
The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism


Center for Medical Genetics, Department of Biomolecular Medicine, Ghent University, 9000 Ghent, Belgium
*
Author to whom correspondence should be addressed.
Genes 2022, 13(2), 265; https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020265
Submission received: 10 January 2022
/
Accepted: 26 January 2022
/
Published: 29 January 2022
(This article belongs to the Special Issue Genetic Research in Metabolic Diseases)
Abstract
:The Ehlers–Danlos syndromes are a group of multisystemic heritable connective tissue disorders with clinical presentations that range from multiple congenital malformations, over adolescent-onset debilitating or even life-threatening complications of connective tissue fragility, to mild conditions that remain undiagnosed in adulthood. To date, thirteen different EDS types have been recognized, stemming from genetic defects in 20 different genes. While initial biochemical and molecular analyses mainly discovered defects in genes coding for the fibrillar collagens type I, III and V or their modifying enzymes, recent discoveries have linked EDS to defects in non-collagenous matrix glycoproteins, in proteoglycan biosynthesis and in the complement pathway. This genetic heterogeneity explains the important clinical heterogeneity among and within the different EDS types. Generalized joint hypermobility and skin hyperextensibility with cutaneous fragility, atrophic scarring and easy bruising are defining manifestations of EDS; however, other signs and symptoms of connective tissue fragility, such as complications of vascular and internal organ fragility, orocraniofacial abnormalities, neuromuscular involvement and ophthalmological complications are variably present in the different types of EDS. These features may help to differentiate between the different EDS types but also evoke a wide differential diagnosis, including different inborn errors of metabolism. In this narrative review, we will discuss the clinical presentation of EDS within the context of inborn errors of metabolism, give a brief overview of their underlying genetic defects and pathophysiological mechanisms and provide a guide for the diagnostic approach.
1. The Ehlers–Danlos Syndromes, a General Introduction
The Ehlers–Danlos syndromes (EDS) are a group of rare heritable connective tissue disorders, clinically hallmarked by generalized joint hypermobility, skin hyperextensibility and fragility, easy bruising, abnormal wound healing and widespread connective tissue friability [1]. The prevalence is estimated to be approximately 1 in 5000 births. Electron microscopy studies on skin biopsies from affected individuals initially suggested EDS to be a disorder of the collagen ‘wickerwork’ [2], a suspicion that was subsequently confirmed by the biochemical and genetic identification of defects in the primary structure of fibrillar collagens, type I, III and V or their modifying enzymes (lysyl hydroxylase 1 and type I procollagen amino-proteinase) in individuals with different types of EDS [3,4].
These discoveries led to the successful proposal of the Villefranche EDS nosology in 1998, which included six EDS types (classical, hypermobility, vascular, kyphoscoliosis, arthrochalasia and dermatosparaxis) and which was used for over two decades [4]. Meanwhile, advances in molecular techniques identified defects in extracellular matrix (ECM) molecules beyond fibrillar collagens (e.g., the glycoprotein tenascin-X, enzymes involved in proteoglycan biosynthesis, the collagen chaperone FKBP22 and the complement factors C1r and C1s).
This broadened the molecular and clinical spectrum of this syndrome over the past two decades and prompted a revision of the EDS classification [5]. The 2017 International EDS Classification recognizes 13 distinct clinical EDS types, 12 of which have been molecularly elucidated, caused by pathogenic variants in 20 different genes (Table 1) [1,5].
2. The Clinical Presentation of EDS, an Overview
Joint hypermobility and skin hyperextensibility are the defining hallmarks of EDS, but a renewed interest in the natural history of these syndromes and the description of several new types has painted an impressive and much more diverse clinical tableau. Depending on the underlying genetic defect, this varies from a mild musculoskeletal and cutaneous phenotype to severe physical disability and life-threatening vascular and visceral complications and can also include such signs and symptoms as skeletal dysplasia, hypotonia, congenital and/or progressive contractures and chronic pain (Table 2).
In general, the autosomal recessive types of EDS are associated with congenital abnormalities, such as hip or other joint dislocations, floppy infant syndrome due to congenital muscle hypotonia and joint hypermobility, kyphoscoliosis, club feet, etc. Although this makes them recognizable entities (very) early on, the diagnosis is often delayed due the fact that EDS is still not a well-known condition.
The diagnosis of the more common and autosomal dominant types (classical (cEDS), hypermobile (hEDS) and vascular EDS (vEDS)) may be missed in childhood due to the presence of only subtle signs of connective tissue fragility that are common in the general population (e.g., joint hypermobility, easy bruising, varicose veins, etc.). This contrasts greatly with most inborn errors of metabolism (IEM) that manifest themselves after a symptom-free period in the first months or years of life [6].
What follows is a concise per system overview of the EDS phenotype keeping a differential diagnosis with the IEM in mind (Table 2).
2.1. Prenatal Findings
Prenatal ultrasound abnormalities sometimes seen in e.g., lysosomal storage diseases (hydrops fetalis) or peroxisome biogenesis disorders (brain anomalies and cystic renal disease), are extremely uncommon in EDS [6]. The lone exception are the congenital anomalies of the kidney and urinary tract (nephroptosis and ureteropelvic junction obstruction) sometimes observed in musculocontractural EDS (mcEDS) [7].
2.2. Integumentary System
The first hallmark of EDS is a hyperextensible skin; i.e., the skin stretches easily and beyond normal but snaps back after release (Figure 1D,R). The skin is often smooth and velvety to the touch. It is fragile and tears easily. This leads to the formation of widened and thin atrophic or so-called ‘cigarette paper scars’, although scar formation may be normal in certain types of EDS (clEDS and pEDS) (Figure 1E,K).
Easy bruising (the spontaneous and recurrent formation of ecchymoses) and hematoma formation are variably present and are most pronounced in vEDS, dEDS, mcEDS and classical-like EDS type 1 (clEDS) [11]. Pretibial hemosiderin deposits (Figure 1D) are a prominent feature of periodontal EDS (pEDS), but can be present in cEDS and vEDS as well. Virtually all dEDS cases reported to date have congenital umbilical hernia. Abdominal herniation (umbilical and inguinal) is occasionally reported in other types of EDS as well [12].
This skin phenotype is markedly different from the one observed in IEM, where pigmentation disorders, lipodystrophy, inverted nipples, aplasia cutis congenita, ichthyosis or dry, thickened scaly skin and xanthomas are observed, all of which are not associated with EDS. The only skin features occasionally shared between rare types of EDS and several types of IEM are hirsutism (dEDS and mcEDS) and a cutis laxa-like skin [13]. The latter is a clinical term for loose, wrinkled, sagging and redundant skin that lacks the excessive elastic recoil typically seen in EDS and can be present particularly in dEDS [10].
2.3. Skeletal System
Joint hypermobility, the second hallmark of EDS, is often the reason for referral both in children and adults. Joint hypermobility is a continuous trait modified by age, ethnicity, sex and environmental factors, such as exercise and past traumas. In the general population, generalized joint hypermobility has a reported prevalence of 6% to 57% in females and 2% to 35% in males [14]. (Generalized) joint hypermobility is also observed in other syndromic conditions, including several congenital disorders of glycosylation (e.g., X-linked SSR4-CDG, Morquio syndrome or mucopolysaccharidosis (MPS) type IV) [15].
The severity is routinely assessed using the Beighton scale. Hypermobility is generally defined by a score ≥ 6 for prepubertal children and adolescents, by a score ≥ 5 for pubertal men and women ≤ 50 years of age and by a score ≥ 4 for men and women > 50 years of age. The Beighton scale is easy-to-use, but has some major limitations, and better methods to measure joint mobility in routine clinical practice are needed [5]. Joint dislocations, joint pain and fatigue are complications of generalized joint hypermobility and are reported in all types of EDS [16].
Congenital hip dysplasia is a universal feature in the athrochalasia type of EDS (aEDS), but can be present in other types as well [17]. A marfanoid habitus is not uncommon in some of the rare autosomal recessive types of EDS (kyphoscoliotic EDS (kEDS), mcEDS and brittle cornea syndrome (BCS)) and is contrary to homocystinuria present in combination with joint hypermobility. Osteoporosis is more prevalent in some types of EDS (kEDS, mcEDS, dEDS, BCS and spondylodysplastic EDS (spEDS)) and is associated with an increased risk of fractures, although seldom as pronounced as in osteogenesis imperfecta. This is similar to what is observed in Gaucher disease and homocystinuria.
Other recurring skeletal features include osteoarthritis, spinal and pectus deformities (Figure 1Q), contractures (Figure 1N,O,S), arachnodactyly (Figure 1L) and foot deformations (Figure 1E,I,O,S).
The most severe skeletal phenotype is reported in spEDS [18]. spEDS-B4GALT7 is predominantly associated with radio-ulnar synostosis, whereas the skeletal findings in spEDS-SLC39A13 and especially in spEDS-B3GALT6 are more pronounced and include widening of the metaphyses and bowing of the diaphysis of long bones, hypoplasia of the iliac bones, acetabular dysplasia, femoral head dysplasia and changes to the shape of the vertebral bodies. Spondylodysplastic EDS thus combines the classic signs of EDS with spondyloepimetaphyseal dysplasia, bone fragility and short stature and evokes a wide differential diagnosis that not only includes other types of EDS but also osteogenesis imperfecta, skeletal dysplasias and MPS (e.g., type IV).
2.4. Neuromuscular System
Hypotonia is a key symptom of many IEM, but it is less frequent and mostly less severe within the group of the Ehlers–Danlos syndromes. Unsurprisingly, it is most frequently reported in types of EDS with a disorganized myomatrix (clEDS type 1 and myopathic EDS (mEDS)). TNX-deficiency (clEDS type 1) is always associated with some degree of muscle weakness (mild to moderate), myalgia and easy fatigability, but all patients reported to date can walk independently.
The combination of congenital proximal joint contractures with distal joint hypermobility should raise a suspicion of mEDS, which shows clinical overlap with Bethlem myopathy and Ullrich congenital muscular dystrophy and is further characterized by profound congenital hypotonia, which appears to improve with age [19,20].
Hypotonia can also be present in cEDS, aEDS, kEDS, mcEDS and spEDS and is variably associated with delayed gross motor development, myopathy with variability in fiber diameter on muscle biopsy, signs of axonal polyneuropathy on electromyography and elevated creatine kinase [12,21,22].
Central nervous system abnormalities have been reported only in mcEDS-CHST14 and spEDS-B3GALT6 and include ventricular enlargement, Dandy–Walker anomalies, hypoplasia of the septum pellucidum, hydrocephalus and a tethered spinal cord [7,12]. Intellectual disability is typically not present but has sporadically been reported in families with AR types of EDS. It remains, however, unclear whether this is truly part of the EDS phenotype or rather caused by environmental or other genetic factors. Encephalopathy is not reported.
2.5. Cardiovascular System
Arterial rupture, occurring spontaneously or preceded by aneurysm, arteriovenous fistulae or dissection, is the predominant feature of vEDS. The median survival for vEDS patients is calculated to be 51 years, albeit with a very large range, and most deaths do indeed result from arterial rupture [9,23,24]. Complications are rare in childhood, but 17% will have a first complication by the age of 20 years, and approximately 70% will have had at least one complication by the age of 40 years [23]. Superficial venous insufficiency is also more prevalent and is often early-onset [23].
Vascular complications are, however, not exclusive to vEDS and can be present in other types of EDS as well albeit with important differences in the number, severity and type of complications [11]. The dissection of medium-sized arteries, for instance, has also been reported in several cases of kEDS and to a lesser extent cEDS, and aortic root dilatation has been reported in cEDS and spEDS-B3GALT6 [8,18]. Another notable example is the risk of intracranial hemorrhage in dEDS and to a lesser extent in mcEDS-CHST14 and kEDS-PLOD1.
This degree of vascular fragility is not seen in IEM; however, recent studies have brought aortic root dilatation to light as an important complication of MPS and more specifically of MPS IVA (Morquio syndrome) [25]. Fabry-like stroke events are not present in EDS.
