
Horizontal Transposon Transfer and Its Implications for the Ancestral Ecology of Hydrophiine Snakes

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Library Construction and Sequencing of Terrestrial Elapids
2.2. Genome Assembly of Terrestrial Elapids
2.3. Genome Completeness and Summary Statistics
2.4. Ab Initio TE Annotation of the Elapid Genomes
2.5. TE Annotation of the Elapid Genomes
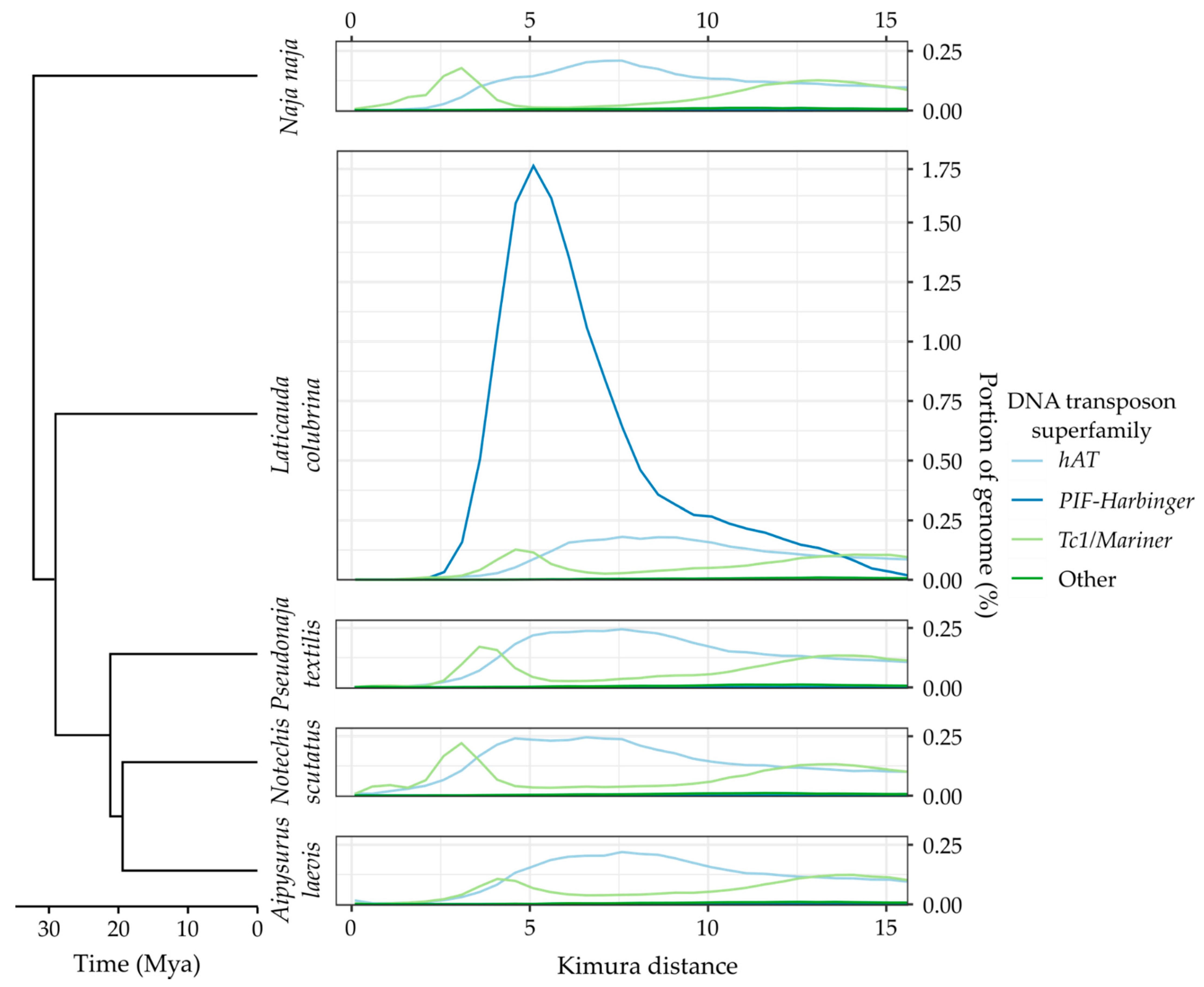
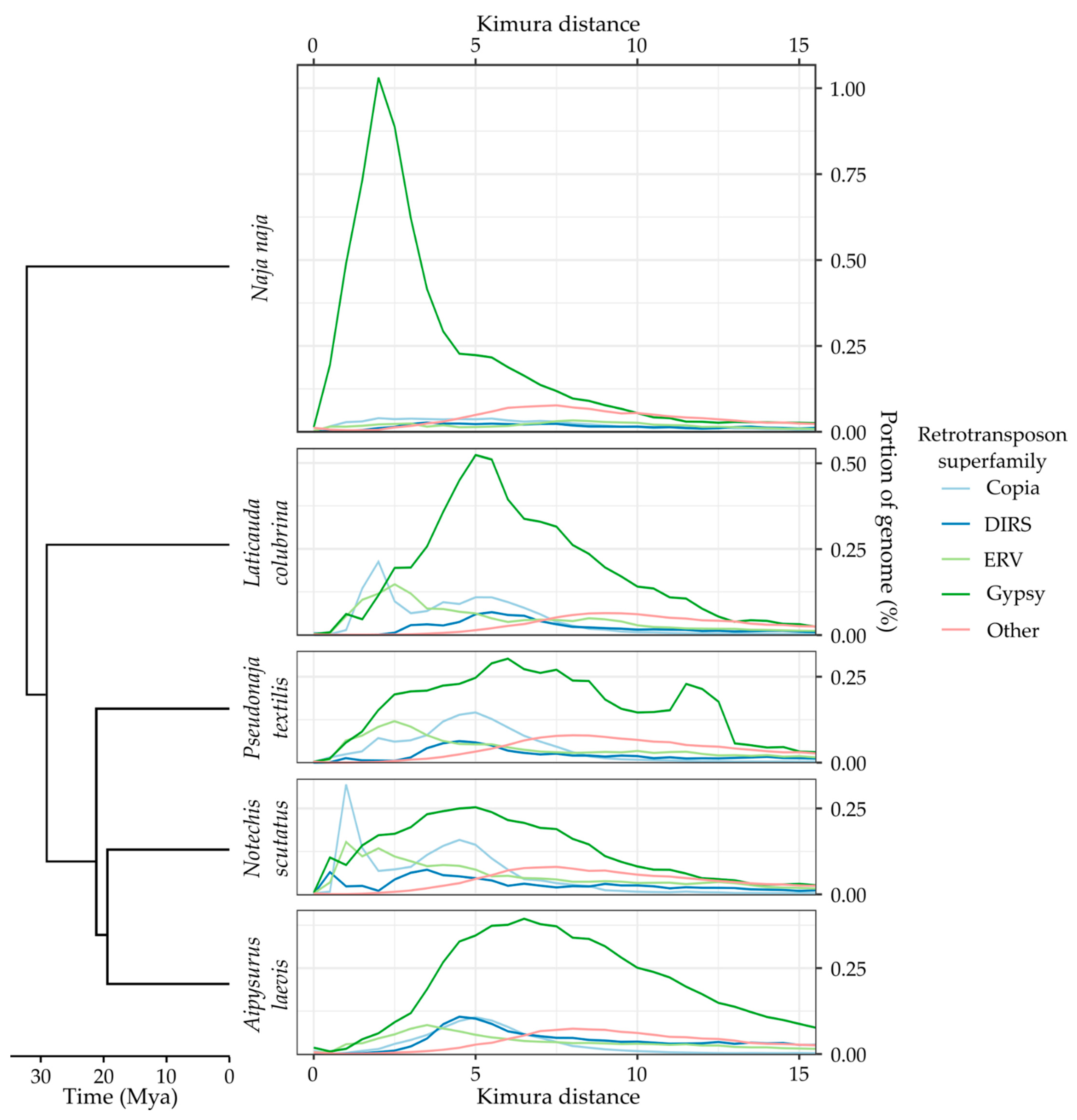
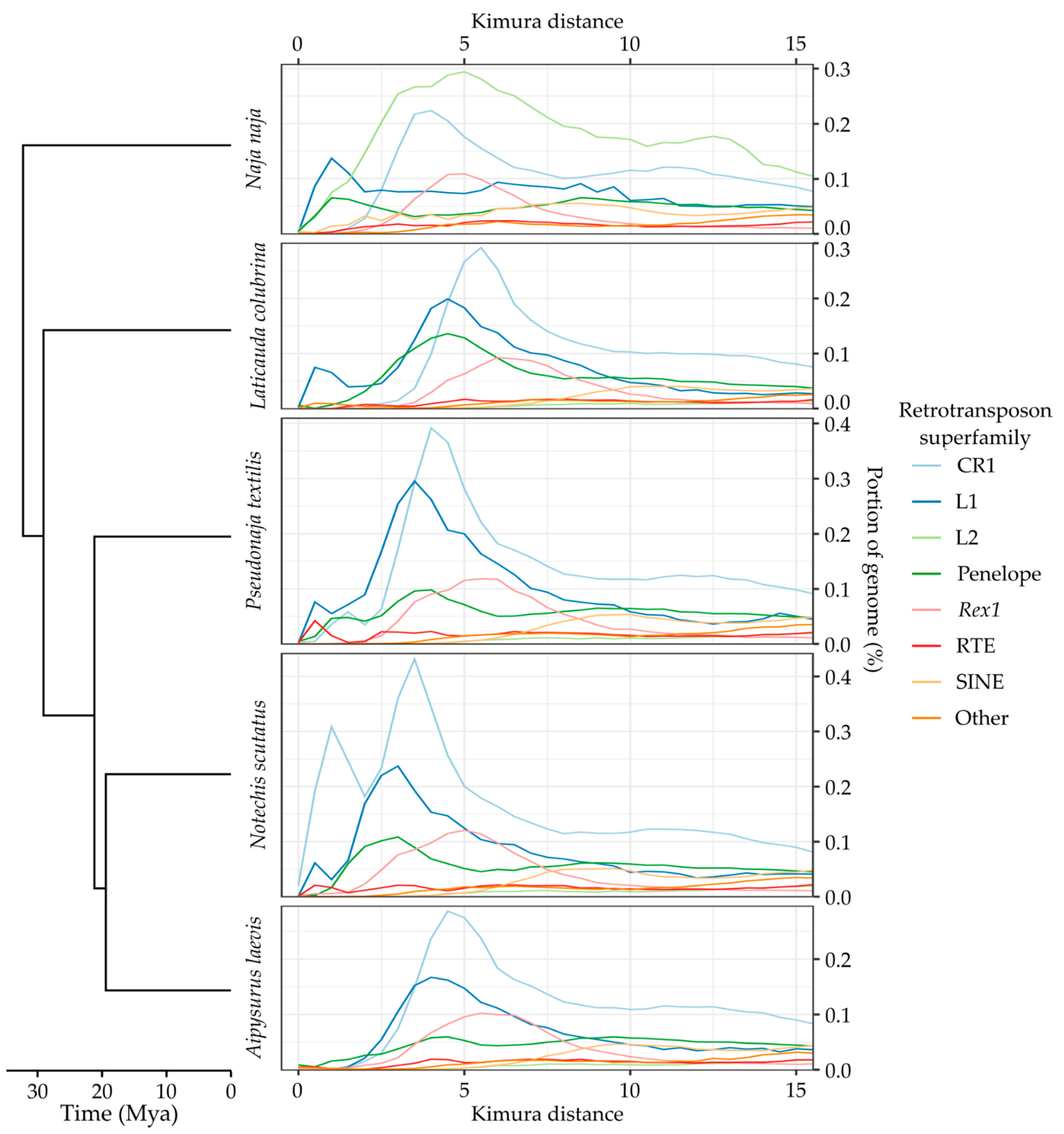
2.6. Estimating Ancestral TE Similarity
2.7. Identifying Recent TE Expansions
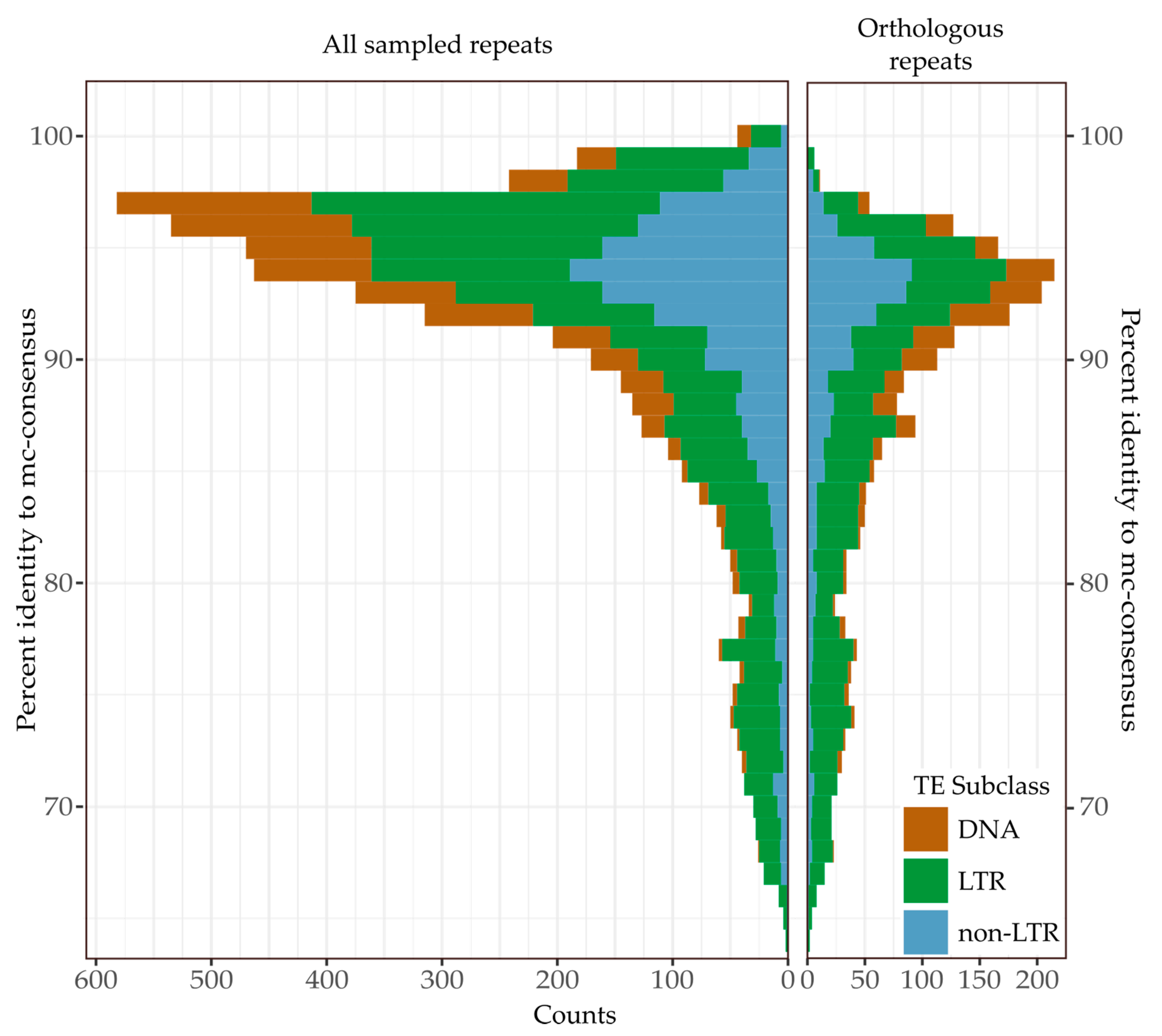
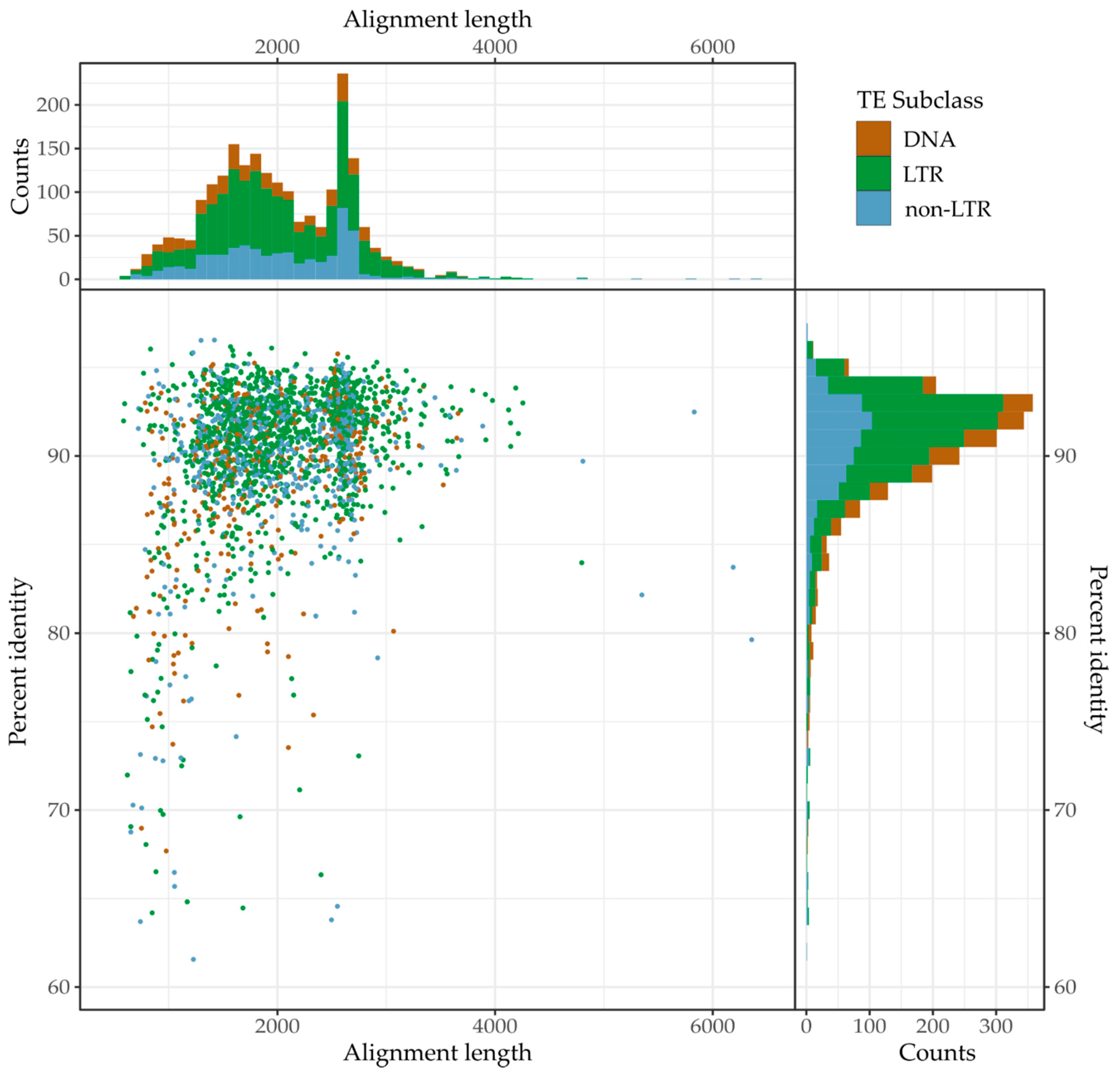
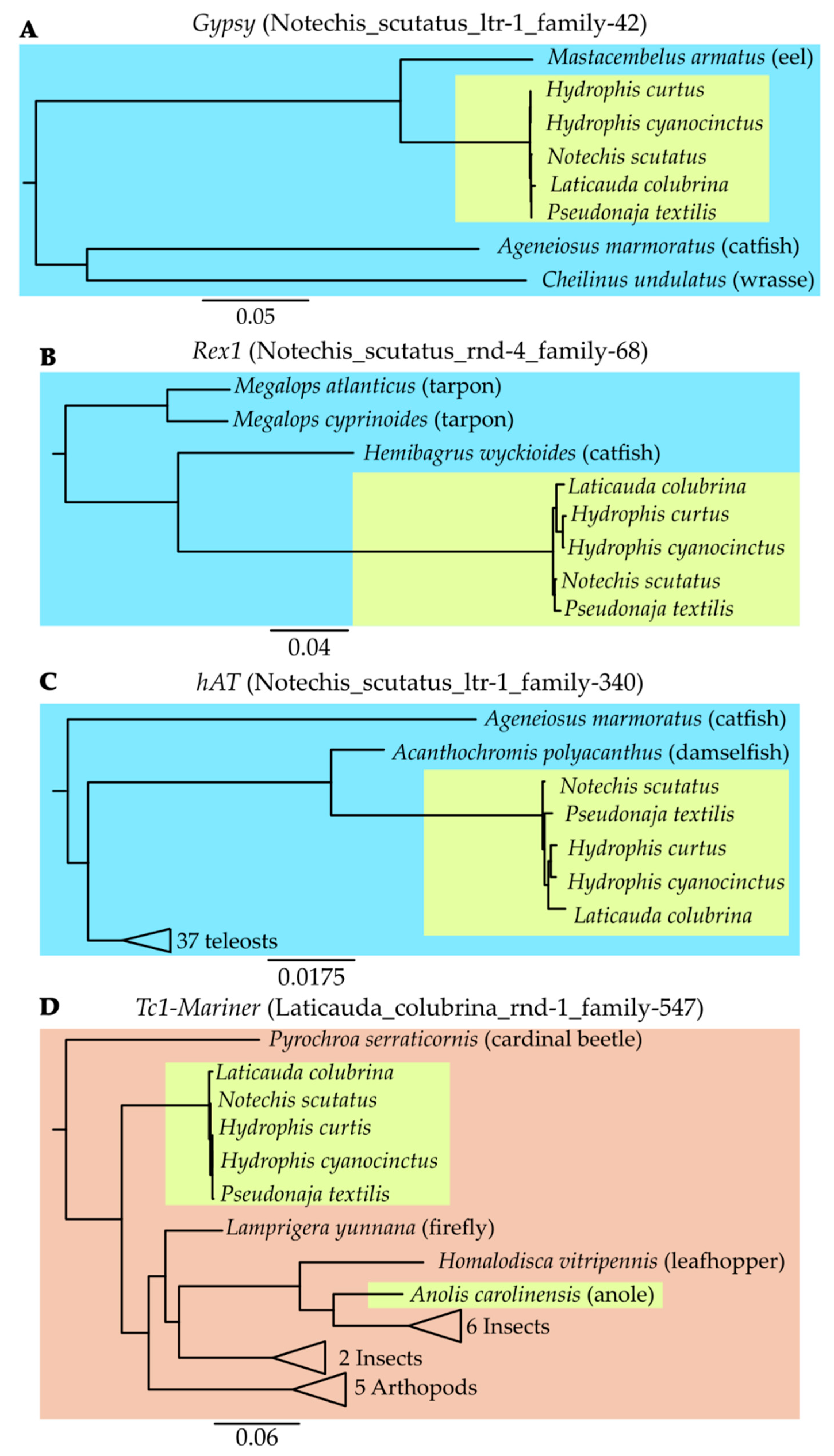
2.8. Continued Expansion or Horizontal Transfer
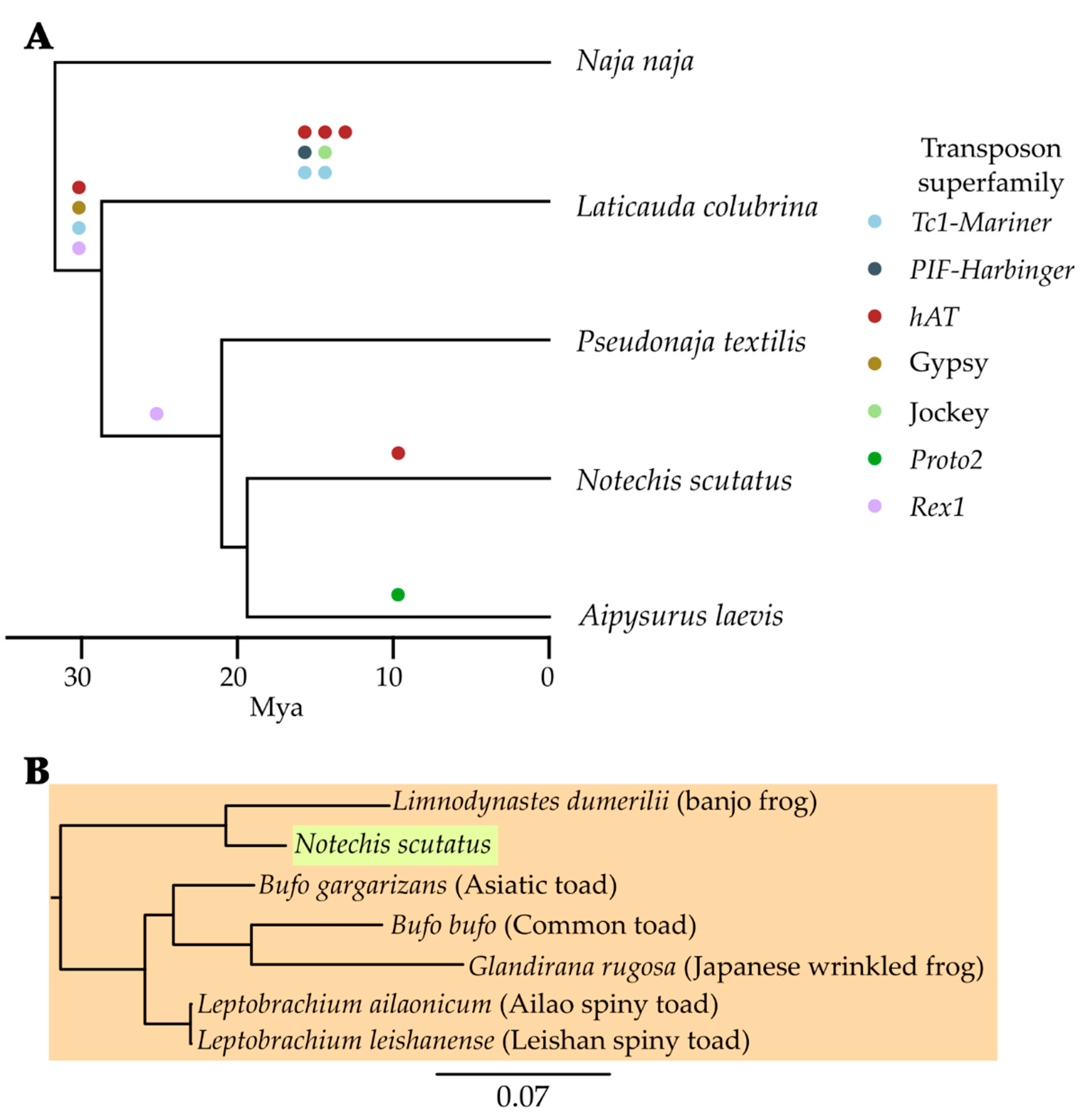
2.9. Identified Potential Sources of Horizontal Transfer Candidates
3. Results and Discussion
3.1. Draft Terrestrial Elapid Genomes Are Fragmented but Show High Completeness
3.2. Genome Quality Affects Repeat Annotation
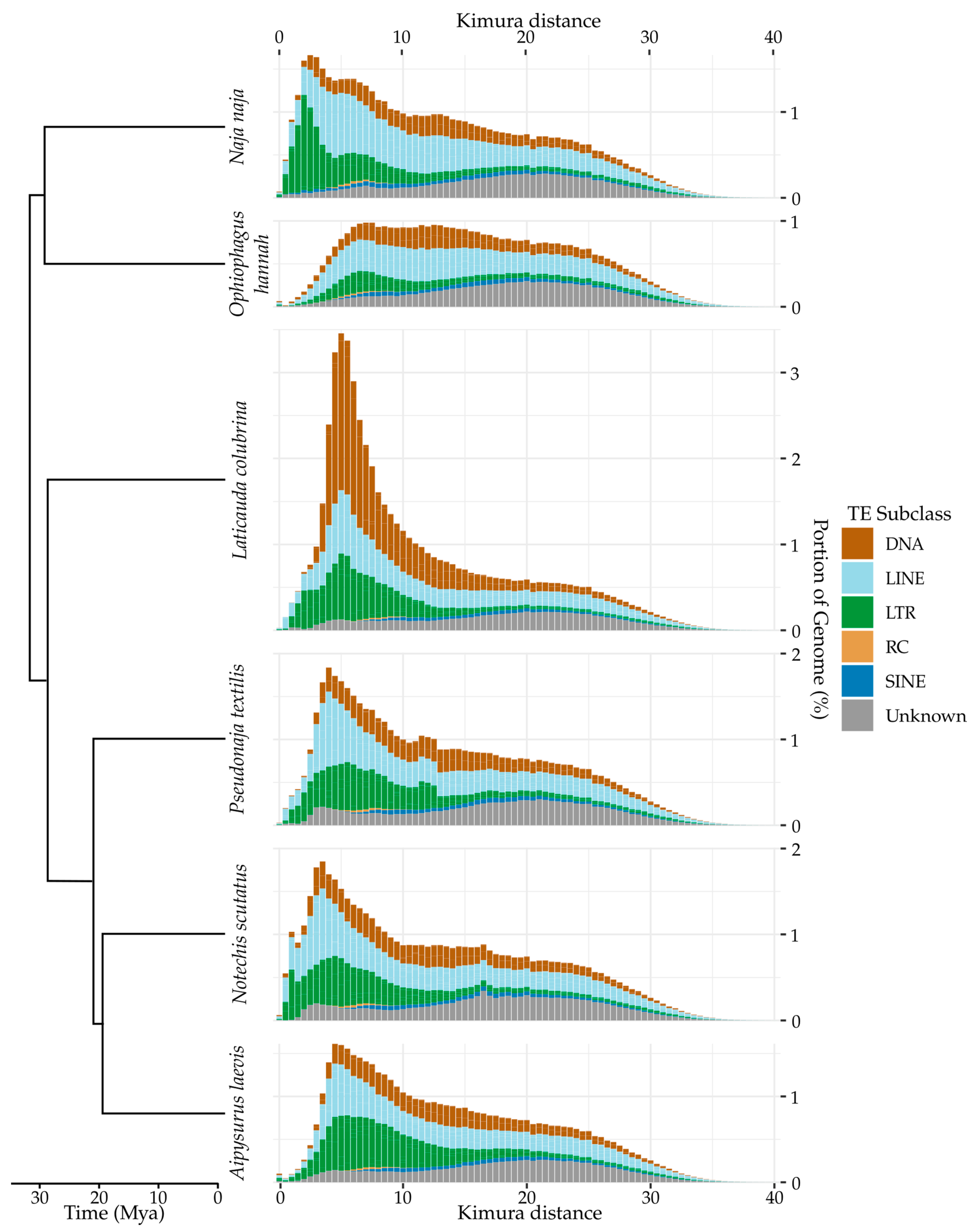
3.3. Recent Insertions vs. Ancestral Insertions
