
Improved DNA Extraction and Illumina Sequencing of DNA Recovered from Aged Rootless Hair Shafts Found in Relics Associated with the Romanov Family

 ,
,
Abstract
:1. Introduction
2. Material
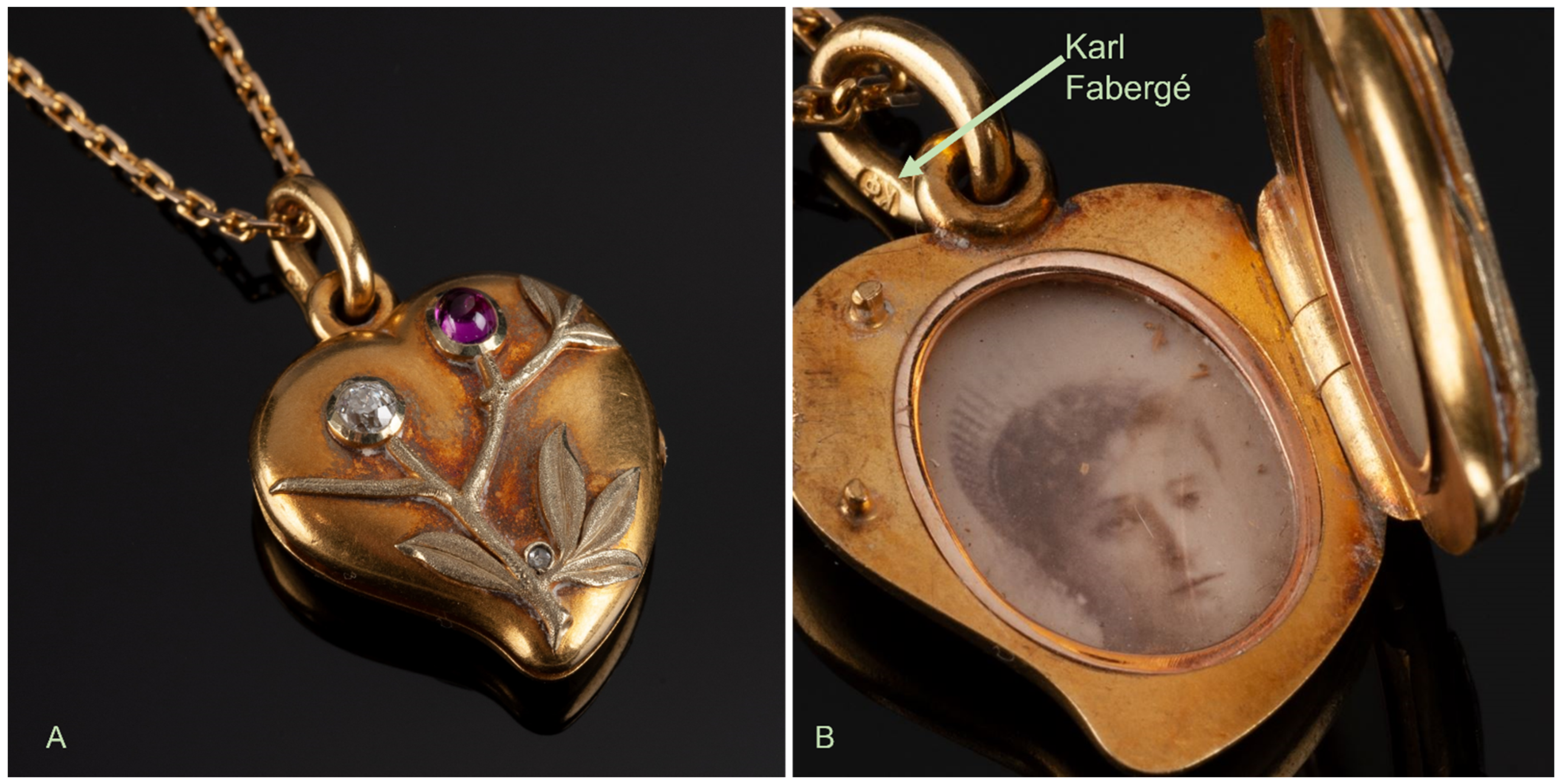
2.1. The Fabergé Locket
2.2. The Picture Frame
2.3. Hair Samples
3. Methods
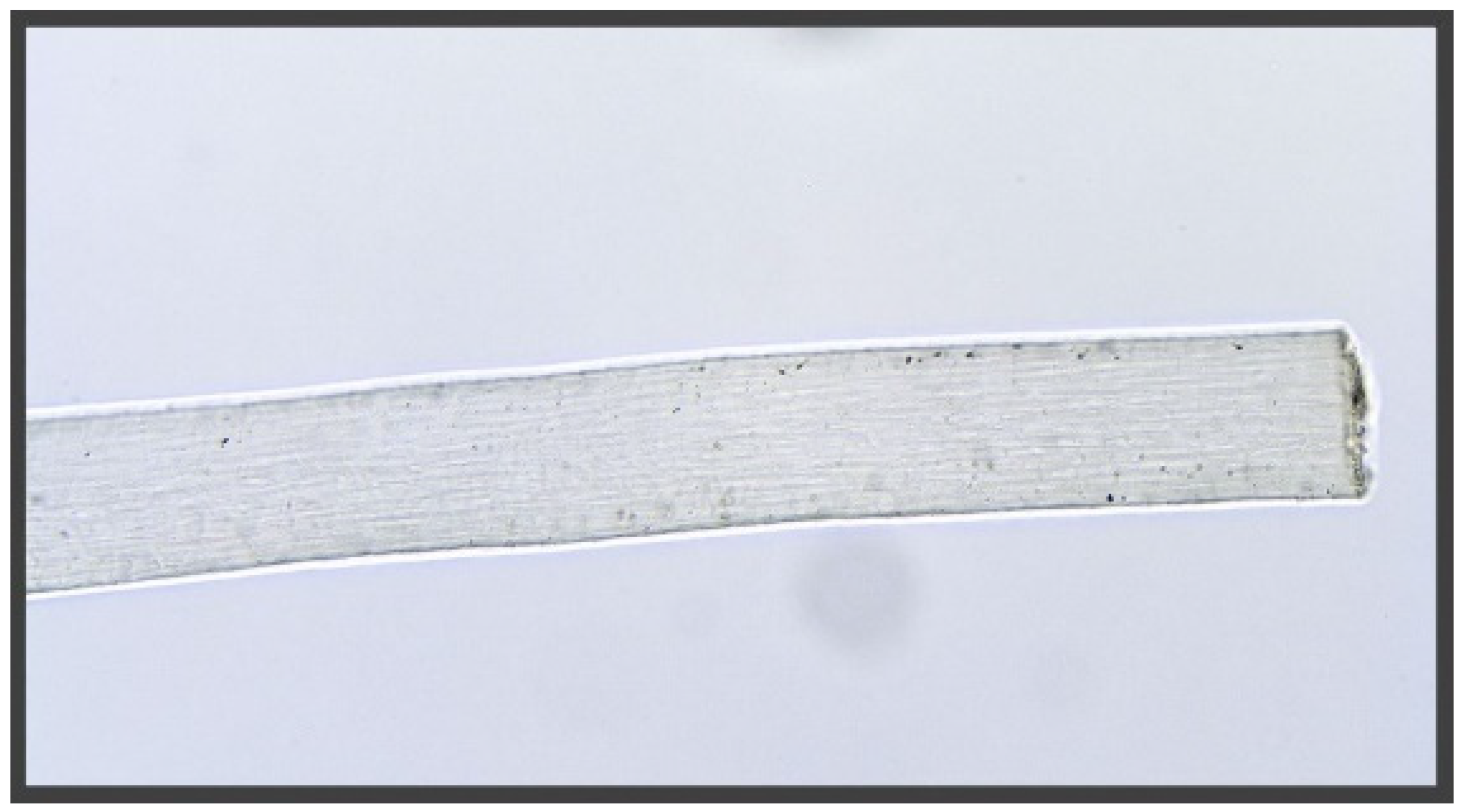
3.1. Microscopic Examination
3.2. DNA Extraction Protocols
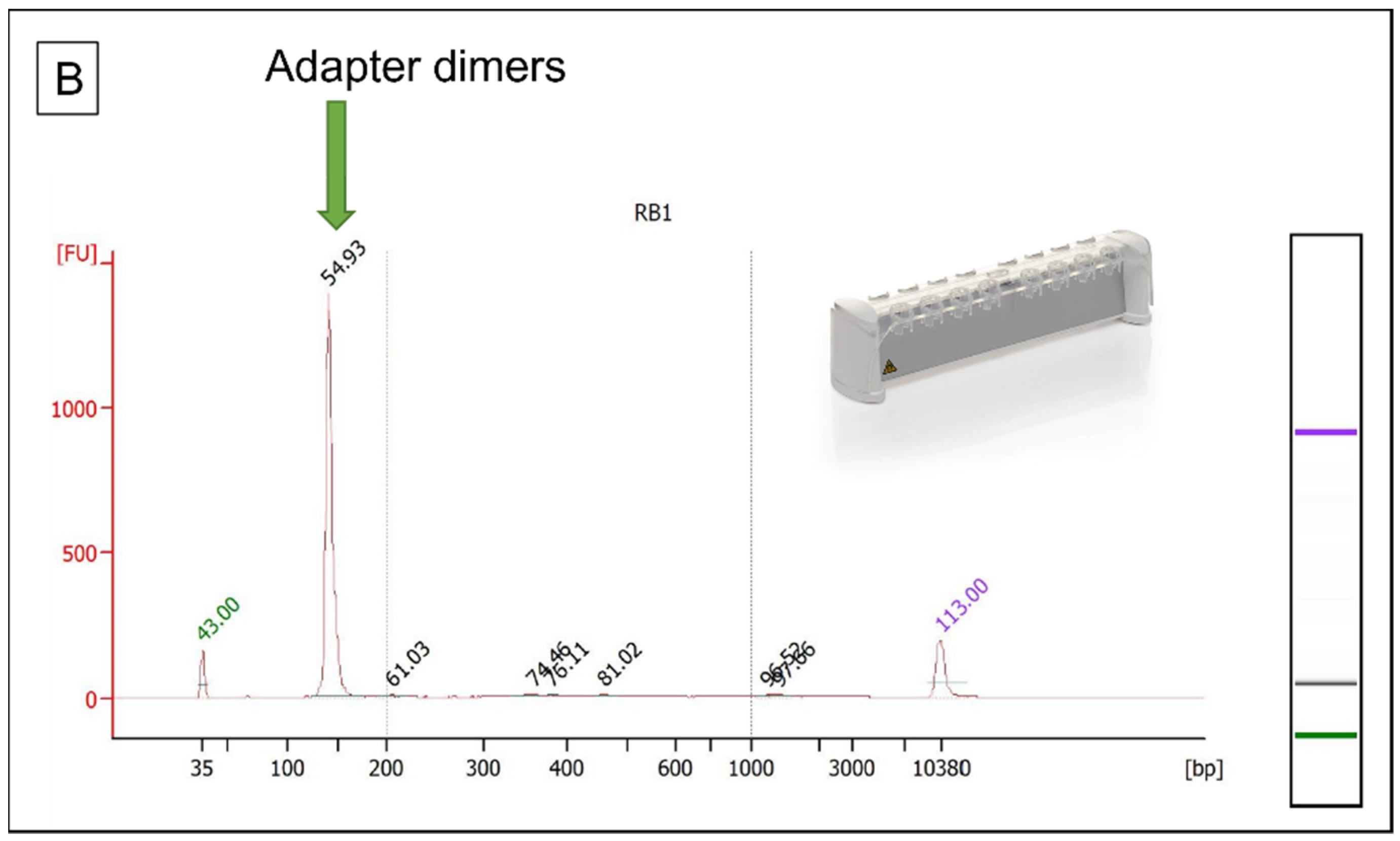
3.3. Library Preparation
3.4. Hybridization Capture
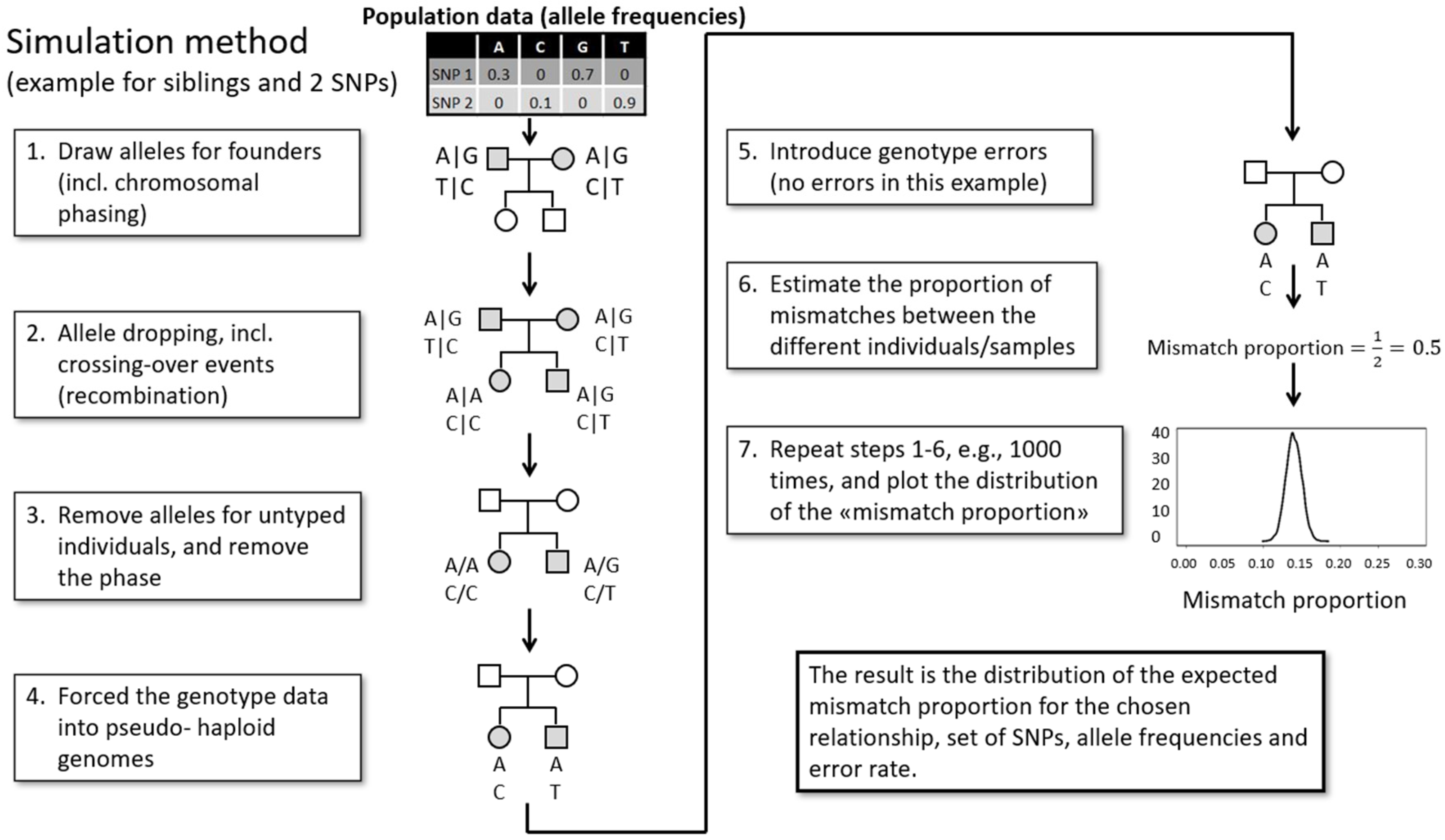
3.5. Data Analyses
4. Results
4.1. Histology Examination
4.2. Illumina Sequencing Results from the Fabergé Locket Hairs
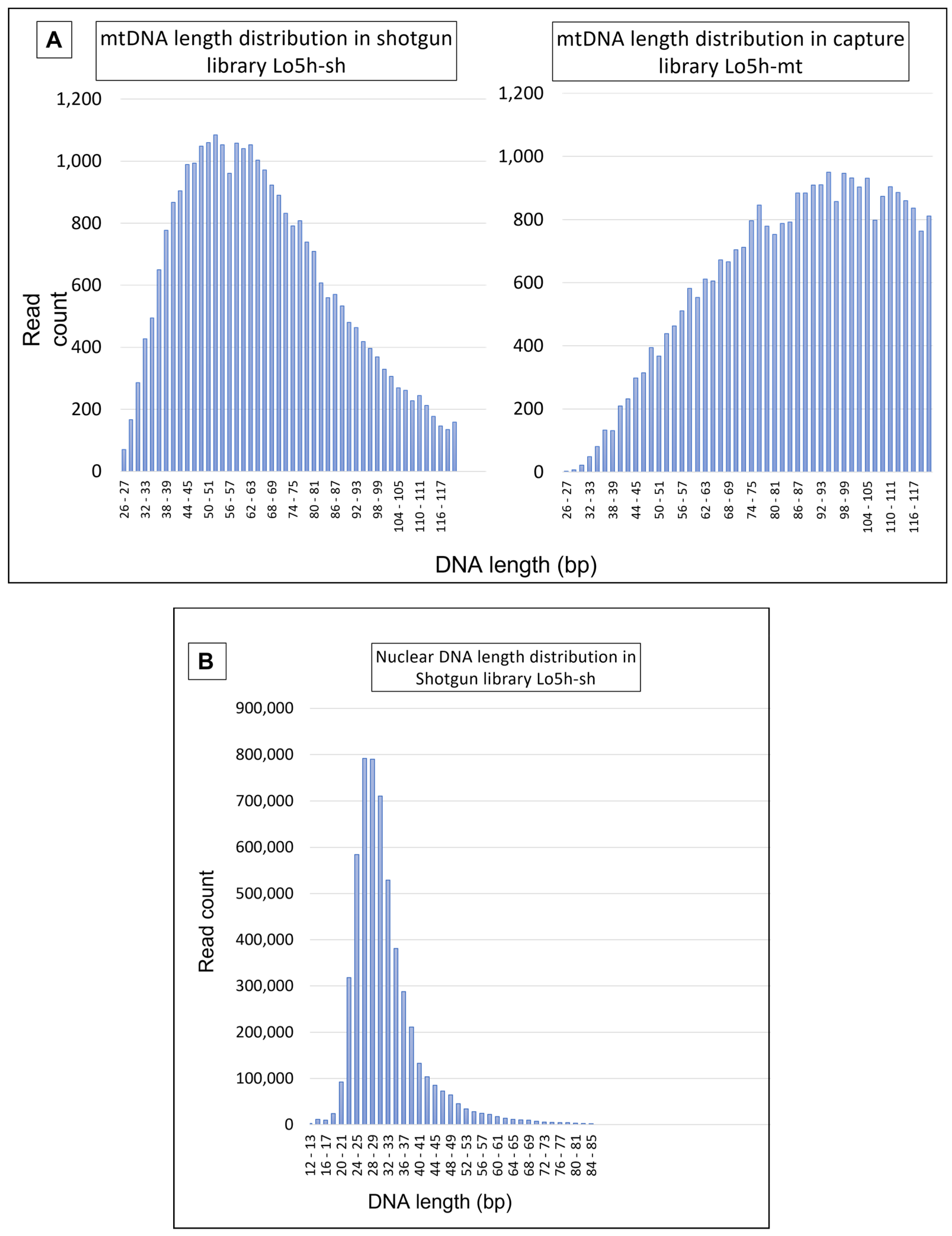
4.2.1. Mitochondrial DNA Results
4.2.2. Nuclear DNA Results