Severe cardiac valvular disease necessitating surgical valve replacement is the hallmark of the extremely rare cardiac valvular type of EDS (cvEDS), whereas mild valvular disease and especially mitral valve prolapse, might be more prevalent in cEDS, clEDS type 1, kEDS, mcEDS and spEDS [12,26]. Cardiomyopathy has only been reported in a very limited number of cases and is not a consistent finding.
2.6. Ocular System
Rupture of the eye globes, either spontaneous or after minor trauma, due to extreme thinning of the cornea is the cardinal feature of brittle cornea syndrome (BCS) and perhaps one of the most debilitating complications within the EDS spectrum [12,27]. It frequently occurs at a young age; however, several adults without ocular rupture have been described. Prior to rupture, corneal thinning will manifest itself by the presence of blue sclerae and corneal ectasia.
In the other types of EDS there is no or a relatively mild ophthalmic phenotype that includes keratoconus (vEDS), blue sclerae (aEDS, dEDS, kEDS, mcEDS and spEDS), microcornea (kEDS-PLOD1 and mcEDS) and rarely corneal clouding (spEDS). The latter is more frequently encountered in IEM, such as MPS, Fabry disease, tyrosinemia type II, cystinosis, mucolipidosis IV and Niemann–Pick disease type A. Refractive errors also appear to be more prevalent in EDS compared to the general population.
2.7. Orocraniofacial
A recognizable facial gestalt has been reported in only a few types of EDS. Perhaps best known is the thin vermilion of the lips, micrognathia, narrow nose, prominent eyes and potentially severe gingival recession, which can lead to tooth loss in some individuals with vEDS [9]. The latter should also raise the concern of the periodontal form of EDS, in which early-onset periodontitis with extensive periodontal destruction and loss of teeth, starting in childhood or adolescence, is universally present and can be accompanied by similar craniofacial dysmorphisms [28].
The facial dysmorphisms in dEDS also combine into a recognizable appearance and include prominent and protuberant eyes with puffy, edematous eyelids and excessive periorbital skin, large fontanels and/or wide cranial sutures, micrognathia and gingival hyperplasia (Figure 1M) [10]. Finally, individuals with mcEDS present with large fontanels, downslanting palpebral fissures, hypertelorism, a short nose with hypoplastic columella, ear deformities, high-arched palate, long philtrum, thin upper lip vermilion, small mouth and microretrognathia (Figure 1J) [7,29].
2.8. Other
The above is only a curated selection from a much more extensive list of anomalies, symptoms and signs described in EDS. To conclude, we will highlight a few clinical pearls that might facilitate early diagnosis.
In addition to vascular rupture, gastrointestinal perforation, usually involving the sigmoid, or hollow organ rupture can be the presenting signs in vEDS [23,30]. Spontaneous pneumothorax is most often associated with Marfan syndrome and Birt–Hogg–Dubé syndrome but is also seen in ~10% of individuals with vEDS [9].
Hearing loss is a key finding in heritable connective tissue disorders, such as osteogenesis imperfecta and Stickler syndrome but is also present in the majority of individuals with kEDS-FKBP14 or mcEDS and a third of the individuals with BCS [7,12,27]. Finally, cryptorchidism is present in the large majority of mcEDS-CHST14 individuals and should raise suspicion of mcEDS in a neonate with signs of connective tissue fragility and contractures [7].
3. The Genetic and Pathophysiological Basis of the EDS
The pleiotropic and multisystemic nature of EDS is caused by alterations in the physical properties of the ECM in many tissues and organs, which, in turn, is the result of a compromised biosynthesis, fibrillogenesis and/or supramolecular organization of collagen fibrils due to pathogenic variants in different genes (Figure 2).
The biosynthetic pathway of fibrillar procollagens is a complex process. Nascent pro-α-chains are heavily post-translationally modified. Three pro-α-chains associate at their carboxy-(C-) terminal propeptides and assemble into a trimeric procollagen molecule propagating to the amino-(N-) terminus in a zipper like fashion. These trimeric proteins consist of either three identical (homotrimer) or genetically distinct (heterotrimer) pro-α-chains, containing repeating Gly--Xaa--Yaa triplets of a glycine residue and two other amino acids and which form a long uninterrupted triple helical domain flanked by globular C- and N-terminal propeptides.
These procollagen molecules are subsequently transported to the extracellular environment. Proteolytic removal of the N- and C-propeptides results in the formation of a mature collagen molecule that can assemble into highly ordered cross-striated fibrils and fibers [31].
Typically, collagen fibrils are composed of different fibrillar collagen types, e.g., collagen I and collagen III (heterotypic collagen I/III fibrils) and the assembly of collagen fibrils occurs in a tissue-specific way, requiring the concerted action of several assisting proteins: organizers (e.g., fibronectin and integrins), nucleators (e.g., type V collagen) and regulators (e.g., small leucine-rich proteoglycans (SLRPs), fibril-associated collagens with interrupted triple helices (FACIT) and glycoproteins) [32].
During fibrillogenesis, fibril growth occurs through linear and lateral fusion of intermediate collagen fibrils, which are subsequently stabilized by the formation of intra- and inter-molecular crosslinks (Figure 2) [33,34,35]. The EDS types can be grouped based on the pathways in which the defective proteins function (Table 1) [5].
3.1. Defects in Fibrillar Collagen Structure and Processing
The first biochemical and/or molecular defects associated with EDS were shown to result from defects in the primary structure, processing, or modification of the fibrillar procollagen types I, III and V leading to qualitative and/or quantitative changes in these collagen types.
Classical EDS—The classical type of EDS (cEDS) is caused by heterozygous pathogenic variants in the COL5A1 or COL5A2 genes, encoding the pro-α1- and pro-α2-chains of type V collagen, respectively. Type V collagen is a low abundant protein in skin, tendons and bone. It is mostly present as a heterotrimer consisting of two pro-α1(V)-chains and one pro-α2(V)-chain, which assembles with type I collagen to form heterotypic type I/V collagen fibrils, where it plays a crucial role during the initiation and regulation of collagen fibrillogenesis [33,36]. Central to the pathogenesis of cEDS is a decreased amount of functional type V collagen in the ECM.
Ultrastructural analysis using transmission electron microcopy (TEM) of skin biopsies from cEDS patients revealed the presence of atypical (cauli)flower-shaped collagen fibrils on cross-section (Figure 3B).
Nevertheless, the molecular consequences remain poorly understood. Studies on dermal fibroblasts from cEDS patients did show decreased migration capacity and alterations in the deposition and organization of several ECM molecules, as well as changes in specific integrin receptors [37]. Subsequent transcriptome-wide analysis on cEDS dermal fibroblasts confirmed a deregulation of genes involved in ECM remodeling and wound healing and indicated a role for altered endoplasmic reticulum (ER) homeostasis and autophagy [38].
Vascular EDS—Vascular EDS (vEDS) is caused by heterozygous pathogenic variants in the COL3A1 gene encoding the pro-α1-chain of type III collagen. The homotrimeric type III collagen is mainly found in tissues with elastic properties (e.g., skin, blood vessels, gastrointestinal tract, uterus etc.) and can also assemble with type I collagen to form heterotypic fibrils.
Like type V collagen, type III collagen is presumed to regulate collagen fibril assembly and diameter [39,40]. Transcriptome profiling of dermal fibroblasts from vEDS patients revealed differential expression of genes encoding ECM molecules as well as genes involved in ER homeostasis [41]. Ultrastructural studies on the skin from vEDS patients showed a thinner dermis, aberrant diameters and distribution of collagen fibrils and variable dilatation of the ER [42].
Type I collagen defects—While most pathogenic defects in the pro-α1- and pro-α2-chains of type I collagen, encoded by the COL1A1 and COL1A2 genes, respectively, result in the brittle bone disorder osteogenesis imperfecta, some specific defects are associated with rare EDS types.
Heterozygous pathogenic variants resulting in partial or complete deletion of exon 6 of the COL1A1 or COL1A2 gene result in the arthrochalasia type of EDS (aEDS). Exon 6 of these genes contains the cleavage site for the N-propeptide and its loss leads to partial processing of type I procollagen with retention of the N-propeptide of either the pro-α1(I)- or the pro-α2(I)-chain in the collagen fibrils [43].
Decreased N-propeptide cleavage of the type I procollagen N-propeptide can also be caused by defects in the type I procollagen N-proteinase, ADAMTS2, due to biallelic loss-of-function variants in the ADAMTS2 gene, which gives rise to the dermatosparaxis EDS type (dEDS). In contrast to aEDS, where either the pro-α1(I)-or the pro-α2(I)chain is affected, cleavage of both pro-α(I)-chains is compromised in dEDS. ADAMTS2 is also known to possess N-proteinase activity for types II, III and V pro-collagens and enzymatic activity for some other molecules [44,45], which likely contributes to the phenotypic differences between aEDS and dEDS.
Incorporation of incompletely processed collagen molecules (still containing N-propeptides) leads to ultrastructural abnormalities of the dermal collagen fibrils, ranging from small-diameter fibrils with irregular contours in aEDS to fibrils that have completely lost their circular dimensions on cross-sections and display a typical hieroglyphic pattern in dEDS (Figure 3C) [10,46].
Biallelic loss-of-function variants in the COL1A2 gene, that lead to unstable mRNA prone to nonsense-mediated mRNA decay and complete lack of the pro-α2(I)-chains, cause the extremely rare cardiac-valvular EDS (cvEDS). The absence of pro-α2(I)-chains forces the formation of α1(I) homotrimers; however, the exact pathomechanisms remain elusive.
Finally, specific heterozygous variants in the COL1A1 gene that substitute an arginine for a cysteine residue in the triple helical region of the pro-α1(I)-chains give rise to a rare phenotype clinically overlapping with cEDS and vEDS [47,48]. In addition to the local destabilization of the type I collagen molecules due to the loss of the helical arginine residue, the introduction of a cysteine residue results in the production of α1(I) dimers and dermal collagen fibrils have variable diameters with irregular interfibrillar spaces [47].
3.2. Defects in Collagen Crosslinking and Folding
Kyphoscoliotic EDS-PLOD1—The first inborn error of collagen metabolism that was elucidated at the biochemical level was kyphoscoliotic EDS (kEDS) with the demonstration of a reduced hydroxylysine content in the dermal collagen of two affected sisters, caused by a lysyl hydroxylase deficiency [49,50]. Subsequently, biallelic pathogenic variants in the PLOD1 gene encoding the collagen modifying enzyme lysyl hydroxylase 1 (LH1) were identified [51].
LH1 catalyzes the co- and posttranslational modification of specific lysine residues in the triple helix to hydroxylysine. These hydroxylysyl residues can either be glycosylated or can form intermolecular crosslinks in the ECM. As a consequence of LH1 deficiency, abnormal crosslink formation in kEDS-PLOD1 patients renders their tissues mechanically instable.
Kyphoscoliotic EDS-FKBP14—More recently, biallelic variants in the FKBP14 gene were found to cause a form of EDS that shows phenotypic overlap with kEDS-PLOD1. FKBP14 encodes the ER-resident protein FKBP22 (FK506 Binding Protein 22 kDa), a peptidyl-prolyl cis-trans isomerase that accelerates procollagen folding in vitro [52,53]. In addition, FKBP22 functions as a molecular chaperone and preferentially interacts with type III, IV, VI and X procollagen but does not bind to type I or V collagen [52,54]. Dermal fibroblasts from FKBP22-deficient patients show enlarged ER, likely reflecting the accumulation of collagen in the ER, as well as altered in vitro assembly of the ECM [55].
Transcriptome profiling of dermal fibroblast cultures of kEDS-PLOD1 and kEDS-FKBP22 patients showed distinct transcriptome signatures with differentially expressed ECM components, which are unique to each kEDS type [56]. The pathophysiological mechanisms linking kEDS-PLOD1 and kEDS-FKBP14 remain currently unknown.
3.3. Defects in ECM Bridging Molecules
Classical-like EDS type 1—The first glycoprotein involved in EDS pathogenesis was tenascin-X (TNX). Biallelic loss-of-function variants in the TNXB gene were shown to lead to a phenotype similar to that caused by defects in type V collagen; hence, this EDS type was coined classical-like EDS (clEDS) [57,58].
