
Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing


1
Department of Neurology, Concord Repatriation General Hospital, Concord, NSW 2139, Australia
2
Sydney Medical School, University of Sydney, Camperdown, NSW 2050, Australia
3
Garvan Institute of Medical Research, Darlinghurst, NSW 2010, Australia
4
Raphael Recanati Genetics Institute, Rabin Medical Center, Beilinson Hospital, Petah Tikva 4941492, Israel
5
Department of Neurology, Rabin Medical Center, Beilinson Hospital, Petah Tikva 4941492, Israel
6
Molecular Medicine Laboratory, Concord Repatriation General Hospital, Concord, NSW 2139, Australia
*
Author to whom correspondence should be addressed.
Genes 2022, 13(3), 471; https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030471
Submission received: 4 February 2022
/
Revised: 24 February 2022
/
Accepted: 2 March 2022
/
Published: 7 March 2022
(This article belongs to the Special Issue Parkinson's Disease: Genetics and Pathogenesis)
Abstract
:Parkinson’s disease may be caused by a single pathogenic variant (monogenic) in 5–10% of cases, but investigation of these disorders provides valuable pathophysiological insights. In this review, we discuss each genetic form with a focus on genotype, phenotype, pathophysiology, and the geographic and ethnic distribution. Well-established Parkinson’s disease genes include autosomal dominant forms (SNCA, LRRK2, and VPS35) and autosomal recessive forms (PRKN, PINK1 and DJ1). Furthermore, mutations in the GBA gene are a key risk factor for Parkinson’s disease, and there have been major developments for X-linked dystonia parkinsonism. Moreover, atypical or complex parkinsonism may be due to mutations in genes such as ATP13A2, DCTN1, DNAJC6, FBXO7, PLA2G6, and SYNJ1. Furthermore, numerous genes have recently been implicated in Parkinson’s disease, such as CHCHD2, LRP10, TMEM230, UQCRC1, and VPS13C. Additionally, we discuss the role of heterozygous mutations in autosomal recessive genes, the effect of having mutations in two Parkinson’s disease genes, the outcome of deep brain stimulation, and the role of genetic testing. We highlight that monogenic Parkinson’s disease is influenced by ethnicity and geographical differences, reinforcing the need for global efforts to pool large numbers of patients and identify novel candidate genes.
1. Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder in which we have an incomplete understanding of the molecular and cellular disease basis and no currently available disease-modifying therapy. A key strategy to understanding the pathogenesis of PD is to investigate the underlying genetic basis. Approximately 5–10% of PD can be attributed to monogenic forms. Other causes are related to a combination of complex genetic susceptibility and environmental factors. For the monogenic forms, there are several well-established genes, with autosomal dominant (SNCA, LRRK2, and VPS35) and autosomal recessive (PRKN, PINK1, DJ1) modes of inheritance. Additionally, there is X-linked inheritance (X-linked dystonia-parkinsonism) and atypical or complex parkinsonian phenotypes due to mutations in the ATP13A2, DCTN1, DNAJC6, FBXO7, PLA2G6, and SYNJ1 genes. Moreover, there are numerous recently reported genes, including CHCHD2, LRP10, TMEM230, UQCRC1, and VPS13C. In some cases, the same gene can be linked with Mendelian forms of PD as well as increased susceptibility (such as SNCA and LRRK2). Furthermore, mutations in genes such as glucocerebrosidase (GBA) fall between a monogenic cause and a genetic susceptibility factor [1]. Further recent discoveries have focused on the clinicogenetic and pathological findings, which will be discussed. Pathophysiological insights will be discussed briefly (for a more detailed discussion, see elsewhere [2]), and a discussion of novel therapeutic candidates can be found elsewhere [3,4]. Recent hot topics include the understanding of the effect of heterozygous variants in recessive PD genes, outcomes in individuals who co-inherit mutations in both GBA and LRRK2, the effect of the underlying genetic form on outcomes from deep brain stimulation, and regional and ethnic differences for mutations in PD genes. In this review, we provide a concise summary of the monogenic origin of PD with a focus on these recent developments.