4.2.3. Biological Sex
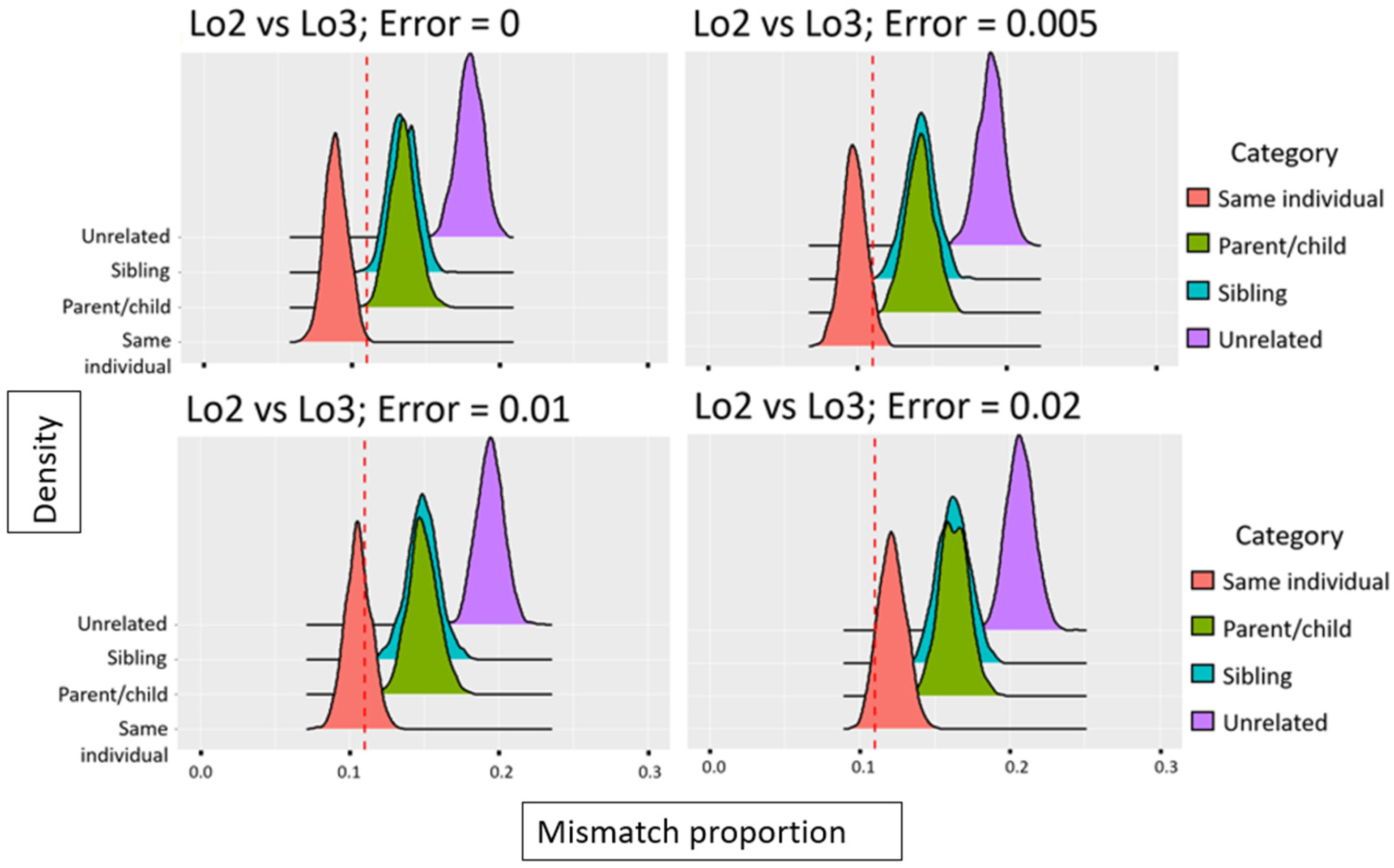
4.2.4. Relatedness Estimates
4.3. Illumina Sequencing Results from the Picture Frame Hairs
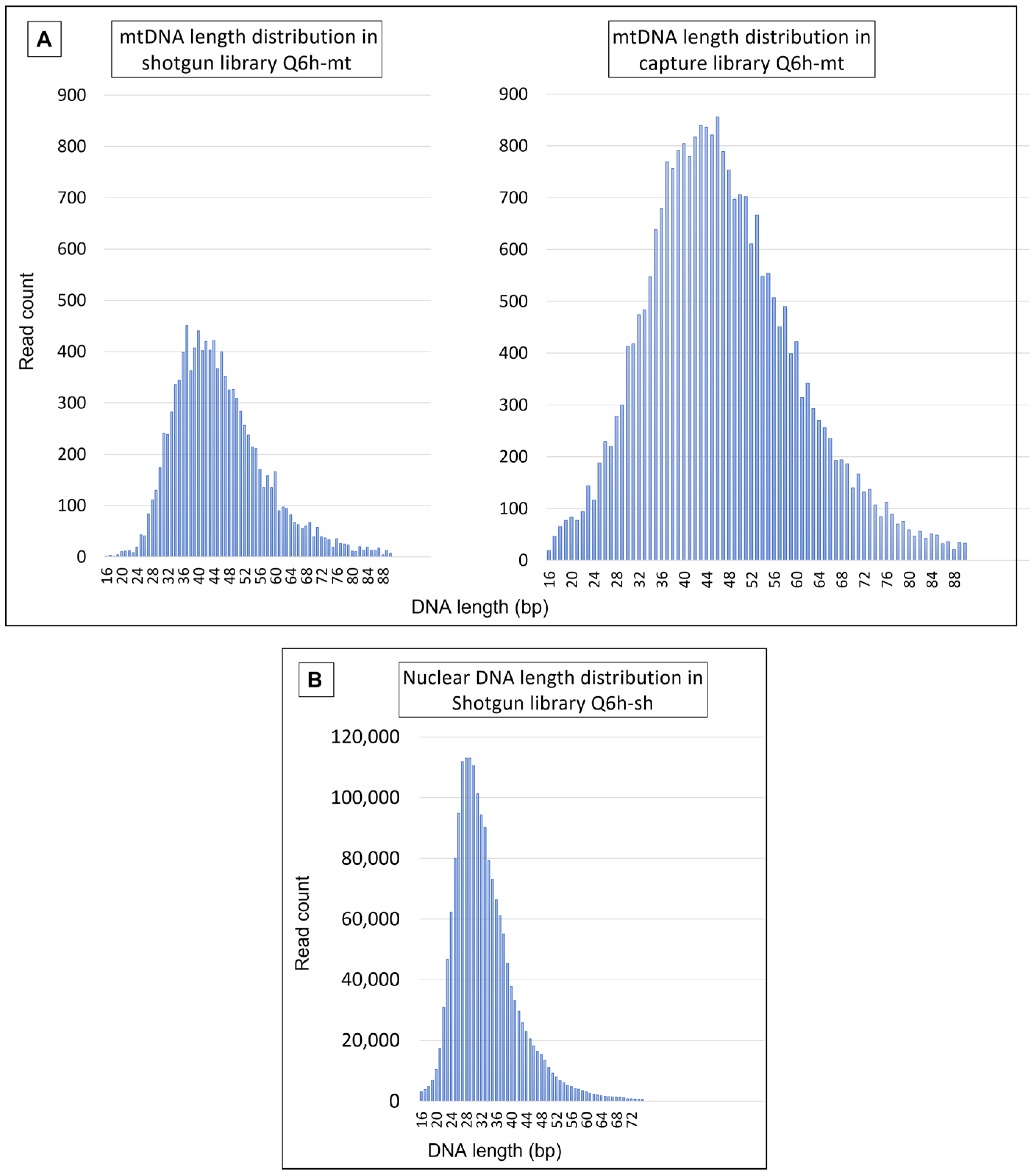
4.3.1. Mitochondrial DNA Results
4.3.2. Nuclear DNA Results
5. Discussion
5.1. DNA Extraction Modifications
5.2. Authenticity of the Items
5.2.1. Fabergé Locket
Historical Evidence of Authenticity
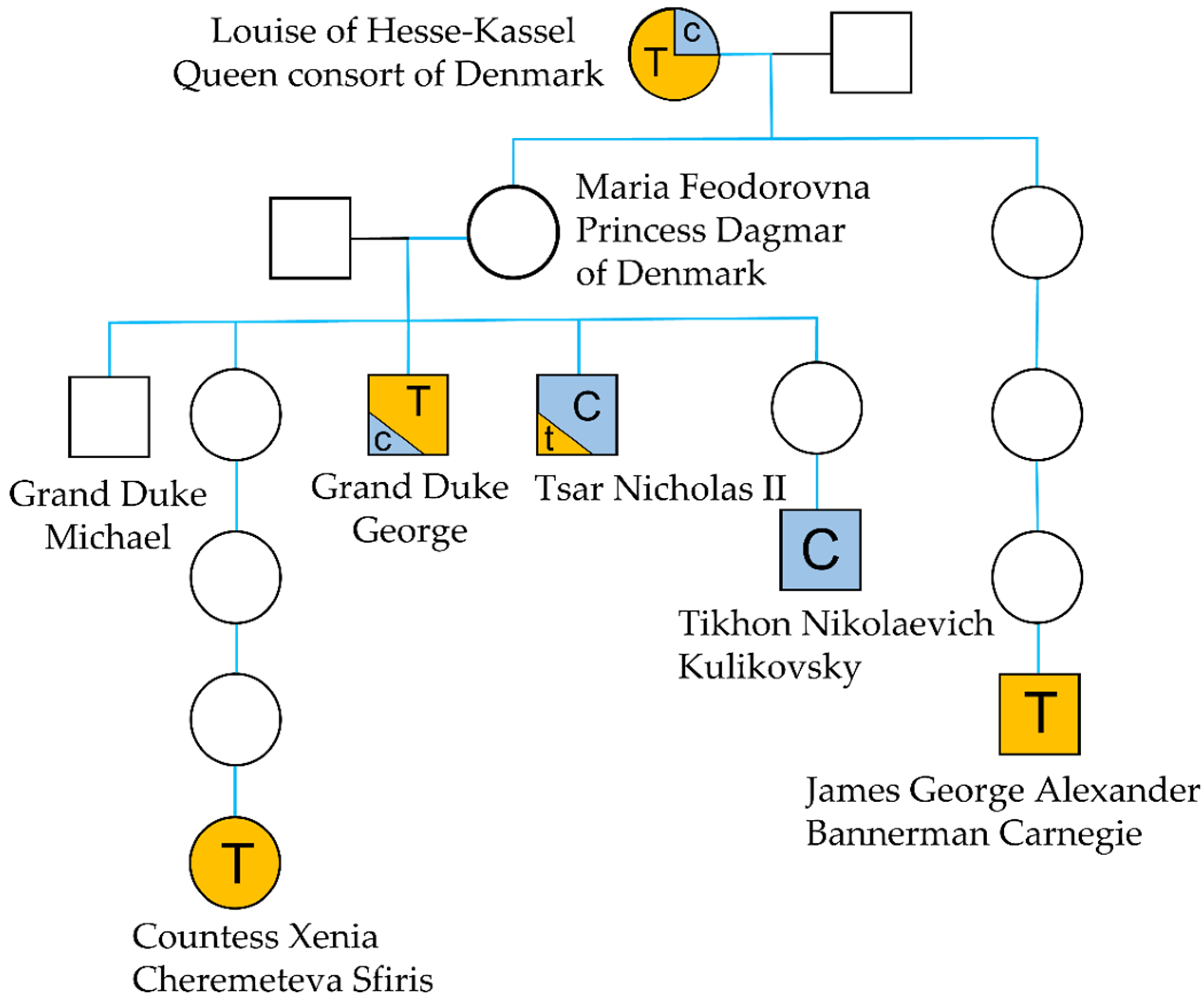
Genetic Evidence
5.2.2. Picture Frame
Historical Evidence
Genetic Evidence and mtDNA Heteroplasmy
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massie, R.K. The Romanovs: The Final Chapter; Random House: New York, NY, USA, 1995. [Google Scholar]