The TNXB gene partially overlaps with the CYP21A2 gene, encoding 21-hydroxylase. Deficiency of 21-hydroxylase leads to congenital adrenal hyperplasia (CAH). A contiguous gene syndrome that combines signs of CAH and signs of EDS that is caused by deletions in CYP21A1 that extend into TNXB has been described [59]. Additionally, in a subset of individuals presenting generalized joint hypermobility, heterozygous TNXB variants were identified that lead to TNXB haploinsufficiency [60].
Myopathic EDS—Biallelic and heterozygous defects in the COL12A1 gene, encoding the pro-α1-chains of type XII collagen, cause the rare myopathic EDS (mEDS).
The homotrimeric type XII collagen is a member of the FACIT family and interacts with TNX [61]. Both molecules can directly or indirectly (e.g., via SLRPs, such as decorin or fibromodulin) bind to fibrillar collagens and form flexible bridges between collagen fibrils and other ECM molecules, thereby, regulating the organization and mechanical properties of collagen fibrils. This is supported by ultrastructural studies of dermal collagen fibrils of both clEDS1 and mEDS patients showing increased interfibrillar space (Figure 3D) [62,63].
Classical-like EDS type 2—More recently, biallelic pathogenic variants were identified in the AEBP1 gene in a few individuals also presenting a phenotype that strongly overlaps with cEDS and clEDS type 1. AEBP1 codes for adipocyte enhancer-binding protein 1 (AEBP1; also known as aortic carboxypeptidase-like protein, ACLP) [64]. AEBP1 is an ECM-associated protein that occurs in tissues with a high collagen content [65,66], binds to type I, III and V collagen, and aids type I collagen polymerization [64].
Ultrastructural studies of patients with biallelic AEBP1 variants revealed dermal collagen fibrils with variable diameters and irregular contours, suggesting the involvement of AEBP1 in collagen fibril formation (Figure 3E) [67]. Furthermore, roles for AEBP1 have been reported in fibroblast-to-myoblast transition via TGFβ-signaling [68] and frizzled-8- and LRP6-mediated activation of canonical Wnt-signaling. [69]. This condition is provisionally referred to as classical-like EDS type 2 (clEDS2).
3.4. Glycosaminoglycan Biosynthesis
Proteoglycans are major ECM constituents that play crucial structural roles and are involved in modulation of cell adhesion and motility, assembly of collagen fibrils and ECM as well as signal transduction. Proteoglycans consist of a core protein containing one or more glycosaminoglycan (GAG)-chains [70,71,72]. The biosynthesis of these GAG chain starts with the addition of four sugar residues to the core protein to form a tetrasaccharide linker, followed by the addition of repeating disaccharide units that define the GAG-chain as heparan sulfate (HS), chondroitin sulfate (CS) and/or dermatan sulfate (DS) (Figure 2).
Spondylodysplastic EDS—The first involvement of proteoglycan defects in EDS pathogenesis was reported in the 1980s with the discovery of GAG-free proteoglycan core proteins produced by fibroblasts of a patient with a condition coined ‘progeroid EDS’ [73,74,75,76,77]. Later, biallelic variants were identified in the B4GALT7 gene, encoding galactosyltransferase-I, the enzyme catalyzing the addition of the second sugar residue during the formation of the tetrasaccharide linker region of proteoglycans [78].
Subsequently, in another form of EDS, biallelic defects were discovered in the B3GALT6 gene, encoding galactosyltransferase-II, catalyzing the addition of the second galactose residue (i.e., the third step) during linker formation. Both conditions are now labelled spondylodysplastic EDS (spEDS). Defective galactosyltransferase-I or -II activity results in variable reduction in HS, CS and DS contents on proteoglycans. Interestingly, studies on b3galt6 knockout zebrafish demonstrated the presence of an unconventional trisaccharide linker region consisting of only three sugar residues instead of four [79].
Musculocontractural EDS—In addition to alterations in linker formation, defects in enzymes specifically responsible for modifying DS GAG-chains have also been linked to EDS pathogenesis. Musculocontractural EDS (mcEDS) is caused by biallelic loss-of-function variants in either the CHST14 or DSE gene, encoding dermatan 4-O-sulfotransferase-1 (D4ST1) and dermatan sulfate epimerase 1 (DS-epi1), respectively [80,81,82,83]. These enzymes catalyze consecutive steps in DS biosynthesis (Figure 2), and their deficiency leads to either complete replacement of DS for CS in D4ST1-deficient patients or severely reduced DS moieties in DS-epi1-deficient patients [83,84].
Altered or lacking GAG chains result in proteoglycans, such as the SLRP decorin, that are not capable of executing their crucial function during collagen fibrillogenesis, hence, leading to abnormal organization of collagen fibrils in the dermis (Figure 3F–H) of individuals with mcEDS or spEDS due to B4GALT7 or B3GALT6 defects.
3.5. Defects in Intracellular Processes
Spondylodysplastic EDS-SLC39A13—A very small subset of EDS patients harbor biallelic pathogenic variants in the SLC39A13 gene, encoding the homodimeric transmembrane Zrt/irt-like protein 13 (ZIP13), which regulates zinc influx in the cytosol. ZIP13 deficiency results in uniform underhydroxylation of lysine and proline residues in collagen and abnormal collagen crosslinking in the ECM [85].
The proposed disease mechanisms include competition of increased Zn2+ levels in the ER with Fe2+, a cofactor for lysyl and prolyl hydroxylases [85], vesicular Zn2+ trapping in the ER resulting in decreased Zn2+ availability [86] and altered activation of BMP/TGFβ signaling [87]. Due to the clinical overlap, this condition is merged with spEDS (spEDS-SLC39A13); however, the mechanisms linking spEDS-B4GALT7, spEDS-B3GALT6 and spEDS-SLC39A13 remain elusive.
Brittle cornea syndrome—Initially suspected to be a form of kEDS, brittle cornea syndrome (BCS) was demonstrated to be caused by biallelic pathogenic variants in either the ZNF469 or the PRDM5 gene [88,89,90]. ZNF469 encodes the zinc finger protein ZNF469 for which the function is largely unknown. PRDM5 encodes the transcription factor PRDM5 (PR domain zinc finger protein 5), which modulates several facets of vertebral tissue development and maintenance and is dependent on Wnt signaling [91,92].
Transcriptome analysis of dermal fibroblasts obtained from patients with pathogenic variants in ZNF469 or PRDM5 demonstrate altered expression of genes involved in ECM regulation. Additionally, immunofluorescent analysis showed altered in vitro deposition of ECM molecules (e.g., type I, III collagen and fibronectin) and some integrin receptors on dermal fibroblasts of BCS patients [89,93]. These overlapping findings point to a role for ZNF469 and PRDM5 in regulating the organization of the ECM.
3.6. Defects in the Complement Pathway
Periodontal EDS—Recently, heterozygous pathogenic gain-of-function variants in the C1R and C1S genes, encoding the C1r and C1s subunits of the first component of the classical complement pathways, were associated with periodontal EDS (pEDS) [28]. Intracellular activation of C1r and/or C1s and the extracellular presence of activated C1s, which can, in turn, activate the classical complement cascade, are central to pEDS pathogenesis [94].
Furthermore, the presence of evolutionarily conserved CUB domains in C1r and C1s suggest that they can also interact with ECM molecules including collagens and/or propeptides [95]. These findings provide opportunities to gain more knowledge about the connection between connective tissues and the immune system.
4. Diagnostic Approach
The 2017 International EDS Classification defined major and minor clinical criteria to guide the diagnosis of the different types of EDS [5]. Nevertheless, the definite diagnosis for all EDS types, except for hEDS, relies on the identification of a molecular defect in the respective genes. For hEDS, the genetic basis remains unknown, and diagnosis relies solely on proper clinical and familial evaluation [5].
The currently used diagnostic workflow mainly applies next-generation sequencing-based approaches focusing on parallel sequencing and subsequent analysis of a panel containing (at least) the 20 known EDS genes, to obtain a time- and cost-effective genetic diagnosis. This analysis should be complemented by a strategy to detect genomic copy number variants, such as array comparative genomic hybridization or shallow whole-genome sequencing.
Special care should be taken when analyzing certain genes. The TNXB gene has a partially duplicated non-functional pseudogene, TNXA, which shares 97% identity to the 3′ region of TNXB. Therefore, exons 32–44 of TNXB should be analyzed using a Sanger sequencing-based method [5]. An 8.9 kb duplication of seven exons (exon 10–16) is the most common pathogenic variant in PLOD1, being found in ~30% of kEDS-PLOD1 cases, and can be detected using a specifically designed PCR or via Multiplex ligation-dependent probe amplification (MLPA).
Despite rigorous genetic testing, there are still patients that present with a clinical EDS diagnosis (hEDS but also other EDS-like phenotypes) in whom no causal genetic defect can be found. This can either be attributed to technical limitations (e.g., deep intronic variants, variants in promotor or untranslated regions (UTRs)) or can point to additional genetic heterogeneity, with novel, yet to be discovered disease genes.
For several EDS types, biochemical and/or ultrastructural analyses can be performed, but they require a skin biopsy, urine or a blood sample. Although several of these analyses were once part of the routine diagnostic work-up, they are now mainly relegated to study the pathogenicity if variants of uncertain significance (VUS) are found, if only one pathogenic variant is detected in a recessive type of EDS or if no pathogenic variants could be detected in a patients with a clear clinical EDS diagnosis.
4.1. Biochemical Analyses
(Pro)collagen analysis—Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of radioactively or fluorescently labeled (pro)collagens isolated from cell and conditioned medium fractions of dermal fibroblast cultures established from a skin biopsy of EDS patients can show quantitative (reduced amounts) and/or qualitative (intracellular retention or altered electrophoretic mobility) alterations in type I and III procollagen. The (pro-)α-chains are partially digested with pepsin (cell and medium fraction) to remove the N- and C-propeptides or left untreated (procollagen analysis) prior to SDS-PAGE.
Qualitative or quantitative alterations in type III (pro)collagen can be detected in about 95% of vEDS patients (Figure 4A). EDS-related defects affecting type I collagen can also be detected. SDS-PAGE analysis of pepsin-digested collagens from aEDS fibroblasts shows an extra band containing the retained N-propeptide, the co-called pNα-chain, in addition to the normal α1(I)- or α2(I)-chains (Figure 4B,C). In contrast, fibroblasts from dEDS patients show an accumulation of type I procollagen chains with a retained N-propeptide when analyzing the procollagen fraction (Figure 4D).
Complete absence of the (pro-)α2(I)-chains is indicative of cvEDS (Figure 4E), and the presence of an additional band in cell and/or medium fractions, representing α1(I) dimers, can point to the presence of specific arginine-to-cysteine substitutions in the pro-α1(I)-chain (Figure 4F). Furthermore, uniform underhydroxylation and underglycosylation of the (pro-)α-chains as a consequence of LH1 deficiency in EDS-PLOD1 patients, can be seen as a faster migration of type I, III and V (pro)collagen chains (Figure 4G). (Pro)collagen analysis of type V collagen is not very sensitive to pick up defects in type V collagen [96].
Urinary analyses—Quantification of lysylpyridinolines (LP) and hydroxylysylpyridinolines (HP) crosslinks in urine using high-performance liquid chromatography is a reliable test to identify LH1 defects. The normal LP/HP ratio in healthy individuals is 0.2, whereas kEDS-PLOD1 patients have increased ratios ranging from 2 to 9 [97]. A mildly increased LP/HP ratio of around 1 is seen in patients with spEDS caused by pathogenic variants in SLC39A13 [85].
Defective DS biosynthesis can be detected by disaccharide analysis measured by anion-exchange chromatography in urine of patients with D4ST1- or DS-epi1-deficient mcEDS. The lack of urinary DS disaccharides is specific for mcEDS [84,98].
Serum measurements—Western blot or enzyme-linked immunosorbent assay (ELISA), developed to detect serum levels of TNX, showed reduced TNX levels in clEDS patients with biallelic variants in the TNXB gene. These tests are not routinely available since it depends on the availability of specific antibodies [58]. More recently, TNX serum levels were determined in clEDS1 patients using nano-liquid chromatography tandem mass spectrometry (nano-LC/MS-MS) [99].