2. Autosomal Dominant Forms
2.1. SNCA
2.1.1. Genotype-Phenotype
SNCA mutations cause autosomal dominant PD and can be due to different mutation types, including missense variants and multiplications (Table 1). So far, there have been eight missense variants identified as causing autosomal dominant PD: p.A30G, p.A30P, p.E46K, p.H50Q, p.G51D, p.A53E, p.A53T, and p.A53V. An MDSGene review identified phenotypic differences between some of these missense variants, with the most common mutation p.A53T having an early age at onset compared to p.A30P and p.E46K [5]. However, these findings are uncertain given that the number of cases for SNCA missense variants other than p.A53T is small [5]. Of note, there is evidence that the p.H50Q variant is not enriched in cases versus controls and thus may not have sufficient evidence to be considered pathogenic [6]. The most recently reported mutation, p.A30G, was found in five affected individuals from three Greek families [7]. The phenotype results in a widely ranging age at onset, an initial good response to medication (Table 2), prominent motor fluctuations, and a range of non-motor manifestations such as orthostatic hypotension, REM-behavior sleep disorder, cognitive impairment, and psychiatric manifestations [7].
Duplications and triplications of the SNCA gene can also cause PD. SNCA duplications cause a phenotype resembling idiopathic PD, whereas SNCA triplications cause rapidly progressive PD with earlier onset and extensive Lewy Body pathology. A recent study highlighted the correlation between SNCA dosage and age at onset, with copy number 3 (heterozygous SNCA duplication) associated with a mean age at onset of 46.9 ± 10.5 years versus copy number 4 (homozygous SNCA duplication, or SNCA triplication) associated with a mean age at onset of 34.5 ± 7.4 [8].
Overall, duplications are more common than missense mutations or triplications [5]. The different mutation types can be stratified according to the age at onset, with early, intermediate, and late onset for triplications, missense mutations, and duplications, respectively [5].

{kind=link}
Table 1.
Genotype-phenotype summary for monogenic forms of Parkinson’s disease.
| Gene | Mode of Inheritance | Frequency | Ethnic Population Distribution | Types of Mutations | Clinical Phenotype | Response to PD Medication | Response to DBS | Pathological Findings |
|---|---|---|---|---|---|---|---|---|
| SNCA | AD | Rare, with a frequency from 0.045% to 1.1% in recent studies [9] | Majority European, then Asians and Hispanics [5] | Missense, duplications, and triplications | Range of age at onset, prominent motor fluctuations, range of complications including cognitive impairment and psychiatric manifestations | Initial good response | Few examples, appears to have a good response for duplications, poor response for missense mutations | α-synuclein-positive and LB pathology [10] |
| LRRK2 | AD | 1% of PD but higher in North African Berber Arab and Ashkenazi Jewish populations | The p.G2019S mutation found in Europeans with high prevalence in North African Berbers and Ashkenazi Jews | 7 missense variants described | Resembles idiopathic PD | Vast majority show a good response to levodopa [11] | DBS is effective [12] | Most patients with the p.G2019S mutation show LB pathology, whereas this finding is rare for other mutations |
| VPS35 | AD | Rare (overall prevalence of 0.115%) | European, Asian, Ashkenazi Jewish [5] | 1 missense mutation described, p.D620N | Resembles idiopathic PD | Good response [11] | Small numbers reported, at least 2 had a good outcome [11] | Not available |
| PRKN | AR | Most common cause of EOPD, 12.5% of recessive PD [13] | Majority Asian, followed by Caucasians and Hispanics [14] | Missense mutations, frameshift mutations, structural variants | EOPD, lower limb dystonia, absence of cognitive impairment | Good response to levodopa therapy, frequent motor fluctuations, and dyskinesias | Good outcome in all patients [11] | Substantia nigra pars compacta loss with the notable absence of LB pathology |
| PINK1 | AR | Second most common cause of EOPD, 1.9% of recessive PD [13] | European, Asian, may be frequent in Arab Berber and Polynesian populations [9,14,15] | Missense mutations, nonsense mutations, structural variants | EOPD, typical PD, dyskinesias, dystonia, and motor fluctuations can occur | Vast majority show a good outcome [11] | Good or moderate [11] | LB pathology may or may not be present in the handful of autopsy cases reported |