- Byard, R.W. The execution of the Romanov family at Yekatarinberg. Forensic Sci. Med. Pathol. 2020, 16, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Ivanov, P.L.; Kimpton, C.P.; Piercy, R.; Benson, N.; Tully, G.; Evett, I.; Hagelberg, E.; Sullivan, K. Identification of the remains of the Romanov family by DNA analysis. Nat. Genet. 1994, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.L.; Wadhams, M.J.; Roby, R.K.; Holland, M.M.; Weedn, V.W.; Parsons, T.J. Mitochondrial DNA sequence heteroplasmy in the Grand Duke of Russia Georgij Romanov establishes the authenticity of the remains of Tsar Nicholas II. Nat. Genet. 1996, 12, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Coble, M.; Loreille, O.; Wadhams, M.J.; Edson, S.; Maynard, K.; Meyer, C.E.; Niederstätter, H.; Berger, C.; Berger, B.; Falsetti, A.B.; et al. Mystery Solved: The Identification of the Two Missing Romanov Children Using DNA Analysis. PLoS ONE 2009, 4, e4838. [Google Scholar] [CrossRef] [Green Version]
- Rogaev, E.I.; Grigorenko, A.P.; Moliaka, Y.K.; Faskhutdinova, G.; Goltsov, A.; Lahti, A.; Hildebrandt, C.; Kittler, E.L.W.; Morozova, I. Genomic identification in the historical case of the Nicholas II royal family. Proc. Natl. Acad. Sci. USA 2009, 106, 5258–5263. [Google Scholar] [CrossRef] [Green Version]
- Melton, T.; Dimick, G.; Higgins, B.; Lindstrom, L.; Nelson, K. Forensic Mitochondrial DNA Analysis of 691 Casework Hairs. J. Forensic Sci. 2005, 50, 73–80. [Google Scholar] [CrossRef]