Western blot analysis of bikunin, a CS-proteoglycan that is abundantly found in blood, showed significantly decreased amounts of bikunin-CS and increased levels of GAG-free bikunin in serum from patients with spEDS-B4GALT7 and spEDS-B3GALT6 [100,101].
Although these serum analyses are sensitive enough to detect the respective EDS types, they are, at present, not routinely available in a diagnostic setting. The classic biochemical derangements observed in IEM, such as metabolic acidosis, hyperammonemia, ketosis or hypoglycemia, are absent in EDS.
4.2. Ultrastructural Collagen Fibril Analyses
Fixed skin biopsies of EDS patients can be processed for TEM studies, which allow ultrastructural analysis of collagen fibrils. Whereas dermal collagen fibrils are usually perfectly round on cross-section and tightly packed, a range of alterations can be observed in EDS patients (Figure 3). Abnormally shaped collagen fibrils, such as irregularly contoured fibrils, so-called (cauli)flower-like fibrils, are seen in cEDS, but occasionally also in mcEDS-CHST14, spEDS-B3GALT6 or clEDS2, and fibrils with a hieroglyphic appearance occur in dEDS [10,29,67,102].
In addition, morphometric analysis showed variable fibril diameters in several EDS types and a shift to larger fibril diameters in clEDS type 2 [67]. Disturbed fibril organization with increased interfibrillar spacing is observed in clEDS type 1, mEDS, mcEDS and spEDS. These ultrastructural findings can be indicative of abnormal collagen fibrillogenesis, but are not specific to EDS, and their presence can support but not confirm an EDS diagnosis.
Author Contributions
Writing—Original Draft Preparation, T.V.D., M.C., D.S. and F.M.; Writing—Review and Editing, T.V.D., M.C., D.S. and F.M.; Visualization, M.C. and D.S.; Supervision, F.M.; Funding Acquisition, M.C., D.S. and F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Research Foundation Flanders (FWO), Belgium (11B7921N to M.C., 12Q5920N to D.S. and 1842318N, G041519N and G0A3322N to F.M.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers–Danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L. The structure of the connective tissue, an explanation of the symptoms of the Ehlers-Danlos syndrome. Dermatology 1955, 110, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; de Paepe, A.; Danks, D.; Finidori, G.; Gedde-Dahl, T.; Goodman, R.; Hall, J.G.; Hollister, D.W.; Horton, W.; McKusick, V.A.; et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am. J. Med. Genet. 1988, 29, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, P.; Regier, D. Inborn Errors of Metabolism: From Preconception to Adulthood. Am. Fam. Physician 2019, 99, 25–32. [Google Scholar]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, T.; Colige, A.; Syx, D.; Giunta, C.; Lindert, U.; Rohrbach, M.; Aryani, O.; Alanay, Y.; Simsek-Kiper, P.O.; Kroes, H.Y.; et al. Expanding the clinical and mutational spectrum of the Ehlers-Danlos syndrome, dermatosparaxis type. Genet. Med. 2016, 18, 882–891. [Google Scholar] [CrossRef] [Green Version]
- D’Hondt, S.; Van Damme, T.; Malfait, F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: A systematic review. Genet. Med. 2018, 20, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rymen, D.; Jaeken, J. Skin manifestations in CDG. J. Inherit. Metab. Dis. 2014, 37, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Remvig, L.; Jensen, D.V.; Ward, R.C. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: Review of the literature. J. Rheumatol. 2007, 34, 804–809. [Google Scholar] [PubMed]
- Castiglioni, C.; Feillet, F.; Barnerias, C.; Wiedemann, A.; Muchart, J.; Cortes, F.; Hernando-Davalillo, C.; Montero, R.; Dupré, T.; Bruneel, A.; et al. Expanding the phenotype of X-linked SSR4-CDG: Connective tissue implications. Hum. Mutat. 2021, 42, 142–149. [Google Scholar] [CrossRef]
- Malfait, F.; Colman, M.; Vroman, R.; De Wandele, I.; Rombaut, L.; Miller, R.E.; Malfait, A.M.; Syx, D. Pain in the Ehlers-Danlos syndromes: Mechanisms, models, and challenges. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 429–445. [Google Scholar] [CrossRef]
- Ayoub, S.; Ghali, N.; Angwin, C.; Baker, D.; Baffini, S.; Brady, A.F.; Giovannucci Uzielli, M.L.; Giunta, C.; Johnson, D.S.; Kosho, T.; et al. Clinical features, molecular results, and management of 12 individuals with the rare arthrochalasia Ehlers-Danlos syndrome. Am. J. Med. Genet. A 2020, 182, 994–1007. [Google Scholar] [CrossRef]
- Van Damme, T.; Pang, X.; Guillemyn, B.; Gulberti, S.; Syx, D.; De Rycke, R.; Kaye, O.; de Die-Smulders, C.E.M.; Pfundt, R.; Kariminejad, A.; et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers-Danlos syndrome. Hum. Mol. Genet. 2018, 27, 3475–3487. [Google Scholar] [CrossRef] [Green Version]
- Hicks, D.; Farsani, G.T.; Laval, S.; Collins, J.; Sarkozy, A.; Martoni, E.; Shah, A.; Zou, Y.; Koch, M.; Bönnemann, C.G.; et al. Mutations in the collagen XII gene define a new form of extracellular matrix-related myopathy. Hum. Mol. Genet. 2014, 23, 2353–2363. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zwolanek, D.; Izu, Y.; Gandhy, S.; Schreiber, G.; Brockmann, K.; Devoto, M.; Tian, Z.; Hu, Y.; Veit, G.; et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum. Mol. Genet. 2014, 23, 2339–2352. [Google Scholar] [CrossRef] [Green Version]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voermans, N.C.; Kempers, M.; Lammens, M.; van Alfen, N.; Janssen, M.C.; Bonnemann, C.; van Engelen, B.G.; Hamel, B.C. Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am. J. Med. Genet. A 2012, 158, 850–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.M.; Bal-Theoleyre, L.; Fauret, A.L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. EJHG 2015, 23, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, M.R.; Lee, C.L.; Lin, S.M.; Hung, C.L.; Niu, D.M.; Chang, T.M.; Chuang, C.K.; Lin, S.P. Aortic Root Dilatation in Taiwanese Patients with Mucopolysaccharidoses and the Long-Term Effects of Enzyme Replacement Therapy. Diagnostics 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Symoens, S.; Coucke, P.; Nunes, L.; De Almeida, S.; De Paepe, A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J. Med. Genet. 2006, 43, e36. [Google Scholar] [CrossRef] [PubMed]
- Dhooge, T.; Van Damme, T.; Syx, D.; Mosquera, L.M.; Nampoothiri, S.; Radhakrishnan, A.; Simsek-Kiper, P.O.; Utine, G.E.; Bonduelle, M.; Migeotte, I.; et al. More than meets the eye: Expanding and reviewing the clinical and mutational spectrum of brittle cornea syndrome. Hum. Mutat. 2021, 42, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Kapferer-Seebacher, I.; Pepin, M.; Werner, R.; Aitman, T.J.; Nordgren, A.; Stoiber, H.; Thielens, N.; Gaboriaud, C.; Amberger, A.; Schossig, A.; et al. Periodontal Ehlers-Danlos Syndrome Is Caused by Mutations in C1R and C1S, which Encode Subcomponents C1r and C1s of Complement. Am. J. Hum. Genet. 2016, 99, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Syx, D.; Van Damme, T.; Symoens, S.; Maiburg, M.C.; van de Laar, I.; Morton, J.; Suri, M.; Del Campo, M.; Hausser, I.; Hermanns-Lê, T.; et al. Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 2015, 36, 535–547. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Birk, D.E.; Brückner, P. Collagens, Suprastructures, and Collagen Fibril Assembly. In The Extracellular Matrix: An Overview; Springer: Berlin/Heidelberg, Germany, 2011; pp. 77–115. [Google Scholar]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Birk, D.E. Type V collagen: Heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 2001, 32, 223–237. [Google Scholar] [CrossRef]
- Canty, E.G.; Kadler, K.E. Procollagen trafficking, processing and fibrillogenesis. J. Cell Sci. 2005, 118, 1341–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenspan, D.S. Biosynthetic processing of collagen molecules. Collagen 2005, 247, 149–183. [Google Scholar]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef] [Green Version]
- Zoppi, N.; Gardella, R.; De Paepe, A.; Barlati, S.; Colombi, M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate alpha2beta1 integrin, and recruit alphavbeta3 Instead of alpha5beta1 integrin. J. Biol. Chem. 2004, 279, 18157–18168. [Google Scholar] [CrossRef] [Green Version]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS ONE 2019, 14, e0211647. [Google Scholar] [CrossRef] [Green Version]
- Keene, D.R.; Sakai, L.Y.; Bächinger, H.P.; Burgeson, R.E. Type III collagen can be present on banded collagen fibrils regardless of fibril diameter. J. Cell Biol. 1987, 105, 2393–2402. [Google Scholar] [CrossRef]
- Romanic, A.M.; Adachi, E.; Kadler, K.E.; Hojima, Y.; Prockop, D.J. Copolymerization of pNcollagen III and collagen I. pNcollagen III decreases the rate of incorporation of collagen I into fibrils, the amount of collagen I incorporated, and the diameter of the fibrils formed. J. Biol. Chem. 1991, 266, 12703–12709. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS ONE 2018, 13, e0191220. [Google Scholar] [CrossRef] [Green Version]
- Crowther, M.A.; Lach, B.; Dunmore, P.J.; Roach, M.R. Vascular collagen fibril morphology in type IV Ehlers-Danlos syndrome. Connect Tissue Res. 1991, 25, 209–217. [Google Scholar] [CrossRef]
- Byers, P.H.; Duvic, M.; Atkinson, M.; Robinow, M.; Smith, L.T.; Krane, S.M.; Greally, M.T.; Ludman, M.; Matalon, R.; Pauker, S.; et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 1997, 72, 94–105. [Google Scholar] [CrossRef]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Bekhouche, M.; Leduc, C.; Dupont, L.; Janssen, L.; Delolme, F.; Vadon-Le Goff, S.; Smargiasso, N.; Baiwir, D.; Mazzucchelli, G.; Zanella-Cleon, I.; et al. Determination of the substrate repertoire of ADAMTS2, 3, and 14 significantly broadens their functions and identifies extracellular matrix organization and TGF-β signaling as primary targets. FASEB J. 2016, 30, 1741–1756. [Google Scholar] [CrossRef] [Green Version]
- Giunta, C.; Chambaz, C.; Pedemonte, M.; Scapolan, S.; Steinmann, B. The arthrochalasia type of Ehlers-Danlos syndrome (EDS VIIA and VIIB): The diagnostic value of collagen fibril ultrastructure. Am. J. Med. Genet. A 2008, 146, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Symoens, S.; De Backer, J.; Hermanns-Le, T.; Sakalihasan, N.; Lapiere, C.M.; Coucke, P.; De Paepe, A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum. Mutat. 2007, 28, 387–395. [Google Scholar] [CrossRef]
- Nuytinck, L.; Freund, M.; Lagae, L.; Pierard, G.E.; Hermanns-Le, T.; De Paepe, A. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am. J. Hum. Genet. 2000, 66, 1398–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krane, S.M.; Pinnell, S.R.; Erbe, R.W. Lysyl-protocollagen hydroxylase deficiency in fibroblasts from siblings with hydroxylysine-deficient collagen. Proc. Natl. Acad. Sci. USA 1972, 69, 2899–2903. [Google Scholar] [CrossRef] [Green Version]
- Pinnell, S.R.; Krane, S.M.; Kenzora, J.E.; Glimcher, M.J. A heritable disorder of connective tissue: Hydroxylysine-deficient collagen disease. N. Engl. J. Med. 1972, 286, 1013–1020. [Google Scholar] [CrossRef]