| PARK7 (DJ1) | AR | 0.16% of recessive PD [13] | Most patients are from Italy, Iran, and Turkey [14] | Missense, splice site, frameshift, and structural variants | EOPD | 50% show a good response, others moderate or minimal [11] | No reports identified [11] | LB pathology [16] |
| TAF1 | X-linked | 0.34 per 100,000 in the Philippines, Island of Panay 5.24 per 100,000 | Philippines, high prevalence on the Island of Panay | Insertion of a SINE-VNTR-Alu type retrotransposon in intron 32 of the TAF1 gene | Parkinsonism, dystonia | May be responsive to levodopa, particularly for those with pure parkinsonism [17] | DBS results in an improvement in dystonia and to a lesser extent parkinsonism [18] | Accumulation of lipofuscin in the neurons and glia, but absence of LB pathology |
| ATP13A2 | AR | Rare | Spread across the globe [19] | Frameshift, missense, and splice site mutations [19] | KRS, clinical triad of spasticity, dementia, and supranuclear gaze palsy [20], facial-faucial-finger mini-myoclonus [21], other phenotypes include HSP | Variable response to levodopa [19] | May respond well, variable [22] | Accumulation of lipofuscin, absence of LB pathology [23] |
| DCTN1 | AD | Rare | Spread across the globe | 10 different heterozygous missense mutations [19] | Perry syndrome—rapidly progressive parkinsonism, depression and mood changes, weight loss, and progressive respiratory changes | May be levodopa-responsive | No reports identified | Selective loss of putative respiratory neurons in the ventrolateral medulla and in the raphe nucleus, no or few LBs, TDP43-positive inclusions [24,25] |
| DNAJC6 | AR | Rare | Mainly found in Middle Eastern populations, although families of European origin have also been found to harbor DNAJC6 mutations | 5 different homozygous mutations, largest family carries a nonsense mutation [19] | Juvenile PD with complicating features, EOPD | Poor | Good outcome [26] | No reports identified |
| FBXO7 | AR | Rare | Reported in the Iranian, Turkish, Italian, Dutch, Pakistani, and Chinese populations | Biallelic missense, splice site, and nonsense mutations | Juvenile PD, EOPD, parknsonian-pyramidal syndrome, can overlap with NBIA [27] | Variable | No reports identified | No reports identified |
| PLA2G6 | AR | Rare | Various ethnic groups, including Indian, Pakistani, European, Japanese, Chinese, and Korean populations | 54 mutations associated with parkinsonism [28] | Adult-onset dystonia-parkinsonism with cognitive and psychiatric symptoms [28], other phenotypes include NBIA | Variable | May benefit from DBS [28] | Mixed Lewy and Tau pathology [28] |
| SYNJ1 | AR | Rare | Reported in Iranian, Italian, German, Algerian, Senegalese, and Chinese populations | Missense, frameshift | Parkinsonism in the third decade of life, complicating features such as dystonia, seizures, or cognitive impairment | Poor | No reports identified | No reports identified |
| CHCHD2 | AD | Rare | Japanese and Chinese patients | Missense, splice site | Typical PD | Good | No reports identified | A brain autopsy revealed widespread α-synuclein pathology with Lewy bodies present in the brainstem, neocortex, and limbic regions [29] |
| LRP10 | AD | Rare | Italy, Taiwan | Loss of function and missense variants | Late onset PD, PD dementia, dementia with Lewy Bodies [30,31,32] | Good [32] | Excellent response for a patient with a LRP10 and GBA variant in trans [33] | Severe LB pathology |
| TMEM230 | AD | Rare | Identified in a Canadian Mennonite family | Missense variant | Typical PD | Responds to levodopa in most cases | No report identified | Typical LB pathology [34] |
| UQCRC1 | AD | Rare | Taiwan, may not be in European populations | Missense variants | Parkinsonism with polyneuropathy | Good | No report identified | No report identified |
| VPS13C | AR | Rare | Turkish, French | Truncating mutations | EOPD, rapid progression, complicating features including dysphagia, cognitive impairment, hyperreflexia [19,35] | Initial good response | Poor | Resembles diffuse LB disease [35] |
AD: autosomal dominant, AR: autosomal recessive, DBS: deep brain stimulation, EOPD: early-onset Parkinson’s disease, HSP: hereditary spastic paraplegia, KRS: Kufor Rakeb syndrome, LB: Lewy body, NBIA: neurodegeneration with brain iron accumulation, PD: Parkinson’s disease, XDP: X-linked dystonia parkinsonism.