- Tobler, R.; Rohrlach, A.; Soubrier, J.; Bover, P.; Llamas, B.; Tuke, J.; Bean, N.; Abdullah-Highfold, A.; Agius, S.; O’Donoghue, A.; et al. Aboriginal mitogenomes reveal 50,000 years of regionalism in Australia. Nature 2017, 544, 180–184. [Google Scholar] [CrossRef]
- Rasmussen, M.; Li, Y.; Lindgreen, S.; Pedersen, J.S.; Albrechtsen, A.; Moltke, I.; Metspalu, M.; Metspalu, E.; Kivisild, T.; Gupta, R.; et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 2010, 463, 757–762. [Google Scholar] [CrossRef]
- Rasmussen, M.; Guo, X.; Wang, Y.; Lohmueller, K.E.; Rasmussen, S.; Albrechtsen, A.; Skotte, L.; Lindgreen, S.; Metspalu, M.; Jombart, T.; et al. An Aboriginal Australian Genome Reveals Separate Human Dispersals into Asia. Science 2011, 334, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Moltke, I.; Korneliussen, T.S.; Seguin-Orlando, A.; Moreno-Mayar, J.V.; LaPointe, E.; Billeck, W.; Willerslev, E. Identifying a living great-grandson of the Lakota Sioux leader Tatanka Iyotake (Sitting Bull). Sci. Adv. 2021, 7, 2013. [Google Scholar] [CrossRef]
- Brandhagen, M.D.; Loreille, O.; Irwin, J.A. Fragmented Nuclear DNA Is the Predominant Genetic Material in Human Hair Shafts. Genes 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef] [PubMed]
- Kavlick, M.F. Development of a triplex mtDNA qPCR assay to assess quantification, degradation, inhibition, and amplification target copy numbers. Mitochondrion 2019, 46, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard. Available online: http://broadinstitute.github.io/picard (accessed on 15 September 2021).