3.4. Recent Expansion of Specific Superfamilies
3.5. Continued Expansion or Horizontal Transfer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. TE Divergence Profiles



References
- Sanders, K.L.; Lee, M.S.Y.; Leys, R.; Foster, R.; Keogh, J.S. Molecular Phylogeny and Divergence Dates for Australasian Elapids and Sea Snakes (hydrophiinae): Evidence from Seven Genes for Rapid Evolutionary Radiations. J. Evol. Biol. 2008, 21, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Mirtschin, P.; Rasmussen, A.; Weinstein, S. Australia’s Dangerous Snakes: Identification, Biology and Envenoming; CSIRO Publishing: Clayton South, VIC, Australia, 2017. [Google Scholar]
- Glodek, G.S.; Voris, H.K. Marine Snake Diets: Prey Composition, Diversity and Overlap. Copeia 1982, 1982, 661–666. [Google Scholar] [CrossRef]
- Voris, H.K.; Voris, H.H. Feeding Strategies in Marine Snakes: An Analysis of Evolutionary, Morphological, Behavioral and Ecological Relationships. Am. Zool. 1983, 23, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Brischoux, F.; Bonnet, X. Life History of Sea Kraits in New Caledonia. Zool. Neocaledonica 2009, 7, 37–51. [Google Scholar]
