
Mammalian SIRT6 Represses Invasive Cancer Cell Phenotypes through ATP Citrate Lyase (ACLY)-Dependent Histone Acetylation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Viral Infection, and Drug Treatment
2.2. Co-Immunoprecipitation (IP)
2.3. Acetyl-CoA Measurement
2.4. RNA Extraction and QPCR
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. Wound Healing Scratch Assay
2.7. Soft Agar Assay
2.8. Fibronectin Adhesion Assay
2.9. Antibodies
3. Results
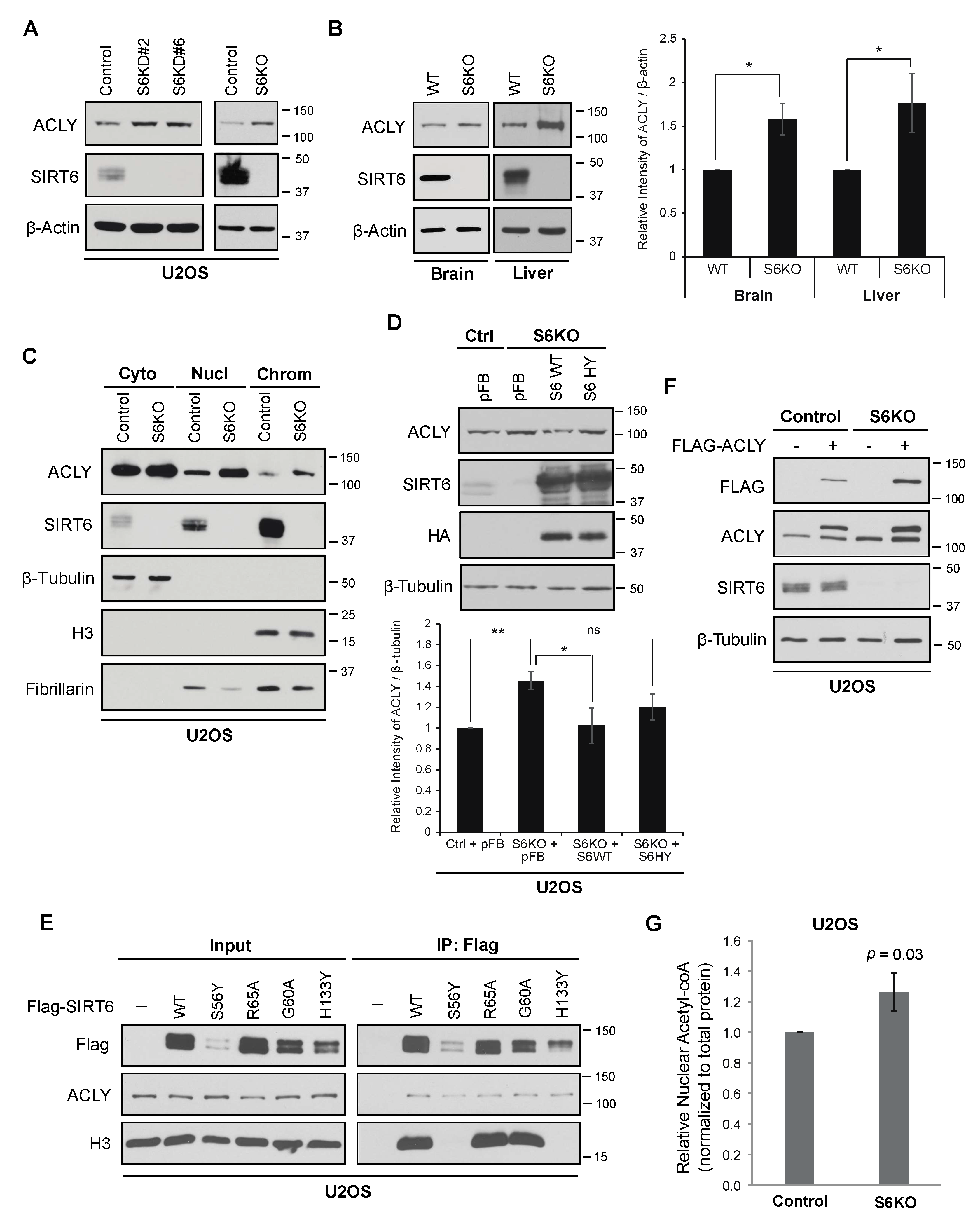
3.1. SIRT6 Deficiency Leads to Increased Levels of ACLY
3.2. ACLY-Dependent Up-Regulation of Acetyl-coA Responsive Genes in SIRT6-Deficient Cells
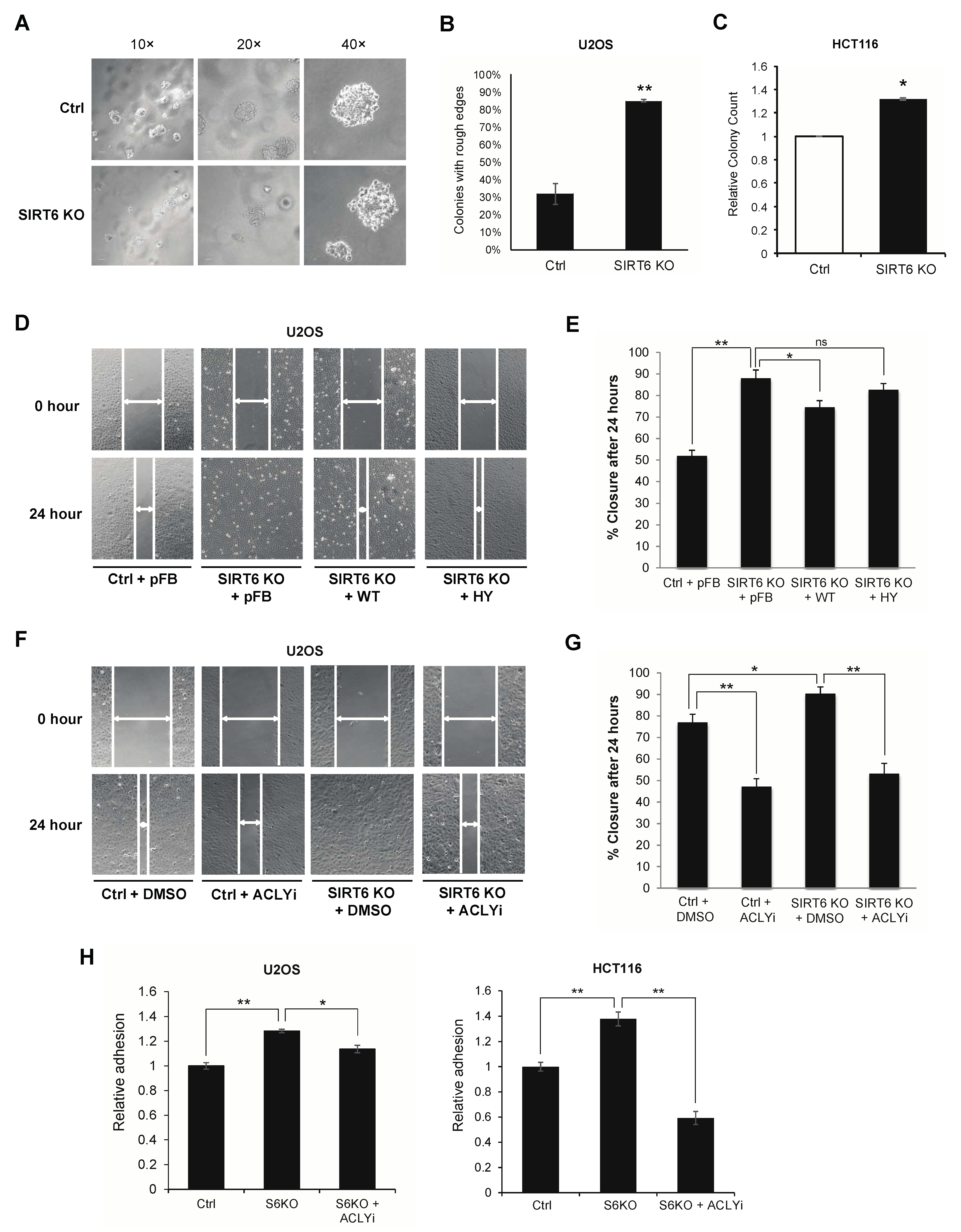
3.3. ACLY-Dependent SIRT6 Regulation of Cancer Migration and Adhesion Phenotypes
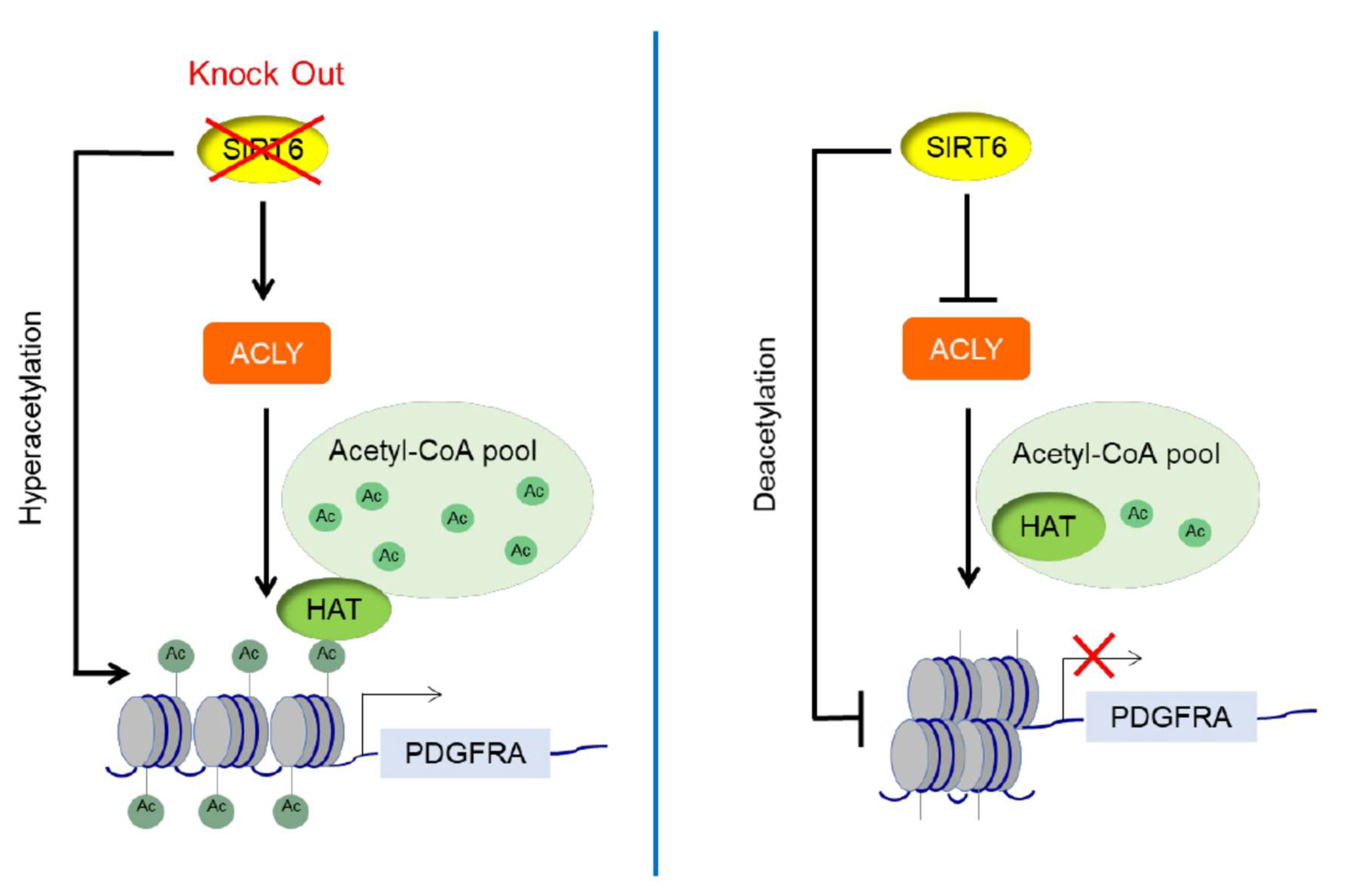
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.V.; Shah, S.; Carrer, A.; Wellen, K.E. A cancerous web: Signaling, metabolism, and the epigenome. Mol. Cell. Oncol. 2014, 2, e965620. [Google Scholar] [CrossRef] [Green Version]
- Ghosh-Choudhary, S.; Liu, J.; Finkel, T. Metabolic Regulation of Cell Fate and Function. Trends Cell Biol. 2020, 30, 201–212. [Google Scholar] [CrossRef]
- Peserico, A.; Simone, C. Physical and Functional HAT/HDAC Interplay Regulates Protein Acetylation Balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [Green Version]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nat. Cell Biol. 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, P.A.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J. Nucleocytosolic Acetyl-Coenzyme A Synthetase Is Required for Histone Acetylation and Global Transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, Z.T.; Voorde, J.V.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Parris, J.L.; Trefely, S.; Henry, R.A.; Montgomery, D.C.; Torres, A.; Viola, J.M.; Kuo, Y.