- Mittnik, A.; Wang, C.C.; Svoboda, J.; Krause, J. A Molecular Approach to the Sexing of the Triple Burial at the Upper Paleolithic Site of Dolni Vestonice. PLoS ONE 2016, 11, e0163019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillmar, A.; Sjölund, P.; Lundqvist, B.; Klippmark, T.; Älgenäs, C.; Green, H. Whole-genome sequencing of human remains to enable genealogy DNA database searches–A case report. Forensic Sci. Int. Genet. 2020, 46, 102233. [Google Scholar] [CrossRef] [PubMed]
- Tillmar, A.; Fagerholm, S.A.; Staaf, J.; Sjölund, P.; Ansell, R. Getting the conclusive lead with investigative genetic genealogy –A successful case study of a 16 year old double murder in Sweden. Forensic Sci. Int. Genet. 2021, 53, 102525. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Nexus. Available online: https://www.snp-nexus.org (accessed on 15 December 2020).
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Ullah, A.Z.D.; Lemoine, N.R.; Chelala, C. SNPnexus: A web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; Souza, M.D.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, M.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enk, J.M.; Devault, A.M.; Kuch, M.; Murgha, Y.E.; Rouillard, J.M.; Poinar, H.N. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 2014, 31, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Arcos, M.C.; Sandoval-Velasco, M.; Schroeder, H.; Carpenter, M.; Malaspinas, A.-S.; Wales, N.; Peñaloza, F.; Bustamante, C.D.; Gilbert, M.T.P. Comparative performance of two whole-genome capture methodologies on ancient DNA Illumina libraries. Methods Ecol. Evol. 2015, 6, 725–734. [Google Scholar] [CrossRef]
- Krasnenko, A.; Tsukanov, K.; Stetsenko, I.; Klimchuk, O.; Plotnikov, N.; Surkova, E.; Ilinsky, V. Effect of DNA insert length on whole-exome sequencing enrichment efficiency: An observational study. Adv. Genom. Genet. 2018, 8, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Arsuaga, J.L.; de Filippo, C.; Nagel, S.; Aximu-Petri, A.; Nickel, B.; Martinez, I.; Gracia, A.; de Castro, J.M.B.; Carbonell, E.; et al. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 2016, 531, 504–507. [Google Scholar] [CrossRef]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef]
- Rathbun, M.M.; McElhoe, J.A.; Parson, W.; Holland, M.M. Considering DNA damage when interpreting mtDNA heteroplasmy in deep sequencing data. Forensic Sci. Int. Genet. 2017, 26, 1–11. [Google Scholar] [CrossRef]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Delser, P.M.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S.; et al. Identification of the remains of King Richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef]
- Parson, W.; Loreille, O.; Niederstätter, H.; Parsons, T.J.; Scheithauer, R. DNA Investigations of the putative Mozart cranium. In Proceedings of the 17th International Symposium on Human Identification, Nashville, TN, USA, 10 October 2006. [Google Scholar]
- Bainbridge, C.; Peter, C.F. Goldsmith and Jeweller to the Russian Imperial Court and the Principal Crowned Heads of Europe; Batsford: London, UK, 1 January 1949. [Google Scholar]
- Available online: https://www.christies.com/features/Faberge-15-things-a-collector-needs-to-know-8353-1.aspx (accessed on 15 September 2021).
- van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, e386–e394. [Google Scholar] [CrossRef]
- FamilyTree DNA. Available online: https://www.familytreedna.com/public/mt-dna-haplotree/H (accessed on 20 December 2021).
- EMPOP. Available online: https://www.empop.org (accessed on 20 December 2021).
- MITOMASTER. Available online: https://www.mitomap.org/foswiki/bin/view/MITOMASTER/WebHome (accessed on 20 December 2021).
- Bachmakov, N.; Expert in Antique Restoration in New York, New York, NY, USA. Personal communication, 2020.
- Rogaev, I.E.; Ovchinnikov, I.V.; Dzhorzh-Khislop, P.; Rogaeva, A.E. Comparison of mitochondrial DNA sequences of T.N. Kulikovski? -Romanov, the nephew of Tsar Nikola? II Romanov, with DNA from the putative remains of the Tsar. Генетика 1996, 32, 1690–1692. [Google Scholar]
- Comas, D.; Pääbo, S.; Bertranpetit, J. Heteroplasmy in the control region of human mitochondrial DNA. Genome Res. 1995, 5, 89–90. [Google Scholar] [CrossRef] [Green Version]
- Bendall, K.E.; Macaulay, V.A.; Sykes, B.C. Variable Levels of a Heteroplasmic Point Mutation in Individual Hair Roots. Am. J. Hum. Genet. 1997, 61, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Salas, A.; Albarrán, C.; Arroyo-Pardo, E.; Castro, A.; Crespillo, M.; di Lonardo, A.M.; Lareu, M.V.; Cubría, C.L.; Soto, M.L.; et al. Results of the 1999–2000 collaborative exercise and proficiency testing program on mitochondrial DNA of the GEP-ISFG: An inter-laboratory study of the observed variability in the heteroplasmy level of hair from the same donor. Forensic Sci. Int. 2002, 125, 1–7. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Sato, H.; Kasai, K. Mitochondrial DNA heteroplasmy among hairs from single individuals. J. Forensic Sci. 2004, 49, 986–991. [Google Scholar] [CrossRef] [Green Version]