- Galbraith, J.D.; Ludington, A.J.; Suh, A.; Sanders, K.L.; Adelson, D.L. New Environment, New Invaders—Repeated Horizontal Transfer of LINEs to Sea Snakes. Genome Biol. Evol. 2020, 12, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, J.D.; Ludington, A.J.; Sanders, K.L.; Suh, A.; Adelson, D.L. Horizontal Transfer and Subsequent Explosive Expansion of a DNA Transposon in Sea Kraits (Laticauda). Biol. Lett. 2021, 17, 20210342. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.N. Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [Green Version]
- Volff, J.-N. Turning Junk into Gold: Domestication of Transposable Elements and the Creation of New Genes in Eukaryotes. Bioessays 2006, 28, 913–922. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [Green Version]
- Cornelis, G.; Funk, M.; Vernochet, C.; Leal, F.; Tarazona, O.A.; Meurice, G.; Heidmann, O.; Dupressoir, A.; Miralles, A.; Ramirez-Pinilla, M.P.; et al. An Endogenous Retroviral Envelope Syncytin and Its Cognate Receptor Identified in the Viviparous Placental Mabuya Lizard. Proc. Natl. Acad. Sci. USA 2017, 114, E10991–E11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, N.; Chijiwa, T.; Matsubara, K.; Oda-Ueda, N.; Hattori, S.; Matsuda, Y.; Ohno, M. Unique Structural Characteristics and Evolution of a Cluster of Venom Phospholipase A2 Isozyme Genes of Protobothrops Flavoviridis Snake. Gene 2010, 461, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A Unified Classification System for Eukaryotic Transposable Elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Kohany, O.; Jurka, M.V. Repetitive Sequences in Complex Genomes: Structure and Evolution. Annu. Rev. Genom. Hum. Genet. 2007, 8, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the Evolution of Eukaryotic Genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [Green Version]
- Kapitonov, V.V.; Tempel, S.; Jurka, J. Simple and Fast Classification of Non-LTR Retrotransposons Based on Phylogeny of Their RT Domain Protein Sequences. Gene 2009, 448, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of Genome Size Evolution in Birds and Mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [Green Version]