2.1.2. Pathophysiology
The discovery of dominant mutations in SNCA as a cause of PD is consistent with the critical role the α-synuclein protein plays in PD pathogenesis. The molecular effects may vary according to the type of SNCA mutation [36]. The p.A30P, p.A53T, and p.E46K mutations all affect the N-terminal domain of the α-synuclein protein [36]. The p.A30P and p.A53T mutations stimulate protofibril formation and smaller to larger aggregates [36]. The p.E46K mutation increases the N-terminal positive charge and enhances N-terminal and C-terminal contacts, whereas the opposite is seen for the p.A30P and p.A53T mutations [36]. A recent study showed impaired mitochondrial respiration, energy deficits, vulnerability to rotenone, and altered lipid metabolism in dopaminergic neurons derived from a patient with the p.A30P mutation in SNCA, with a comparison to gene-corrected clones, highlighting the numerous effects of these mutations [37].
Table 2.
Levodopa-responsiveness stratified according to Parkinson’s disease monogenic forms.
| Good Response to Levodopa | Poor, Variable, or Uncertain Response to Levodopa |
|---|---|
| SNCA | TAF1 |
| LRRK2 | ATP13A2 |
| VPS35 | DCTN1 |
| PRKN | DNAJC6 |
| PINK1 | FBXO7 |
| DJ1 | PLA2G6 |
| CHCHD2 | SYNJ1 |
| LRP10 | |
| TMEM230 | |
| UQCRC1 | |
| VPS13C |
2.2. LRRK2
2.2.1. Genotype-Phenotype
At least seven missense variants in LRRK2 have been described as causing PD (p.N1437H, p.R1441C/G/H, p.Y1699C, p.G2019S, and p.I2020T) [3]. On an individual level, LRRK2-PD is clinically indistinguishable from idiopathic PD. However, as a group, it may be considered as having a milder phenotype [38,39]. For example, LRRK2 mutation carriers are less likely to have non-motor symptoms such as olfactory impairment, cognitive features, and REM-behavior sleep disorder [39]. Furthermore, patients with LRRK2-PD may be susceptible to certain cancers [40,41,42]. A very recent study provides evidence that LRRK2-PD is associated with a significantly higher risk of stroke [43]. Additionally, recent evidence suggests that regular use of non-steroidal anti-inflammatory drugs may be associated with reduced penetrance of PD in both pathogenic and risk variant carriers [44].
The most common and well-characterized LRRK2 mutation is the p.G2019S mutation. It has a prevalence of 1% in the PD population with a high prevalence in North African Berber Arab (39%) and Ashkenazi Jewish (approximately 18%) populations [45,46,47]. The penetrance of this mutation is incomplete and variable and influenced by age, environment, and genetic background [48].
Other mutations in LRRK2 may be relevant to different ethnic and regional populations. For example, the p.R1441C variant has a founder effect in Basque populations and may be higher in Southern Italy and Belgium [38]. The p.G2019S mutation is very rare in Chinese populations, whereas the p.G2385R and p.R1628P variants are common (5–10% in patients, 2–5% in controls) [49,50,51].
Recent reports suggest that loss of function variants in LRRK2 are not associated with PD, arguing that haploinsufficiency is neither causative nor protective of PD [52].
2.2.2. Pathophysiology
All the definite LRRK2 mutations are in the catalytic domains and may result in hyperactivation of the kinase domain [3,53]. LRRK2 is involved in a large array of cell biological processes, and the disease mechanism may reflect important roles in microtubule function and Rab proteins as phosphorylation substrates [2,54].