- Tully, G.; Barritt, S.; Bender, K.; Brignon, E.; Capelli, C.; Dimo-Simonin, N.; Eichmann, C.; Ernst, C.; Lambert, C.; Lareu, M.; et al. Results of a collaborative study of the EDNAP group regarding mitochondrial DNA heteroplasmy and segregation in hair shafts. Forensic Sci. Int. 2004, 140, 1–11. [Google Scholar] [CrossRef]
- Van Der Gaag, K.J.; Desmyter, S.; Smit, S.; Prieto, L.; Sijen, T. Reducing the Number of Mismatches between Hairs and Buccal References When Analysing mtDNA Heteroplasmic Variation by Massively Parallel Sequencing. Genes 2020, 11, 1355. [Google Scholar] [CrossRef]
- Desmyter, S.; Bodner, M.; Huber, G.; Dognaux, S.; Berger, C.; Noël, F.; Parson, W. Hairy matters: MtDNA quantity and sequence variation along and among human head hairs. Forensic Sci. Int. Genet. 2016, 25, 1–9. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated Item | Hair Shafts Tested | Experiment | Purification Protocol |
|---|---|---|---|
| Single hair of ~5 cm (light brown). Lo1a | Microscopic examination | NA | |
| Single hair of ~2.5 cm (light brown). Lo1b | Microscopic examination | ||
| Single hair of ~3.8 cm (light brown). Lo1c | Microscopic examination | ||
| Fabergé Locket | 5 hairs for a total of ~16 cm. Lo5h | DNA | MinElute column |
| Single hair of 4 cm (light brown). Lo2 | DNA | Magnetic silica beads | |
| Single hair of 3.2 cm (light brown). Lo3 | DNA | Magnetic silica beads | |
| Single hair of 6 cm (white). Q3 | Microscopic examination | NA | |
| Frame | 6 hairs for a total of ~27 cm. Q6h | DNA | MinElute column |
| Single hair of 5.5 cm (white). Q1 | DNA | Magnetic silica beads | |
| Single hair of 7 cm (brown). Q2 | DNA | Magnetic silica beads |
| Shotgun Libraries | Platforms Used | Number of Mapped Reads | Percentage of Mapped Reads | Number of Unique Reads |
| Lo5h-sh (5 hairs) | MiSeq FGxTM | 6,229,985 | 48% | 5,500,712 |
| Lo2-sh (1 hair) | NextSeq 500 | 199,948,284 | 66.54% | 38,036,020 |
| Lo3-sh (1 hair) | NextSeq 500 | 151,630,120 | 78.91% | 33,388,550 |
| mtDNA-Enriched Library | Platforms Used | Number of Mapped Reads | Percentage of Mapped Reads | Number of Unique mtDNA Reads |
| Lo5h-mt (5 hairs) | NextSeq 500 | 21,109,044 | 97.50% | 35,188 |
| Library | Mapped mtDNA Reads Average Length (bp) |
|---|---|
| Lo2-sh | 67.82/67.89 |
| Lo3-sh | 84.99/85.17 |
| Lo5h-sh | 66.04/69.16 |
| Lo5h-mt | 93.46 |
| Library | Mapped Nuclear DNA Reads Average Length (bp) |
|---|---|
| Lo2-sh | 33.05 |
| Lo3-sh | 33.13 |
| Lo5h-sh | 31.96 |
| Library | Rx | SD | CI | CI Low | CI Sup |
|---|---|---|---|---|---|
| Lo2-sh | 0.965 | 0.231 | 0.097 | 0.869 | 1.062 |
| Lo3-sh | 0.935 | 0.229 | 0.097 | 0.838 | 1.031 |
| Shotgun Libraries | Platforms Used | Number of Mapped Raw Reads | Percentage of Raw Mapped Reads | Number of Unique Human Reads |
| Q6h-sh (6 hairs) | MiSeq FGx | 2,072,278 | 21.17% | 1,817,968 |
| Q1-sh (1 hair) | MiSeq FGx | 267,392 | 70.12% | 239,692 |
| Q2-sh (1 hair) | NextSeq 500 | 278,531,240 | 89% | 56,390,706 |
| mtDNA-Enriched Libraries | Platforms Used | Number of Mapped Raw Reads | Percentage of Raw Mapped mtDNA Reads | Number of Unique mtDNA Reads |
| Q6h-mt (6 hairs) | MiSeq FGx | 7,059,775 | 90.68% | 26,038 |
| Q1-mt (1 hair) | MiSeq FGx | 3,586,989 | 96.69% | 14,514 |
| Q2-mt (1 hair) | MiSeq FGx | 8,104,414 | 88% | 24,636 |
| Sequencing Platform | Library | Number of Unique mtDNA Reads | Average mtDNA Size (bp). hg19/rCRS |
|---|---|---|---|
| MiSeq FGxTM | Q6h-sh | 11,052 | 45.78/45.56 |
| MiSeq FGxTM | Q6h-mt | 26,038 | 47.26 |
| MiSeq FGxTM | Q1-sh | 1281 | NA |
| MiSeq FGxTM | Q1-mt | 14,514 | 38.80 |
| NextSeq 500 | Q2-sh | 15,267 | 42.32/43.9 |
| MiSeq FGxTM | Q2-mt | 24,636 | 45.33 |
| Library | Rx | SD | CI | CI Low | CI Sup |
|---|---|---|---|---|---|
| Q1-sh | 0.903 | 0.201 | 0.084 | 0.819 | 0.987 |
| Q2-sh | 0.964 | 0.228 | 0.095 | 0.868 | 1.059 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loreille, O.; Tillmar, A.; Brandhagen, M.D.; Otterstatter, L.; Irwin, J.A. Improved DNA Extraction and Illumina Sequencing of DNA Recovered from Aged Rootless Hair Shafts Found in Relics Associated with the Romanov Family. Genes 2022, 13, 202. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020202
Loreille O, Tillmar A, Brandhagen MD, Otterstatter L, Irwin JA. Improved DNA Extraction and Illumina Sequencing of DNA Recovered from Aged Rootless Hair Shafts Found in Relics Associated with the Romanov Family. Genes. 2022; 13(2):202. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020202
Chicago/Turabian StyleLoreille, Odile, Andreas Tillmar, Michael D. Brandhagen, Linda Otterstatter, and Jodi A. Irwin. 2022. "Improved DNA Extraction and Illumina Sequencing of DNA Recovered from Aged Rootless Hair Shafts Found in Relics Associated with the Romanov Family" Genes 13, no. 2: 202. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020202