-M.; Blair, I.A.; Meier, J.L.; et al. Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. J. Biol. Chem. 2017, 292, 3312–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Wu, Z.; Fournel, L.; Coquerel, A.; Lincet, H.; Alifano, M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett. 2020, 471, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.-F.; Lim, H.-W.; Liu, S.; et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.V.; Berry, C.T.; Kim, K.; Sen, P.; Kim, T.; Carrer, A.; Trefely, S.; Zhao, S.; Fernandez, S.; Barney, L.E.; et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes Dev. 2018, 32, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265.e6. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Presegué, L.; Vaquero, A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2014, 282, 1745–1767. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 Promotes DNA Repair Under Stress by Activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Spiegelman, N.A.; Nelson, O.D.; Jing, H.; Lin, H. SIRT6 regulates Ras-related protein R-Ras2 by lysine defatty-acylation. eLife 2017, 6, 39. [Google Scholar] [CrossRef]
- Tasselli, L.; Zheng, W.; Chua, K.F. SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol. Metab. 2017, 28, 168–185. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; Mccord, R.A.; Boxer, L.; Barber, M.; Hong, T.; Gozani, O.; Chua, K.F. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009, 8, 2664–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zeng, Y.; Wu, B.; Deiters, A.; Liu, W.R. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem. Biol. 2016, 11, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, Y.X.W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.A.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nat. Cell Biol. 2008, 452, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zwaans, B.M.M.; Eckersdorff, M.; Lombard, D.B. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 2009, 8, 2662–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Dai, Y.; Faller, D.V. Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl. Oncogenom. 2008, 3, 53–65. [Google Scholar]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic Instability and Aging-like Phenotype in the Absence of Mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Xiao, C.; Wang, R.-H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.-I.; Park, O.; Ki, S.H.; et al. Hepatic-Specific Disruption of SIRT6 in Mice Results in Fatty Liver Formation Due to Enhanced Glycolysis and Triglyceride Synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor that Controls Cancer Metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Durso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S.; Rigor, P.; Cervantes, M.; Ceglia, N.; Sebastian, C.; Xiao, C.; Roqueta-Rivera, M.; Deng, C.; Osborne, T.F.; Mostoslavsky, R.; et al. Partitioning Circadian Transcription by SIRT6 Leads to Segregated Control of Cellular Metabolism. Cell 2014, 158, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Wang, R.-H.; Lahusen, T.J.; Park, O.; Bertola, A.; Maruyama, T.; Reynolds, D.; Chen, Q.; Xu, X.; Young, H.A.; et al. Progression of Chronic Liver Inflammation and Fibrosis Driven by Activation of c-JUN Signaling in Sirt6 Mutant Mice. J. Biol. Chem. 2012, 287, 41903–41913. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Yang, H.; Tan, C.; Li, J.; Liu, Z.; Quan, Q.; Kong, S.; Ye, J.; Gao, B.; Fang, D. USP10 Antagonizes c-Myc Transcriptional Activation through SIRT6 Stabilization to Suppress Tumor Formation. Cell Rep. 2013, 5, 1639–1649. [Google Scholar] [CrossRef] [Green Version]