- Hyland, J.; Ala-Kokko, L.; Royce, P.; Steinmann, B.; Kivirikko, K.I.; Myllylä, R. A homozygous stop codon in the lysyl hydroxylase gene in two siblings with Ehlers–Danlos syndrome type VI. Nat. Genet. 1992, 2, 228–231. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Bachinger, H.P. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J. Biol. Chem. 2014, 289, 18189–18201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Mizuno, K.; Bachinger, H.P. Ziploc-ing the structure 2.0: Endoplasmic reticulum-resident peptidyl prolyl isomerases show different activities toward hydroxyproline. J. Biol. Chem. 2017, 292, 9273–9282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Mizuno, N.; Holden, P.; Lim, P.J.; Gould, D.B.; Rohrbach, M.; Giunta, C.; Bächinger, H.P. The novel missense mutation Met48Lys in FKBP22 changes its structure and functions. Sci. Rep. 2020, 10, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.; Giunta, C.; Krabichler, B.; Ruschendorf, F.; Zoppi, N.; Colombi, M.; Bittner, R.E.; Quijano-Roy, S.; Muntoni, F.; Cirak, S.; et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 2012, 90, 201–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, P.J.; Lindert, U.; Opitz, L.; Hausser, I.; Rohrbach, M.; Giunta, C. Transcriptome Profiling of Primary Skin Fibroblasts Reveal Distinct Molecular Features Between PLOD1- and FKBP14-Kyphoscoliotic Ehlers-Danlos Syndrome. Genes 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, M.L.; Khattab, A. Genetics of congenital adrenal hyperplasia and genotype-phenotype correlation. Fertil. Steril. 2019, 111, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Hansen, U.; Keene, D.R.; Bruckner, P.; Chiquet-Ehrismann, R.; Chiquet, M.; Koch, M. Collagen XII interacts with avian tenascin-X through its NC3 domain. J. Biol. Chem. 2006, 281, 27461–27470. [Google Scholar] [CrossRef] [Green Version]
- Bristow, J.; Carey, W.; Egging, D.; Schalkwijk, J. Tenascin-X, Collagen, Elastin, and the Ehlers–Danlos Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2005, 139C, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbaere, S.; Dhooge, T.; Syx, D.; Petit, F.; Goemans, N.; Destrée, A.; Vanakker, O.; De Rycke, R.; Symoens, S.; Malfait, F. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet. Med. 2020, 22, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ith, B.; Wei, J.; Yet, S.F.; Perrella, M.A.; Layne, M.D. Aortic carboxypeptidase-like protein is expressed in collagen-rich tissues during mouse embryonic development. Gene Expr. Patterns 2005, 5, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Layne, M.D.; Yet, S.F.; Maemura, K.; Hsieh, C.M.; Bernfield, M.; Perrella, M.A.; Lee, M.E. Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell. Biol. 2001, 21, 5256–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 28, 1853–1864. [Google Scholar] [CrossRef]
- Gagnon, A.; Landry, A.; Proulx, J.; Layne, M.D.; Sorisky, A. Aortic carboxypeptidase-like protein is regulated by transforming growth factor beta in 3T3-L1 preadipocytes. Exp. Cell Res. 2005, 308, 265–272. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Furuhashi, H.; Irie, R.; Nishikawa, M.; Yamamoto, J.; Hibi, T.; Miura, S.; Minamino, T.; et al. Aortic carboxypeptidase-like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 128, 1581–1596. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Couchman, J.R.; Pataki, C.A. An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 2012, 60, 885–897. [Google Scholar] [CrossRef]
- Prydz, K.; Dalen, K.T. Synthesis and sorting of proteoglycans. J. Cell Sci. 2000, 113 Pt 2, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Aguirre-Negrete, M.G.; González-Flores, S.; Reynoso-Luna, M.C.; Fragoso, R.; Nazará, Z.; Tapia-Arizmendi, G.; Cantú, J.M. Ehlers-Danlos features with progeroid facies and mild mental retardation. Further delineation of the syndrome. Clin. Genet. 1986, 30, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Aguirre-Negrete, M.G.; Liparoli, J.C.; Cantú, J.M. Third case of a distinct variant of the Ehlers-Danlos Syndrome (EDS). Clin. Genet. 1981, 20, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Aguirre-Negrete, M.; Ramirez-Soltero, S.; González-Mendoza, A.; Martínez, R.M.; Velázquez-Cabrera, A.; Cantu, J. A distinct variant of the Ehlers-Danlos syndrome. Clin. Genet. 1979, 16, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Rosthøj, S.; Quentin, E.; Hollmann, J.; Glössl, J.; Okada, S.; Tønnesen, T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am. J. Hum. Genet. 1987, 41, 436–453. [Google Scholar] [PubMed]
- Quentin, E.; Gladen, A.; Rodén, L.; Kresse, H. A genetic defect in the biosynthesis of dermatan sulfate proteoglycan: Galactosyltransferase I deficiency in fibroblasts from a patient with a progeroid syndrome. Proc. Natl. Acad. Sci. USA 1990, 87, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Fukumoto, S.; Furukawa, K.; Urano, T. Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J. Biol. Chem. 1999, 274, 28841–28844. [Google Scholar] [CrossRef] [Green Version]
- Delbaere, S.; De Clercq, A.; Mizumoto, S.; Noborn, F.; Bek, J.W.; Alluyn, L.; Gistelinck, C.; Syx, D.; Salmon, P.L.; Coucke, P.J.; et al. b3galt6 Knock-Out Zebrafish Recapitulate β3GalT6-Deficiency Disorders in Human and Reveal a Trisaccharide Proteoglycan Linkage Region. Front. Cell Dev. Biol. 2020, 8, 597857. [Google Scholar] [CrossRef]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. A 2010, 152, 1333–1346. [Google Scholar] [CrossRef]
- Kosho, T.; Takahashi, J.; Ohashi, H.; Nishimura, G.; Kato, H.; Fukushima, Y. Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A 2005, 138, 282–287. [Google Scholar] [CrossRef]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Lê, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Hatamochi, A.; Honda, T.; Yamaguchi, T.; Okamoto, N.; Miyake, N.; Yamada, S.; Sugahara, K. Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 2017, 50, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome--an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Walker, J.M.; Wang, F.; Park, J.G.; Palmer, A.E.; Giunta, C.; Rohrbach, M.; Steinmann, B.; Eide, D.J. Promotion of vesicular zinc efflux by ZIP13 and its implications for spondylocheiro dysplastic Ehlers-Danlos syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, E3530–E3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Abu, A.; Frydman, M.; Marek, D.; Pras, E.; Nir, U.; Reznik-Wolf, H.; Pras, E. Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am. J. Hum. Genet. 2008, 82, 1217–1222. [Google Scholar] [CrossRef] [Green Version]
- Burkitt Wright, E.M.M.; Spencer, H.L.; Daly, S.B.; Manson, F.D.C.; Zeef, L.A.H.; Urquhart, J.; Zoppi, N.; Bonshek, R.; Tosounidis, I.; Mohan, M.; et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am. J. Hum. Genet. 2011, 88, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.K.; Gohil, K.; Packer, L.; Burk, R.F. Selenium deficiency, endurance exercise capacity, and antioxidant status in rats. J. Appl. Physiol. 1987, 63, 2532–2535. [Google Scholar] [CrossRef]
- Meani, N.; Pezzimenti, F.; Deflorian, G.; Mione, M.; Alcalay, M. The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS ONE 2009, 4, e4273. [Google Scholar] [CrossRef] [Green Version]
- Porter, L.F.; Gallego-Pinazo, R.; Keeling, C.L.; Kamieniorz, M.; Zoppi, N.; Colombi, M.; Giunta, C.; Bonshek, R.; Manson, F.D.; Black, G.C. Bruch’s membrane abnormalities in PRDM5-related brittle cornea syndrome. Orphanet J. Rare Dis. 2015, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbach, M.; Spencer, H.L.; Porter, L.F.; Burkitt-Wright, E.M.; Bürer, C.; Janecke, A.; Bakshi, M.; Sillence, D.; Al-Hussain, H.; Baumgartner, M.; et al. ZNF469 frequently mutated in the brittle cornea syndrome (BCS) is a single exon gene possibly regulating the expression of several extracellular matrix components. Mol. Genet. Metab. 2013, 109, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, R.; Kapferer-Seebacher, I.; Amberger, A.; Redolfi, R.; Dalonneau, F.; Björck, E.; Milnes, D.; Bally, I.; Rossi, V.; Thielens, N.; et al. C1R Mutations Trigger Constitutive Complement 1 Activation in Periodontal Ehlers-Danlos Syndrome. Front. Immunol. 2019, 10, 2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bork, P.; Beckmann, G. The CUB domain. A widespread module in developmentally regulated proteins. J. Mol. Biol. 1993, 231, 539–545. [Google Scholar] [CrossRef]
- Malfait, F.; Coucke, P.; Symoens, S.; Loeys, B.; Nuytinck, L.; De Paepe, A. The molecular basis of classic Ehlers-Danlos syndrome: A comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 2005, 25, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, M.; Vandersteen, A.; Yiş, U.; Serdaroglu, G.; Ataman, E.; Chopra, M.; Garcia, S.; Jones, K.; Kariminejad, A.; Kraenzlin, M.; et al. Phenotypic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): Clinical, molecular and biochemical delineation. Orphanet J. Rare Dis. 2011, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Lautrup, C.K.; Teik, K.W.; Unzaki, A.; Mizumoto, S.; Syx, D.; Sin, H.H.; Nielsen, I.K.; Markholt, S.; Yamada, S.; Malfait, F.; et al. Delineation of musculocontractural Ehlers-Danlos Syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1197. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Watanabe, A.; Takeshita, H.; Matsumoto, K.I. A method for quantification of serum tenascin-X by nano-LC/MS/MS. Clin. Chim. Acta 2016, 459, 94–100. [Google Scholar] [CrossRef]
- Bruneel, A.; Dubail, J.; Roseau, C.; Prada, P.; Haouari, W.; Huber, C.; Dupré, T.; Poüs, C.; Cormier-Daire, V.; Seta, N. Serum bikunin is a biomarker of linkeropathies. Clin. Chim. Acta 2018, 485, 178–180. [Google Scholar] [CrossRef]
- Haouari, W.; Dubail, J.; Lounis-Ouaras, S.; Prada, P.; Bennani, R.; Roseau, C.; Huber, C.; Afenjar, A.; Colin, E.; Vuillaumier-Barrot, S.; et al. Serum bikunin isoforms in congenital disorders of glycosylation and linkeropathies. J. Inherit. Metab. Dis. 2020, 43, 1349–1359. [Google Scholar] [CrossRef]
- Vogel, A.; Holbrook, K.A.; Steinmann, B.; Gitzelmann, R.; Byers, P.H. Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab. Investig. 1979, 40, 201–206. [Google Scholar] [PubMed]
Figure 1.
Clinical characteristics of different EDS types. (A–E): Individuals with classical Ehlers–Danlos syndrome with pathogenic variants in COL5A1 presenting epicanthal folds (A,C), a somewhat flattened facial appearance (B), skin hyperextensibility (D) and atrophic scars and molluscoid pseudotumors (E). (F–I): Individuals with brittle cornea syndrome in whom pathogenic defects in ZNF469 were identified. Clinical characteristics include hypertelorism, downslanting palpebral fissures, (variable) blue sclerae (F–H), synophrys (H) and deformities of the feet (I). (J–L): Individuals with musculocontractural Ehlers–Danlos syndrome with pathogenic variants in CHST14 presenting atrophic scars (K); facial features including flattened profile, malar hypoplasia, downslanting palpebral fissures, blue sclerae, long philtrum with thin upper lip and protruding jaw with pointed chin (J); and characteristic hand deformities (L). (M): Individual with dermatosparaxis Ehlers–Danlos syndrome and characteristic facial appearance including downslanting palpebral fissures, mild telecanthus, palpebral edema, epicanthic folds, blue sclerae, low-set and floppy ears, saggy cheeks and prominent lips. (N–S): Individuals with spondylodysplastic Ehlers–Danlos syndrome with pathogenic variants in B3GALT6 presenting flexion contractures (N–S), short and deformed extremities (N–S), muscle atrophy (S), severe kyphoscoliosis with pectus deformities (N–Q) and hyperextensible skin (R). Facial features include midfacial hypoplasia with frontal bossing, blue sclerae, downslanting of the palpebral fissures, a short nose with anteverted nares, a long philtrum and low-set ears (N,P).