- Suh, A.; Paus, M.; Kiefmann, M.; Churakov, G.; Franke, F.A.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Mesozoic Retroposons Reveal Parrots as the Closest Living Relatives of Passerine Birds. Nat. Commun. 2011, 2, 443. [Google Scholar] [CrossRef]
- Ivancevic, A.M.; Kortschak, R.D.; Bertozzi, T.; Adelson, D.L. Horizontal Transfer of BovB and L1 Retrotransposons in Eukaryotes. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A.; et al. Squamate Reptiles Challenge Paradigms of Genomic Repeat Element Evolution Set by Birds and Mammals. Nat. Commun. 2018, 9, 2774. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; Hall, K.T.; Guibotsy Mboulas, M.L.; Gu, W.; de Koning, A.P.J.; Fox, S.E.; Poole, A.W.; Vemulapalli, V.; Daza, J.M.; Mockler, T.; et al. Discovery of Highly Divergent Repeat Landscapes in Snake Genomes Using High-Throughput Sequencing. Genome Biol. Evol. 2011, 3, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.C.; Edwards, R.J.; Cheng, Y.; Warren, W.C.; Burt, D.W.; Sherwin, W.B.; Hofmeister, N.R.; Werner, S.J.; Ball, G.F.; Bateson, M.; et al. Transcript- and Annotation-Guided Genome Assembly of the European Starling. bioRxiv 2021. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Levy Karin, E.; Mirdita, M.; Söding, J. MetaEuk-Sensitive, High-Throughput Gene Discovery, and Annotation for Large-Scale Eukaryotic Metagenomics. Microbiome 2020, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mirarab, S.; Nguyen, N.; Warnow, T. SEPP: SATé-Enabled Phylogenetic Placement. Pac. Symp. Biocomput. 2012, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional Classification of Proteins via Subfamily Domain Architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, Submission and Screening of Repetitive Elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- NCBI Resource Coordinators Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [CrossRef] [Green Version]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Simões, B.F.; Gower, D.J.; Rasmussen, A.R.; Sarker, M.A.R.; Fry, G.C.; Casewell, N.R.; Harrison, R.A.; Hart, N.S.; Partridge, J.C.; Hunt, D.M.; et al. Spectral Diversification and Trans-Species Allelic Polymorphism during the Land-to-Sea Transition in Snakes. Curr. Biol. 2020, 30, 2608–2615.e4. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 6 January 2022).