- Bauer, I.; Grozio, A.; Lasigliè, D.; Basile, G.; Sturla, L.; Magnone, M.; Sociali, G.; Soncini, D.; Caffa, I.; Poggi, A.; et al. The NAD+-dependent Histone Deacetylase SIRT6 Promotes Cytokine Production and Migration in Pancreatic Cancer Cells by Regulating Ca2+ Responses. J. Biol. Chem. 2012, 287, 40924–40937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Tian, H.; Xue, L.; Wang, L. SIRT6 abrogation promotes adrenocortical carcinoma through activation of NF-kappaB signaling. Mol. Cell Biochem. 2019, 458, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeoni, F.; Tasselli, L.; Tanaka, S.; Villanova, L.; Hayashi, M.; Kubota, K.; Isono, F.; Garcia, B.A.; Michishita-Kioi, E.; Chua, K.F. Proteomic analysis of the SIRT6 interactome: Novel links to genome maintenance and cellular stress signaling. Sci. Rep. 2013, 3, 3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, A.A.; Siddik, Z.H. Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape. Cell Biochem. Funct. 2015, 33, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Disel, U.; Madison, R.; Abhishek, K.; Chung, J.H.; Trabucco, S.E.; Matos, A.O.; Frampton, G.M.; Albacker, L.A.; Reddy, V.; Karadurmus, N.; et al. The Pan-Cancer Landscape of Coamplification of the Tyrosine Kinases KIT, KDR, and PDGFRA. Oncologist 2019, 25, e39–e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes, S.; Angulo-Ibanez, M.; Tasselli, L.; Carlson, S.M.; Zheng, W.; Li, T.-M.; Chua, K.F. The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability. J. Biol. Chem. 2018, 293, 11242–11250. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.-P.; Uddin, B.; Voit, R.; Schiebel, E. Human phosphatase CDC14A is recruited to the cell leading edge to regulate cell migration and adhesion. Proc. Natl. Acad. Sci. USA 2016, 113, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Mangan, S.; Zaslaver, A.; Alon, U. The Coherent Feedforward Loop Serves as a Sign-sensitive Delay Element in Transcription Networks. J. Mol. Biol. 2003, 334, 197–204. [Google Scholar] [CrossRef]