Figure 1.
Clinical characteristics of different EDS types. (A–E): Individuals with classical Ehlers–Danlos syndrome with pathogenic variants in COL5A1 presenting epicanthal folds (A,C), a somewhat flattened facial appearance (B), skin hyperextensibility (D) and atrophic scars and molluscoid pseudotumors (E). (F–I): Individuals with brittle cornea syndrome in whom pathogenic defects in ZNF469 were identified. Clinical characteristics include hypertelorism, downslanting palpebral fissures, (variable) blue sclerae (F–H), synophrys (H) and deformities of the feet (I). (J–L): Individuals with musculocontractural Ehlers–Danlos syndrome with pathogenic variants in CHST14 presenting atrophic scars (K); facial features including flattened profile, malar hypoplasia, downslanting palpebral fissures, blue sclerae, long philtrum with thin upper lip and protruding jaw with pointed chin (J); and characteristic hand deformities (L). (M): Individual with dermatosparaxis Ehlers–Danlos syndrome and characteristic facial appearance including downslanting palpebral fissures, mild telecanthus, palpebral edema, epicanthic folds, blue sclerae, low-set and floppy ears, saggy cheeks and prominent lips. (N–S): Individuals with spondylodysplastic Ehlers–Danlos syndrome with pathogenic variants in B3GALT6 presenting flexion contractures (N–S), short and deformed extremities (N–S), muscle atrophy (S), severe kyphoscoliosis with pectus deformities (N–Q) and hyperextensible skin (R). Facial features include midfacial hypoplasia with frontal bossing, blue sclerae, downslanting of the palpebral fissures, a short nose with anteverted nares, a long philtrum and low-set ears (N,P).

Figure 2.
Collagen and proteoglycan biosynthesis in the context of EDS. Defective molecules associated with EDS are indicated in bold, and the respective EDS type is indicated. Fibrillar collagen biosynthesis is initiated by transcription and translation of pro-α-chains (step 1). Nascent pro-α-chains are co- and post-translationally modified by several modifying enzymes in the endoplasmic reticulum (ER), such as proline and lysine hydroxylases and galactosyltransferases (step 2). Triple helix formation starts by the association of the C-terminal propeptides of three pro-α-chains and propagates towards the N-terminus in a zipperlike fashion during which several molecular chaperones assist (step 3). Trimeric procollagen molecules aggregate laterally, are transported in secretory vesicles and are secreted into the extracellular environment (step 4). Collagen molecules are formed by removal of the N- and C-propeptides by ADAMTS-2 and BMP-1/mTLD, respectively (step 5). These collagens subsequently assemble into highly ordered striated fibrils. The assembly of collagen fibrils is tissue-specific and requires several assisting proteins (step 6). Fibronectin and integrins serve as organizers of fibril assembly at the plasma membrane. At the cell surface, some collagens, including type V collagen, function as nucleators and initiate immature fibril assembly. Type V collagen co-assembles with type I collagen to form heterotypic fibrils with the entire triple helical domain of type V collagen embedded within the fibril. The partially processed N-propeptide domain of type V collagen protrudes to the fibril surface where it controls fibrillogenesis by sterically hindering the addition of collagen monomers. Intermediate fibrils are deposited into the ECM and stabilized by interactions with regulators, such as the small leucine-rich proteoglycan (SLRP) decorin, tenascin-X and type XIII collagen. These molecules influence the rate of assembly, size and structure of the collagen fibrils. Subsequent fibril growth occurs through linear and lateral fusion of intermediate collagen fibrils, which are stabilized by intra- and inter-molecular crosslinks. Proteoglycan biosynthesis is initiated by the synthesis of a core protein, which is then modified by several Golgi-resident enzymes. First, a common linker region in formed by the addition of four monosaccharides. Formation of this tetrasaccharide linker region begins with the stepwise addition of a xylose (Xyl) residue to a serine residue of the core protein, catalyzed by xylosyltransferase-I and -II (XylT-I/-II). Subsequently, two galactose (Gal) residues are added by galactosyltransferase-I (GalT-I or β4GalT7) and galactosyltransferase-II (GalT-II or β3GalT6). Finally, the addition of a glucuronic acid (GlcA), catalyzed by glucuronosyltransferase-I (GlcAT-I) completes the formation of the linker region. The alternating addition of either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) and GlcA to the nascent GAG-chain result in the formation of heparan sulfate (HS) proteoglycans and chondroitin sulfate (CS)/dermatan sulfate (DS) proteoglycans. The GAG-chains are further modified by epimerization and sulfation reactions. The epimerization of GlcA towards iduronic acid (IdoA), which is catalyzed by DS epimerases-1 and -2 (DS-epi1 and DS-epi2) is necessary for the formation of DS. Subsequently, dermatan 4-O-sulfotransferase 1 (D4ST1) is able to catalyze 4-O-sulfation of GalNAc, which prevents back-epimerization of the adjacent IdoA.
Figure 2.
Collagen and proteoglycan biosynthesis in the context of EDS. Defective molecules associated with EDS are indicated in bold, and the respective EDS type is indicated. Fibrillar collagen biosynthesis is initiated by transcription and translation of pro-α-chains (step 1). Nascent pro-α-chains are co- and post-translationally modified by several modifying enzymes in the endoplasmic reticulum (ER), such as proline and lysine hydroxylases and galactosyltransferases (step 2). Triple helix formation starts by the association of the C-terminal propeptides of three pro-α-chains and propagates towards the N-terminus in a zipperlike fashion during which several molecular chaperones assist (step 3). Trimeric procollagen molecules aggregate laterally, are transported in secretory vesicles and are secreted into the extracellular environment (step 4). Collagen molecules are formed by removal of the N- and C-propeptides by ADAMTS-2 and BMP-1/mTLD, respectively (step 5). These collagens subsequently assemble into highly ordered striated fibrils. The assembly of collagen fibrils is tissue-specific and requires several assisting proteins (step 6). Fibronectin and integrins serve as organizers of fibril assembly at the plasma membrane. At the cell surface, some collagens, including type V collagen, function as nucleators and initiate immature fibril assembly. Type V collagen co-assembles with type I collagen to form heterotypic fibrils with the entire triple helical domain of type V collagen embedded within the fibril. The partially processed N-propeptide domain of type V collagen protrudes to the fibril surface where it controls fibrillogenesis by sterically hindering the addition of collagen monomers. Intermediate fibrils are deposited into the ECM and stabilized by interactions with regulators, such as the small leucine-rich proteoglycan (SLRP) decorin, tenascin-X and type XIII collagen. These molecules influence the rate of assembly, size and structure of the collagen fibrils. Subsequent fibril growth occurs through linear and lateral fusion of intermediate collagen fibrils, which are stabilized by intra- and inter-molecular crosslinks. Proteoglycan biosynthesis is initiated by the synthesis of a core protein, which is then modified by several Golgi-resident enzymes. First, a common linker region in formed by the addition of four monosaccharides. Formation of this tetrasaccharide linker region begins with the stepwise addition of a xylose (Xyl) residue to a serine residue of the core protein, catalyzed by xylosyltransferase-I and -II (XylT-I/-II). Subsequently, two galactose (Gal) residues are added by galactosyltransferase-I (GalT-I or β4GalT7) and galactosyltransferase-II (GalT-II or β3GalT6). Finally, the addition of a glucuronic acid (GlcA), catalyzed by glucuronosyltransferase-I (GlcAT-I) completes the formation of the linker region. The alternating addition of either N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) and GlcA to the nascent GAG-chain result in the formation of heparan sulfate (HS) proteoglycans and chondroitin sulfate (CS)/dermatan sulfate (DS) proteoglycans. The GAG-chains are further modified by epimerization and sulfation reactions. The epimerization of GlcA towards iduronic acid (IdoA), which is catalyzed by DS epimerases-1 and -2 (DS-epi1 and DS-epi2) is necessary for the formation of DS. Subsequently, dermatan 4-O-sulfotransferase 1 (D4ST1) is able to catalyze 4-O-sulfation of GalNAc, which prevents back-epimerization of the adjacent IdoA.

Figure 3.
Ultrastructural findings of dermal collagen fibrils in different EDS types. (A) Control showing tightly packed collagen fibrils with uniform diameters. (B) Presence of some very large fibril diameters with irregular contours, called ‘collagen cauliflowers’ in classical EDS (cEDS) due to a heterozygous pathogenic variant in COL5A2. (C) Hieroglyphic aspect of the collagen fibrils in dermatosparaxis EDS (dEDS) due to biallelic pathogenic variants in ADAMTS2. (D) Increased interfibrillar spacing myopathic EDS (mEDS) due to a heterozygous pathogenic variant in COL12A1. (E) Collagen fibrils with large and small collagen fibril diameters and irregular contours in classical EDS-like type 2 (clEDS2) due to biallelic pathogenic variants in AEBP1. (F) Dispersed collagen fibrils with variable collagen fibril diameters, sporadic fibrils with very irregular contours and granulofilamentous deposits between collagen fibrils in spondylodysplastic EDS (spEDS) due to biallelic pathogenic variants in B3GALT6. (G) Collagen fibrils with variable diameters and the intermittent presence of small flower-like fibrils and irregular interfibrillar spaces filled with granulofilamentous deposits in musculocontractural EDS (mcEDS) due to biallelic pathogenic variants in CHST14. (H) Grossly normal collagen fibril architecture with mildly increased interfibrillar spacing in musculocontractural EDS (mcEDS) due to biallelic pathogenic variants in DSE. Scale bars: 500 nm.
Figure 3.
Ultrastructural findings of dermal collagen fibrils in different EDS types. (A) Control showing tightly packed collagen fibrils with uniform diameters. (B) Presence of some very large fibril diameters with irregular contours, called ‘collagen cauliflowers’ in classical EDS (cEDS) due to a heterozygous pathogenic variant in COL5A2. (C) Hieroglyphic aspect of the collagen fibrils in dermatosparaxis EDS (dEDS) due to biallelic pathogenic variants in ADAMTS2. (D) Increased interfibrillar spacing myopathic EDS (mEDS) due to a heterozygous pathogenic variant in COL12A1. (E) Collagen fibrils with large and small collagen fibril diameters and irregular contours in classical EDS-like type 2 (clEDS2) due to biallelic pathogenic variants in AEBP1. (F) Dispersed collagen fibrils with variable collagen fibril diameters, sporadic fibrils with very irregular contours and granulofilamentous deposits between collagen fibrils in spondylodysplastic EDS (spEDS) due to biallelic pathogenic variants in B3GALT6. (G) Collagen fibrils with variable diameters and the intermittent presence of small flower-like fibrils and irregular interfibrillar spaces filled with granulofilamentous deposits in musculocontractural EDS (mcEDS) due to biallelic pathogenic variants in CHST14. (H) Grossly normal collagen fibril architecture with mildly increased interfibrillar spacing in musculocontractural EDS (mcEDS) due to biallelic pathogenic variants in DSE. Scale bars: 500 nm.

Figure 4.
Illustration of representative (pro)collagen electrophoretic mobility patterns for different EDS types. Metabolically labelled (pro)collagen chains isolated from cells and conditioned medium of dermal fibroblast cultures were either partially digested with pepsin (cell and medium fraction) or left untreated (procollagen) prior to SDS-PAGE. All individual intermediate and mature (pro)collagen chains are indicated on the left side of panel A as a reference. At the right side of the gels, (pro)collagen chains displaying a difference are highlighted. pNα- and pCα-chain denote pro-α-chains that contain only the N- or C-terminal propeptide, respectively. C: control and P: patient. (A) Severely reduced amounts of the type III (pro)collagen homotrimer in the cell and medium fractions in a vascular EDS (vEDS) patient. (B) The presence of additional mutant pNα1(I)-chains, in the cell and medium fractions of patients with arthrochalasia EDS (aEDS) caused by a defect in COL1A1. (C) The presence of additional mutant pNα2(I)-chains, in the cell and medium fractions of patients with arthrochalasia EDS (aEDS) caused by a defect in COL1A2. (D) Accumulation of procollagen chains with a retained N-propeptide (pNα1(I) and pNα2(I)) and nearly complete absence of bands representing the pCα1(I) and pCα2(I) procollagen chains in a patient with dermatosparaxis EDS (dEDS) due to biallelic ADAMTS2 defects. Of note, a normal electrophoretic mobility of the collagen chains is seen in the cell and medium fractions because propeptides are enzymatically removed with pepsin during sample preparation. (E) Complete absence of the α2(I) procollagen chains in cell and medium fractions in a patient with cardiac-valvular EDS (cvEDS). (F) Abnormal disulfide-bonded α1(I) dimers are present in the cell layer but not in the medium fraction of a patient with a pathogenic variant in the COL1A1 gene leading to an arginine-to-cysteine substitution in the α1(I)-chain (c.934C>T, p.(Arg312Cys)). (G) Type I, III and V (pro)collagen chains from a kyphoscoliotic EDS (kEDS) patient with biallelic PLOD1 mutations show a uniformly faster migration in both cell and medium fractions and on procollagen gels, thereby, demonstrating underhydroxylation and underglycosylation of lysyl residues.
