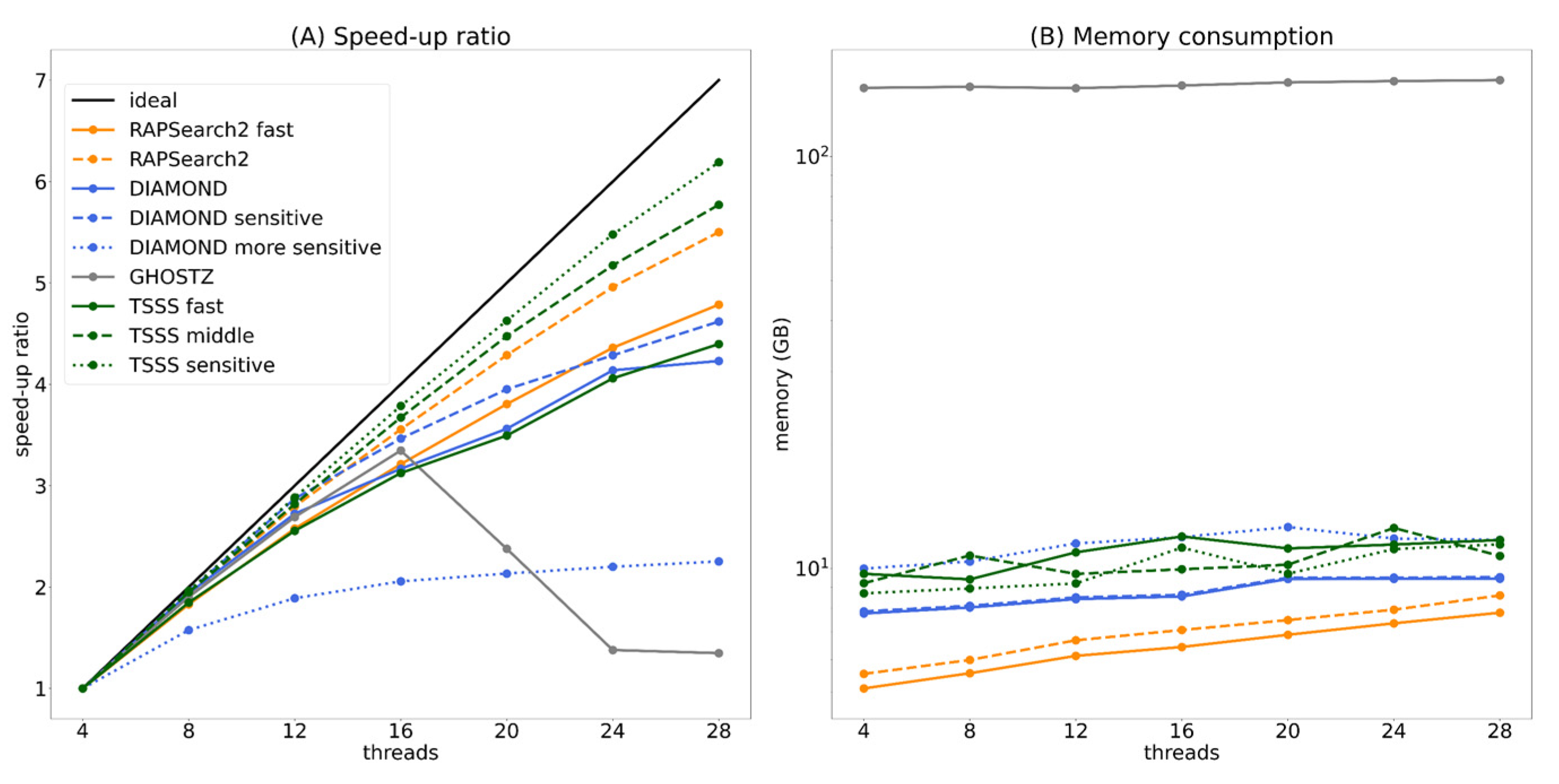
Herein, we proposed a homology search algorithm that employed TSSS with different reduced amino acid alphabets and compared the implementation of this method against several existing homology search tools. TSSS was 5.0 and 2.2 times faster than RAPSearch2 and GHOSTZ, respectively, using parameters providing the same accuracy. In addition, the same parameters achieved 336 times faster search than that with BLAST. These results indicate that the TSSS’s two-step seed search strategy is more effective than the traditional one-step seed search strategy widely used among homology search tools such as DIAMOND and GHOSTZ. However, the implementation of TSSS has scope for improvement in terms of the pre-calculation of the seed search. DIAMOND was faster than TSSS when the number of queries was sufficiently large and up to 4.9 times faster using the parameter set providing the same accuracy. The most important reason for its faster speed is the double indexing technique that DIAMOND uses to construct a data structure for all query sequences as well as for all database sequences, thereby shortening the execution time per query. Therefore, double indexing or the construction of a suitable data structure specialized for TSSS must accelerate our implementation. Furthermore, TSSS combined with the spaced seed technique utilized in DIAMOND instead of enumeration of all words will enhance the performance. The clustering of database sequences technique utilized in GHOSTZ is another option to enhance the performance.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}