- Roake, C.M.; Chen, L.; Chakravarthy, A.L.; Ferrell, J.E.; Raffa, G.D.; Artandi, S.E. Disruption of Telomerase RNA Maturation Kinetics Precipitates Disease. Mol. Cell 2019, 74, 688–700.e3. [Google Scholar] [CrossRef]
- Dominy, J.E.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.; Camporez, J.P.; Ruan, H.-B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The Deacetylase Sirt6 Activates the Acetyltransferase GCN5 and Suppresses Hepatic Gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhanati, S.; Ben-Hamo, R.; Kanfi, Y.; Varvak, A.; Glazz, R.; Lerrer, B.; Efroni, S.; Cohen, H.Y. Reciprocal Regulation between SIRT6 and miR-122 Controls Liver Metabolism and Predicts Hepatocarcinoma Prognosis. Cell Rep. 2016, 14, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Tao, R.; Gao, X.; Li, T.; Zhou, X.; Guan, K.-L.; Xiong, Y.; Lei, Q.-Y. Acetylation Stabilizes ATP-Citrate Lyase to Promote Lipid Biosynthesis and Tumor Growth. Mol. Cell 2013, 51, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Tasselli, L.; Li, T.-m.; Chua, K.F. Mammalian SIRT6 Represses Invasive Cancer Cell Phenotypes through ATP Citrate Lyase (ACLY)-Dependent Histone Acetylation. Genes 2021, 12, 1460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091460
Zheng W, Tasselli L, Li T-m, Chua KF. Mammalian SIRT6 Represses Invasive Cancer Cell Phenotypes through ATP Citrate Lyase (ACLY)-Dependent Histone Acetylation. Genes. 2021; 12(9):1460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091460
Chicago/Turabian StyleZheng, Wei, Luisa Tasselli, Tie-mei Li, and Katrin F. Chua. 2021. "Mammalian SIRT6 Represses Invasive Cancer Cell Phenotypes through ATP Citrate Lyase (ACLY)-Dependent Histone Acetylation" Genes 12, no. 9: 1460. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12091460