Figure 4.
Illustration of representative (pro)collagen electrophoretic mobility patterns for different EDS types. Metabolically labelled (pro)collagen chains isolated from cells and conditioned medium of dermal fibroblast cultures were either partially digested with pepsin (cell and medium fraction) or left untreated (procollagen) prior to SDS-PAGE. All individual intermediate and mature (pro)collagen chains are indicated on the left side of panel A as a reference. At the right side of the gels, (pro)collagen chains displaying a difference are highlighted. pNα- and pCα-chain denote pro-α-chains that contain only the N- or C-terminal propeptide, respectively. C: control and P: patient. (A) Severely reduced amounts of the type III (pro)collagen homotrimer in the cell and medium fractions in a vascular EDS (vEDS) patient. (B) The presence of additional mutant pNα1(I)-chains, in the cell and medium fractions of patients with arthrochalasia EDS (aEDS) caused by a defect in COL1A1. (C) The presence of additional mutant pNα2(I)-chains, in the cell and medium fractions of patients with arthrochalasia EDS (aEDS) caused by a defect in COL1A2. (D) Accumulation of procollagen chains with a retained N-propeptide (pNα1(I) and pNα2(I)) and nearly complete absence of bands representing the pCα1(I) and pCα2(I) procollagen chains in a patient with dermatosparaxis EDS (dEDS) due to biallelic ADAMTS2 defects. Of note, a normal electrophoretic mobility of the collagen chains is seen in the cell and medium fractions because propeptides are enzymatically removed with pepsin during sample preparation. (E) Complete absence of the α2(I) procollagen chains in cell and medium fractions in a patient with cardiac-valvular EDS (cvEDS). (F) Abnormal disulfide-bonded α1(I) dimers are present in the cell layer but not in the medium fraction of a patient with a pathogenic variant in the COL1A1 gene leading to an arginine-to-cysteine substitution in the α1(I)-chain (c.934C>T, p.(Arg312Cys)). (G) Type I, III and V (pro)collagen chains from a kyphoscoliotic EDS (kEDS) patient with biallelic PLOD1 mutations show a uniformly faster migration in both cell and medium fractions and on procollagen gels, thereby, demonstrating underhydroxylation and underglycosylation of lysyl residues.


{kind=link}
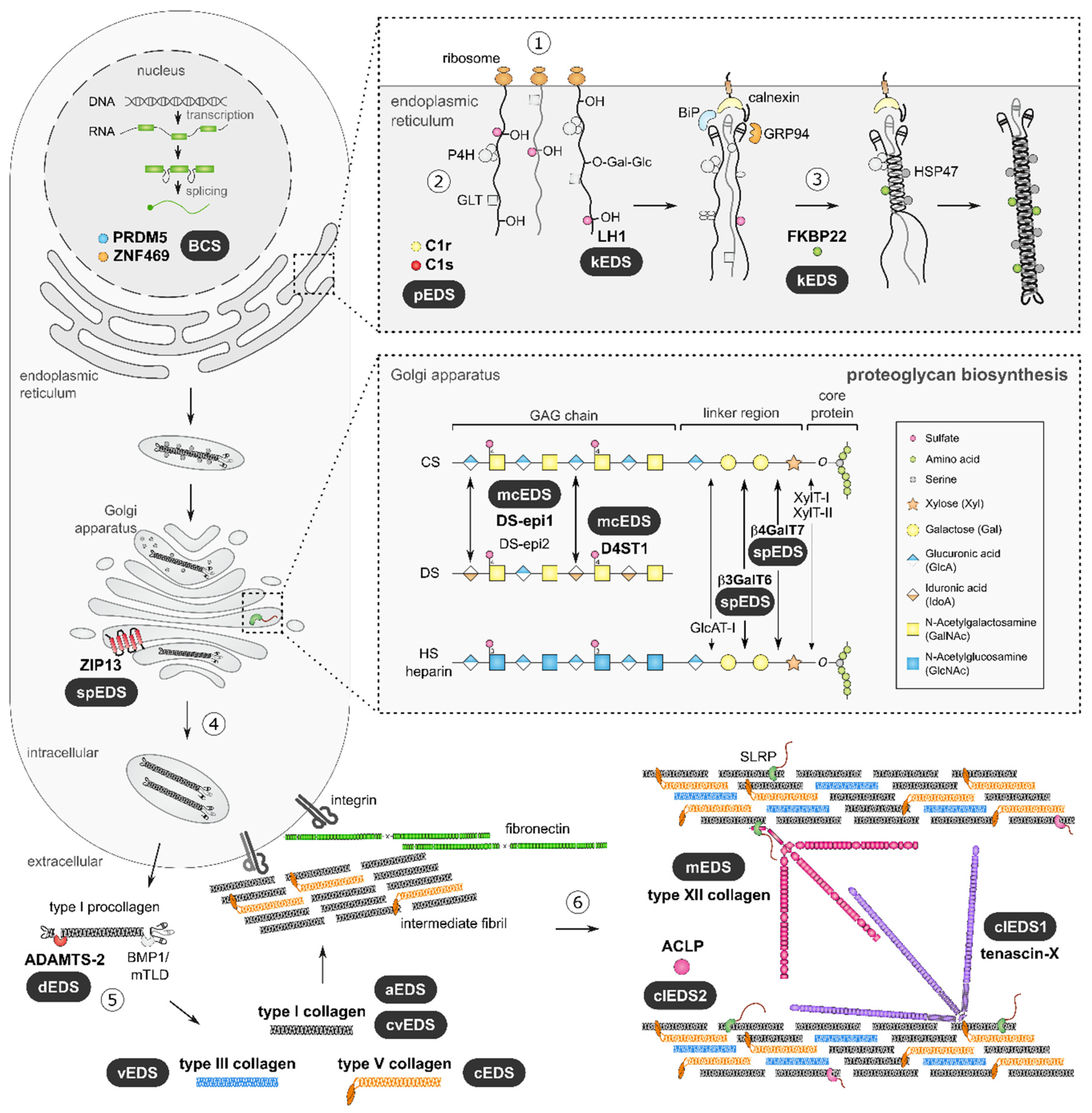{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of the EDS types, genes and proteins, and inheritance pattern (IP) as defined by the 2017 International EDS Classification with indication of the estimated prevalence, the associated pathophysiological mechanisms and available biochemical tests. AD: autosomal dominant, AR: autosomal recessive, NA: not available, ?: unknown.
Table 1.
Overview of the EDS types, genes and proteins, and inheritance pattern (IP) as defined by the 2017 International EDS Classification with indication of the estimated prevalence, the associated pathophysiological mechanisms and available biochemical tests. AD: autosomal dominant, AR: autosomal recessive, NA: not available, ?: unknown.
| EDS Type | Gene | Protein | IP | Estimated Incidence/Reported Individuals | Pathophysiological Mechanism | Biochemical Testing |
|---|---|---|---|---|---|---|
| Defects in Collagen Structure and Collagen Processing | ||||||
| Classical (cEDS) | COL5A1 COL5A2 (COL1A1 p.(Arg312Cys)) | α1-chain of type V procollagen α2-chain of type V procollagen α1-chain of type I procollagen | AD | 1:20,000 | Decreased type V collagen amounts affecting the initiation and assembly of heterotypic type I/V collagen fibrils Decreased type V collagen amounts affecting the initiation and assembly of heterotypic type I/V collagen fibrils Local destabilization of the type I collagen molecules and production of α1(I) dimers | (Pro)collagen biochemistry |
| Vascular (vEDS) | COL3A1 (COL1A1 p.(Arg312Cys), p.(Arg574Cys), p.(Arg1093Cys)) | α1-chain of type III procollagen α1-chain of type I procollagen | AD | 1:50,000–1:200,000 | Decreased type III collagen amounts affecting the initiation and assembly of heterotypic type I/III collagen fibrils Local destabilization of the type I collagen molecules and production of α1(I) dimers | (Pro)collagen biochemistry (Pro)collagen biochemistry |
| Arthrochalasia (aEDS) | COL1A1 COL1A2 | α1-chain of type I procollagen α2-chain of type I procollagen | AD | <60 reported individuals | (Partial or complete) deletion of exon 6 leading to partial processing of type I procollagen with retention of the N-propeptide of either the pro-α1(I)- or the pro-α2(I)-chain | (Pro)collagen biochemistry (Pro)collagen biochemistry |
| Dermatosparaxis (dEDS) | ADAMTS2 | A Disintegrin And Metalloproteinase with Thrombospondin Motifs 2 (ADAMTS-2) | AR | 15 reported individuals (14 families) | Absent N-propeptide cleavage of both pro-α(I)-chains | Procollagen biochemistry |
| Cardiac-valvular (cvEDS) | COL1A2 | α2-chain of type I procollagen | AR | 6 reported individuals (5 families) | Total absence of pro-α2(I)-chains leading to the formation of α1(I) homotrimers | (Pro)collagen biochemistry |
| Defects in Collagen Folding and Collagen Cross-Linking | ||||||
| Kyphoscoliotic (kEDS) | PLOD1 FKBP14 | Lysyl hydroxylase 1 (LH1) FK506 Binding Protein, 22 kDa (FKBP22) | AR | <100 reported individuals <35 reported individuals | Deficient post-translational hydroxylation of lysyl residues causing impaired crosslink formation in the collagen triple helix Deficiency of the molecular chaperone (possibly) leads to premature interaction and accumulation of collagen molecules in the endoplasmic reticulum | (Pro)collagen biochemistry Urinary crosslink analysis: increased LP/HP ratio (between 2–9) |
| Defects in Extracellular Matrix Bridging Molecules | ||||||
| Classical-like (clEDS) | TNXB | Tenascin X (TNX) | AR | <65 reported individuals | Interference with the normal organization and mechanical properties of collagen fibrils in the ECM | TNX serum levels |
| Myopathic (mEDS) | COL12A1 | α1-chain of type XII procollagen | AD AR | <20 reported individuals | Interference with the normal organization and mechanical properties of collagen fibrils in the ECM | NA |
| Defects in Intracellular Processes | ||||||
| Brittle cornea syndrome (BCS) | ZNF469 PRDM5 | Zinc Finger Protein 469 (ZNF469) PR/SET Domain 5 (PRDM5) | AR | <55 reported individuals <35 reported individuals | Disturbed ECM regulation, but exact pathophysiological mechanism remains unclear Disturbed ECM regulation, but exact pathophysiological mechanism remains unclear | NA NA |
| Spondylodysplastic (spEDS) | SLC39A13 | Zrt/Irt-Like Protein 13 (ZIP13) | AR | 13 reported individuals (7 families) | Generalized underhydroxylation of lysyl and prolyl residues of collagen and abnormal crosslinking of collagen in the ECM, but exact pathophysiological mechanism remains unclear | Urinary crosslink analysis: increased LP/HP ratio (around 1) |
| Defects in Glycosaminoglycan Biosynthesis | ||||||
| Musculocontractural (mcEDS) | CHST14 DSE | Dermatan 4-O-sulfotranferase-1 (D4ST1) Dermatan sulfate epimerase-1 (DS-epi1) | AR | <70 reported individuals | Defective biosynthesis of dermatan sulfate (DS) resulting in depletion of DS | Urinary disaccharide analysis Urinary disaccharide analysis |
| Spondylodysplastic (spEDS) | B4GALT7 B3GALT6 | Galactosyltransferase I (b4GalT7) Galactosyltransferase II (b3GalT6) | AR | <15 reported individuals <50 reported individuals | Absence of the first galactose residue of the tetrasacharide linker region of proteoglycans Absence of the second galactose residue of the tetrasacharide linker region of proteoglycans | Serum bikunin analysis Serum bikunin analysis |
| Defects in the Complement Pathway | ||||||
| Periodontal (pEDS) | C1S C1R | Complement C1s (C1s) Complement C1r (C1r) | AD | <150 reported individuals | Gain of function variants possibly leading to abnormal interactions with components of the ECM Gain of function variants possibly leading to abnormal interactions with components of the ECM | NA |
| Molecularly Unresolved | ||||||
| Hypermobile (hEDS) | ? | unknown | ? | ? | ? | NA |
| Novel Type of EDS (Identified After the 2017 Classification) | ||||||
| Classical-like II (clEDS II) | AEBP1 | Adipocyte enhancer-binding protein 1 (AEBP1) | AR | 9 reported individuals (9 families) | Interference with normal collagen fibril formation, but exact pathophysiological mechanism remains unclear | NA |
Table 2.