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian Cobra Reference Genome and Transcriptome Enables Comprehensive Identification of Venom Toxins. Nat. Genet. 2020, 52, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The King Cobra Genome Reveals Dynamic Gene Evolution and Adaptation in the Snake Venom System. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishida, T.; Toda, M.; Go, Y.; Tatsumoto, S.; Sasai, T.; Hikida, T. Population History and Genomic Admixture of Sea Snakes of the Genus Laticauda in the West Pacific. Mol. Phylogenet. Evol. 2021, 155, 107005. [Google Scholar] [CrossRef] [PubMed]
- Ludington, A.J.; Sanders, K.L. Demographic Analyses of Marine and Terrestrial Snakes (Elapidae) Using Whole Genome Sequences. Mol. Ecol. 2021, 30, 545–554. [Google Scholar] [CrossRef]
- Rhie, A.; Mccarthy +2, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Gedman, G.L.; et al. Towards Complete and Error-Free Genome Assemblies of All Vertebrate Species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Peona, V.; Blom, M.P.K.; Xu, L.; Burri, R.; Sullivan, S.; Bunikis, I.; Liachko, I.; Haryoko, T.; Jønsson, K.A.; Zhou, Q.; et al. Identifying the Causes and Consequences of Assembly Gaps Using a Multiplatform Genome Assembly of a Bird-of-Paradise. Mol. Ecol. Resour. 2021, 21, 263–286. [Google Scholar] [CrossRef]
- Smit, A.F. Repeat-Masker Open-4.0 2013–2015. Available online: http://www.repeatmasker.org (accessed on 6 January 2022).
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Wang, Z.-J.; Li, Q.-Y.; Lian, J.-M.; Zhou, Y.; Lu, B.-Z.; Jin, L.-J.; Qiu, P.-X.; Zhang, P.; Zhu, W.-B.; et al. Evolutionary Trajectories of Snake Genes and Genomes Revealed by Comparative Analyses of Five-Pacer Viper. Nat. Commun. 2016, 7, 13107. [Google Scholar] [CrossRef]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal Transfer of Transposable Elements and Why It Matters for Eukaryotic Evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Minton, S.A., Jr.; Minton, M.R. Toxicity of Some Australian Snake Venoms for Potential Prey Species of Reptiles and Amphibians. Toxicon 1981, 19, 749–755. [Google Scholar] [CrossRef]
- Melville, J.; Wilson, S.K. Dragon Lizards of Australia: Evolution, Ecology and a Comprehensive Field Guide; Museums Victoria Publishing (Australia): Melbourne, Australia, 2019; ISBN 9781921833496. [Google Scholar]
- Heatwole, H.; Powell, J. Resistance of Eels (Gymnothorax) to the Venom of Sea Kraits (Laticauda Colubrina): A Test of Coevolution. Toxicon 1998, 36, 619–625. [Google Scholar] [CrossRef]
- Shine, R.; Shine, T.; Goiran, C. Morphology, Reproduction and Diet of the Greater Sea Snake, Hydrophis Major (Elapidae, Hydrophiinae). Coral Reefs 2019, 38, 1057–1064. [Google Scholar] [CrossRef]
- O’Shea, M. The Book of Snakes: A Life-Size Guide to Six Hundred Species from around the World; University of Chicago Press: Chicago, IL, USA, 2018; ISBN 9780226459424. [Google Scholar]
- Smith, M.A. Monograph of the Sea-Snakes (Hydrophiidae); Printed by Order of the Trustees of the British Museum: London, UK, 1926. [Google Scholar]
- Hoffstetter, R. Contribution à L’étude Des Elapidae Actuels et Fossiles et de L’ostéologie Des Ophidiens. Publ. Musée Conflu. 1939. [Google Scholar] [CrossRef]
- Storr, G.M. Some Aspects of the Geography of Australian Reptiles. Senckenb. Biol. 1964, 45, 577–589. [Google Scholar]
- Keogh, J.S. Molecular Phylogeny of Elapid Snakes and a Consideration of Their Biogeographic History. Biol. J. Linn. Soc. Lond. 1998, 63, 177–203. [Google Scholar] [CrossRef]
- Sanders, K.L.; Lee, M.S.Y. Molecular Evidence for a Rapid Late-Miocene Radiation of Australasian Venomous Snakes (Elapidae, Colubroidea). Mol. Phylogenet. Evol. 2008, 46, 1165–1173. [Google Scholar] [CrossRef]
- Hall, R. The Palaeogeography of Sundaland and Wallacea since the Late Jurassic. J. Limnol. 2013, 72, e1. [Google Scholar] [CrossRef]
- Yang, T.; Gurnis, M.; Zahirovic, S. Mantle-induced Subsidence and Compression in SE Asia since the Early Miocene. Geophys. Res. Lett. 2016, 43, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- Esquerré, D.; Donnellan, S.; Brennan, I.G.; Lemmon, A.R.; Moriarty Lemmon, E.; Zaher, H.; Grazziotin, F.G.; Keogh, J.S. Phylogenomics, Biogeography, and Morphometrics Reveal Rapid Phenotypic Evolution in Pythons After Crossing Wallace’s Line. Syst. Biol. 2020, 69, 1039–1051. [Google Scholar] [CrossRef] [Green Version]
- Heinicke, M.P.; Greenbaum, E.; Jackman, T.R.; Bauer, A.M. Phylogeny of a Trans-Wallacean Radiation (Squamata, Gekkonidae, Gehyra) Supports a Single Early Colonization of Australia. Zool. Scr. 2011, 40, 584–602. [Google Scholar] [CrossRef]