Clinical signs and symptoms of the different EDS types. Major clinical criteria according to the 2017 International EDS Classification are written in bold, minor clinical criteria are written in normal font and additional phenotypic features are written in italics. (G)JH: (generalized) joint hypermobility, MVP: mitral valve prolaps.
Table 2.
Clinical signs and symptoms of the different EDS types. Major clinical criteria according to the 2017 International EDS Classification are written in bold, minor clinical criteria are written in normal font and additional phenotypic features are written in italics. (G)JH: (generalized) joint hypermobility, MVP: mitral valve prolaps.
| EDS Type | Integumentary System | Skeletal System | Neuromuscular | Craniofacial | Ophthalmological | Vascular | Cardiac | Other |
|---|---|---|---|---|---|---|---|---|
| cEDS (COL5A1; COL5A2; COL1A1 p.(Arg312Cys)) | skin hyperextensibility with atrophic scarring, easy bruising soft doughy skin skin fragility, molluscoid pseudotumours, subcutaneous spheroids, hernia (or history thereof) | GJH, complications of JH | (mild) muscle hypotonia, delayed motor development | epicanthal folds | Rarely aortic root dilatation, rarely arterial dissection/rupture | MVP (non-progressive) | ||
| vEDS (COL3A1; COL1A1 p.(Arg312Cys, p.(Arg574Cys), p.(Arg1093Cys) | bruising unrelated to identified trauma and/or in unusual sites, translucent skin, acrogeria | talipes equinovarus, congenital hip dislocation, small joint hypermobility, tendon and muscle rupture | characteristic facial features (large eyes, periorbital pigmentation, small chin, sunken cheeks, thin nose and lips and lobeless ears), gingival recession and gingival fragility | keratoconus | arterial rupture at young age, carotid-cavernous sinus fistula, early-onset varicose veins | spontaneous sigmoid colon perforation, uterine rupture during third trimester of pregnancy, spontaneous pneumothorax | ||
| aEDS (COL1A1; COL1A2) | skin hyperextensibility, tissue fragility including atrophic scars, easy bruising, umbilical hernia | congenital bilateral hip dislocation, severe generalized JH with multiple dislocations, kyphoscoliosis, radiologically mild osteopenia, fractures, foot and hand deformities, pectus deformity | muscle hypotonia, delayed motor development | large fontanelle, frontal bossing, hypertelorism, blue sclerae, epicanthal folds, depressed nasal ridge, midface hypoplasia, micrognathia, dentinogenesis imperfecta | pregnancy-related complications (breech, PPROM, polyhydramnios, decreased fetal movements) | |||
| dEDS (ADAMTS2) | extreme skin fragility with congenital or postnatal tears, progressively redundant, almost lax skin with excessive skin folds at wrists and ankles, increased palmar wrinkling, severe bruisability with risk of subcutaneous haematoma, umbilical hernia, soft and doughy skin texture, skin hyperextensibility, atrophic scars, hirsutism | postnatal growth retardation with short limbs, GJH, osteopenia, fractures | delayed motor development | large fontanel, puffy eyelids, excessive peri-orbital skin, downslanting palpebral fissures, blue sclerae, hypoplastic chin, tooth abnormalities | refractive errors, strabismus, glaucoma | Intracerebral hemorrhage | perinatal complications related to tissue fragility, complications of visceral fragility, preterm birth (PPROM), | |
| cvEDS (COL1A1; COL1A2) | skin involvement, inguinal hernia | JH, pectus deformity, joint dislocations, foot deformities | blue sclerae, refractive errors | severe progressive cardiac valvular insufficiency | ||||
| kEDS (PLOD1; FKBP14) | skin hyperextensibility, easy bruising, umbilical or inguinal hernia PLOD1: skin fragility FKBP14: hyperkeratosis follicularis, umbilical skin redundancy | congenital or early-onset kyphoscoliosis, GJH with (sub)luxations, osteopenia/osteoporosis, pectus deformity, marfanoid habitus, talipes equinovarus | congenital muscle hypotonia, delayed motor development FKBP14: muscle atrophy | blue sclerae PLOD1: characteristic craniofacial features: low-set ears, epicanthal folds, down-slanting palpebral fissures, synophrys and high palate | refractive errors PLOD1: microcornea | rupture/aneurysm of medium-sized artery, antenatal/neonatal brain hemorrhage | pregnancy-related complications (breech, PPROM, oligohydramnios, decreased fetal movements) FKBP14: congenital hearing impairment, bladder diverticula, learning disabilities | |
| clEDS-1 (TNXB) | skin hyperextensibility with velvety skin texture and absence of (extensive) atrophic scarring, easily bruisable skin/spontaneous ecchymoses, acrogeric hands | GJH, foot deformities, mallet fingers, clino- or brachydactyly, (sub)luxations | mild proximal and distal muscle weakness, axonal polyneuropathy, atrophy of muscle in hands and feet | narrow/high arched palate | rarely arterial aneurysms | valvular abnormalities | oedema in legs in absence of cardiac failure, vaginal, uterine or rectal prolapse, gastrointestinal complications (perforation, diverticular disease, …), postpartum hemorrhage | |
| clEDS2 (AEBP1) | skin hyperextensibility with atrophic scarring, translucent skin, easy bruising, hernia, aged appearance | GJH, foot deformities, early-onset osteopenia, joint dislocations (mostly hip), arachnodactyly, (kypho)scoliosis, osteopenia | bad tooth quality, dental abnormalities, high narrow palate, | varicose veins, aorta dilatation | MVP | bowel rupture | ||
| mEDS (COL12A1) | soft, doughy skin, atrophic scars, hypertrophic scars | proximal joint contractures, (general/distal) JH, congenital hip dislocation, congenital (kypho)scoliosis, pectus deformity | congenital muscle hypotonia and/or muscle atrophy, motor developmental delay, myopathy on muscle biopsy | |||||
| BCS (ZNF469; PRDM5) | soft, velvety and/or translucent skin, mild skin hyperextensibility, easy bruising, hernia | developmental dysplasia of hip, scoliosis, arachnodactyly, hypermobility of distal joints, pes planus, hallux valgus, mild finger contractures, fractures, osteopenia/osteoporosis | hypotonia in infancy (usually mild) | blue sclerae, frontal bossing, high palate, depressed nasal bridge and/or prominent chin | thin cornea with/without rupture, early-onset progressive keratoconus and/or keratoglobus, enucleation or corneal scarring because of previous rupture, progressive loss of corneal stromal depth, high myopia, retinal detachment | MVP (non-progressive) | deafness (often mixed conductive and sensorineural), hypercompliant tympanic membranes | |
| mcEDS (CHST14; DSE) | skin hyperextensibility, easy bruising, skin fragility with atrophic scars, increased palmar wrinkling, large subcutaneous hematomas, hernia | Recurrent/chronic dislocations, pectus deformities, spinal deformities, peculiar fingers, progressive talipes deformities, mild postnatal growth restriction, marfanoid habitus | congenital multiple contractures (typically adduction/flexion contractures and talipes equinovarus), hypotonia, motor developmental delay, ventricular abnormalities on brain imaging, tethered spinal cord | large fontanelle, short downslanting palpebral fissures, blue sclerae, hypertelorism, short nose with hypoplastic columella, low-set and rotated ears, long philtrum with thin upper lip vermillion, small mouth and hypoplastic chin, crowded teeth | strabismus, refractive errors, glaucoma, retinal detachment | congenital heart defects (typically ASD), valve abnormalities, aortic root dilatation | chronic constipation, colonic diverticulae, pneumo(haemo)thorax, nephrolithiasis/cystolithiasis, hydronephrosis, cryptorchidism in males, hearing impairment, constipation, diverticula, poor breast development in females | |
| spEDS (B3GALT6; B4GALT7; SLC39A13) | skin hyperextensibility, soft and doughy, thin and translucent skin B4GALT7: single transverse palmar crease SLC39A13: hands with finely wrinkled palms | short stature (progressive in childhood), bowing of limbs, pes planus, osteopenia, (characteristic X-ray findings of) skeletal dysplasia, JH B4GALT7: radioulnar synostosis B3GALT6: kyphoscoliosis (congenital or early-onset), JH (generalized or restricted to distal joints), peculiar fingers, osteoporosis with spontaneous fractures SLC39A13: tapering fingers, hypermobility of distal joints | muscle hypotonia (ranging from severe congenital to mild later-onset), delayed motor development B4GALT7: bilateral elbow contractures B3GALT6: joint contractures (congenital or progressive) SLC39A13: atrophy of thenar muscles | B4GALT7: triangular face, wide-spaced eyes, proptosis, narrow mouth, low-set ears, sparse scalp hair, abnormal dentition, flat face, wide forehead, blue sclerae and cleft palate/bifid uvula B3GALT6: midfacial hypoplasia, frontal bossing, proptosis, or prominent eyes, blue sclerae, downslanting palpebral fissures, depressed nasal bridge, long upper lip, low-set ears, micrognathia, abnormal dentition, cleft palate, sparse hair, tooth discoloration, dysplastic teeth SLC39A13: protuberant eyes with bluish sclerae, hypodontia of one or few teeth | B4GALT7: clouded cornea, refractive errors B3GALT6: hypermetropia, rarely corneal clouding SLC39A13: refractive errors | Rarely aortic aneurysm SLC39A13: varicose veins | B3GALT6: MVP, congenital heart defects | cognitive impairment B3GALT6: lung hypoplasia, restrictive lung disease |
| pEDS (C1R; C1S) | pretibial plaques, easy bruising, skin hyperextensibility and fragility, wide or atrophic scarring, hernias, acrogeria, skin translucency | JH (mostly distal), kypho (scoliosis) | Severe and intractable early-onset periodontitis, lack of attached gingiva, marfanoid facial features | prominent vasculature, rarely arterial dissection/rupture | family history of first-degree relative who meets clinical criteria, increased infection rate | |||
| hEDS (genetic defect unknown) | unusually soft or velvety skin, mild skin hyperextensibility, unexplained striae, bilateral piezogenic papules, hernia, mild atrophic scarring, | GJH, arachnodactyly, arm span to height ratio ≥1.05, | dental crowding and high or narrow palate | aortic root dilatation with z score > +2 | MVP | pelvic floor, rectal and/or uterine prolapse, positive family history of hEDS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Van Damme, T.; Colman, M.; Syx, D.; Malfait, F. The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes 2022, 13, 265. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020265
AMA Style
Van Damme T, Colman M, Syx D, Malfait F. The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism. Genes. 2022; 13(2):265. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020265
Chicago/Turabian StyleVan Damme, Tim, Marlies Colman, Delfien Syx, and Fransiska Malfait. 2022. "The Ehlers–Danlos Syndromes against the Backdrop of Inborn Errors of Metabolism" Genes 13, no. 2: 265. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13020265
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.