- Scanlon, J.D.; Lee, M.S.Y.; Archer, M. Mid-Tertiary Elapid Snakes (Squamata, Colubroidea) from Riversleigh, Northern Australia: Early Steps in a Continent-Wide Adaptive Radiation. Geobios Mem. Spec. 2003, 36, 573–601. [Google Scholar] [CrossRef]
- Heatwole, H.; Grech, A.; Marsh, H. Paleoclimatology, Paleogeography, and the Evolution and Distribution of Sea Kraits (Serpentes; Elapidae; Laticauda). Herpetol. Monographs. 2017, 31, 1–17. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Naja naja | Ophiophagus hannah | Laticauda colubrina | Pseudonaja textilis | Notechis scutatus | Aipysurus laevis | |
|---|---|---|---|---|---|---|
| No. of scaffolds | 1897 | 296,399 | 20,583 | 28,550 | 52,414 | 196,089 |
| Assembly size | 1.77 Gb | 1.59 Gb | 2.04 Gb | 1.59 Gb | 1.67 Gb | 1.85 Gb |
| Min. scaffold | 585 bp | 200 bp | 1000 bp | 1000 bp | 1000 bp | 179 bp |
| Max. scaffold | 375.0 Mb | 2.845 Mb | 114.7 Mb | 55.11 Mb | 50.92 Mb | 529.6 kb |
| Scaffold N50 | 224.1 Mb | 241.5 kb | 39.96 Mb | 14.69 Mb | 5.997 Mb | 24.81 kb |
| Scaffold L50 | 3 | 1750 | 16 | 31 | 66 | 22,027 |
| No. contigs | 13,805 | 816,633 | 105,298 | 88,022 | 131,892 | 310,523 |
| Contig N50 | 277.2 kb | 4.170 kb | 34.05 kb | 48.75 kb | 29.62 kb | 10.89 kb |
| Contig L50 | 1587 | 94,154 | 16,388 | 8,636 | 14,776 | 43,489 |
| % Gaps | 6.20% | 13.4% | 8.88% | 2.47% | 4.84% | 13.6% |
| % GC | 40.46% | 39.46% | 41.25% | 39.84% | 39.72% | 39.90% |
| BUSCO v5 Completeness (sauropsida_odb10, n = 7840) | 90.0% | 84.5% | 90.9% | 91.0% | 87.9% | 59.9% |
| 88.6% | 83.6% | 89.9% | 89.3% | 86.3% | 58.9% |
| 1.4% | 0.9% | 1.0% | 1.7% | 1.6% | 1.0% |
| 1.9% | 5.5% | 2.3% | 2.1% | 3.3% | 18.7% |
| 8.1% | 10.0% | 6.8% | 6.9% | 8.8% | 21.4% |
| GenBank Accession | GCA_009733165.1 | GCA_000516915.1 | GCA_015471245.1 | GCA_900518735.1 | GCA_900518725.1 | - |
| Ensembl Identifier | Nana_v5 | - | - | EBS10Xv2-PRI | TS10Xv2-PRI | - |
| First published | Suryamohan et al. [43] | Vonk et al. [44] | Kishida et al. [45] | This study | This study | Ludington and Sanders [46] |
| Naja naja | Ophiophagus hannah | Laticauda colubrina | Pseudonaja textilis | Notechis scutatus | Aipysurus laevis | |
|---|---|---|---|---|---|---|
| Class I (Retrotransposons) | 30.58 | 19.73 | 23.00 | 27.81 | 27.29 | 25.91 |
| Penelope | 1.72 | 1.37 | 2.08 | 2.06 | 1.98 | 1.64 |
| LINE/CR1 | 4.37 | 4.12 | 4.12 | 5.54 | 6.28 | 4.67 |
| LINE/L1 | 3.24 | 2.21 | 2.94 | 4.04 | 3.40 | 2.72 |
| LINE/L2 | 7.14 | 3.39 | 1.05 | 1.41 | 1.4 | 1.27 |
| LINE/Rex-Babar | 1.17 | 1.11 | 1.08 | 1.44 | 1.42 | 1.26 |
| LINE/RTE | 1.40 | 1.52 | 1.07 | 1.50 | 1.43 | 1.25 |
| LINE/Other | 0.62 | 0.69 | 0.54 | 0.66 | 0.64 | 0.59 |
| SINE | 0.37 | 0.43 | 0.30 | 0.40 | 0.39 | 0.36 |
| LTR/Copia | 0.71 | 0.47 | 1.02 | 1.27 | 1.65 | 0.89 |
| LTR/DIRS | 0.81 | 0.61 | 0.86 | 1.04 | 1.19 | 1.57 |
| LTR/ERV | 0.60 | 0.42 | 1.23 | 1.38 | 1.69 | 1.16 |
| LTR/Gypsy | 7.01 | 1.97 | 5.6 | 5.61 | 4.38 | 7.22 |
| LTR/Other | 1.42 | 1.42 | 1.11 | 1.46 | 1.44 | 1.31 |
| Class II (DNA Transposons) | 7.81 | 7.45 | 18.08 | 9.02 | 9.20 | 7.95 |
| DNA/hAT | 4.25 | 4.13 | 3.78 | 5.09 | 5.13 | 4.47 |
| DNA/PIF-Harbinger | 0.02 | 0.03 | 11.19 | 0.02 | 0.02 | 0.02 |
| DNA/Tc1-Mariner | 3.13 | 2.88 | 2.79 | 3.47 | 3.63 | 3.08 |
| DNA/Other | 0.22 | 0.24 | 0.17 | 0.24 | 0.22 | 0.20 |
| RC/Helitron | 0.19 | 0.17 | 0.15 | 0.20 | 0.20 | 0.18 |
| Unknown | 7.93 | 8.57 | 6.46 | 9.18 | 9.26 | 7.89 |
| Total interspersed repeats | 46.32 | 35.75 | 47.54 | 46.01 | 45.75 | 41.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbraith, J.D.; Ludington, A.J.; Sanders, K.L.; Amos, T.G.; Thomson, V.A.; Enosi Tuipulotu, D.; Dunstan, N.; Edwards, R.J.; Suh, A.; Adelson, D.L. Horizontal Transposon Transfer and Its Implications for the Ancestral Ecology of Hydrophiine Snakes. Genes 2022, 13, 217. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020217
Galbraith JD, Ludington AJ, Sanders KL, Amos TG, Thomson VA, Enosi Tuipulotu D, Dunstan N, Edwards RJ, Suh A, Adelson DL. Horizontal Transposon Transfer and Its Implications for the Ancestral Ecology of Hydrophiine Snakes. Genes. 2022; 13(2):217. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020217
Chicago/Turabian StyleGalbraith, James D., Alastair J. Ludington, Kate L. Sanders, Timothy G. Amos, Vicki A. Thomson, Daniel Enosi Tuipulotu, Nathan Dunstan, Richard J. Edwards, Alexander Suh, and David L. Adelson. 2022. "Horizontal Transposon Transfer and Its Implications for the Ancestral Ecology of Hydrophiine Snakes" Genes 13, no. 2: 217. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020217