2.3. VPS35
2.3.1. Genotype-Phenotype
VPS35 is implicated in autosomal dominant PD [55,56], with the missense variant p.D620N being the only mutation confirmed to date. This variant appears to be a mutational hotspot identified in different ethnic populations [57]. The mutation has an overall prevalence of 0.115% from the reported studies but may be as high as 1% in autosomal dominant PD [57,58,59]. The phenotype resembles idiopathic PD with a median age at onset of 49 years, levodopa responsiveness, and predominant tremor [5,58]. A recent study suggests that disease progression may be slow, with minimal cognitive impairment even after more than 10 years of disease onset [60].
2.3.2. Pathophysiology
VPS35 plays a critical role in endosomal trafficking, but there is emerging evidence for a role in mitochondrial function [61]. The p.D620N mutation impairs the sorting function of the retromer complex, resulting in a disturbance of maturation of endolysosomes and autophagy, membrane receptor recycling, and mitochondrial-derived vesicle formation [2,59,62]. There may also be a role in neurotransmission and an interaction with other genes causing monogenic PD (such as SNCA, LRRK2, and PRKN) [62].
3. Autosomal Recessive Forms
3.1. PRKN
3.1.1. Genotype-Phenotype
Mutations in PRKN are the most common cause of early-onset PD (EOPD), particularly in European populations. A recent study by Lesage and colleagues demonstrated that PRKN mutations account for 27.6% of autosomal recessive families [13]. They found that the proportion of probands with PRKN mutations is higher the younger the age at onset (AAO), as follows: 42.2% for AAO less than or equal to 20 years, 29% for 21 to 30 years, 13% for 31 to 40 years, but only 4.4% for 41 to 60 years [13].
A variety of different mutation types are described, including structural variants (43.2%, including exonic deletions, duplications, and triplications), missense mutations (22.3%), and frameshift mutations (16.5%) [14,63]. Deletions in exon 3 are the most common mutation [14]. Furthermore, a deletion of the PRKN and PACRG gene promoter has also been described in autosomal recessive PD [63].
PD-PRKN is characterized phenotypically by an early age at disease onset, lower limb dystonia at presentation, absence of cognitive impairment, a good and sustained response to levodopa, and frequent motor fluctuations and dyskinesias [64].
3.1.2. Pathophysiology
Mutations in PRKN and PINK1 likely disturb PINK1/parkin-mediated mitophagy, which is the selective degradation of mitochondria, a function essential for mitochondrial homeostasis [65]. In brief, parkin is a E3 ubiquitin ligase that ubiquinates outer mitochondrial membrane proteins such as mitofusin 1 and 2 [66]. PINK1 phosphorylates parkin and maintains its mitochondrial stabilization and translocation, mediating parkin activation [2,66].
3.2. PINK1
3.2.1. Genotype-Phenotype
PINK1 is the second most common cause of autosomal recessive PD and is characterized by typical Parkinson’s features such as tremor, bradykinesia, and rigidity, with a median age of onset of 32 [14,67]. Additional phenotypic features include dyskinesias in 39%, dystonia in 21%, and motor fluctuations in 34%, with cognitive impairment and psychosis occurring rarely (14% and 9%, respectively) [14]. The disease is slowly progressive, with a sustained response to levodopa therapy, although with an increased tendency for levodopa-induced dyskinesias.
The main mutation type was missense mutations (47.6%), then structural variants (19.1%), followed by nonsense mutations (14.3%) [14]. The most common specific mutation was a missense mutation, c.1040T>C (p.Leu347Pro) [14].
A recent paper suggests that the c.1040T>C mutation is frequently found in patients from the Pacific Islands [15]. The allele frequency was particularly high in West Polynesians (2.8%), which would translate to a homozygosity of 1 in 5000 people, suggesting that this could have a major contribution to EOPD in the region [15].
3.2.2. Pathophysiology
See PRKN above.
3.3. PARK7
3.3.1. Genotype-Phenotype
Mutations in PARK7 can cause early-onset autosomal recessive parkinsonism, with at least 20 mutations in the PARK7 gene identified. The majority of PARK7 mutation carriers have EOPD (83%), whereas 13% have juvenile onset and 4% have late onset [14]. Recently, a Turkish family with juvenile PD was found to have a novel deletion of the neighboring genes of PARK7 and TNFRSF9, raising the possibility of TNFRSF9 as a disease modifier [68].
3.3.2. Pathophysiology
DJ-1 is ubiquitously expressed and is highly expressed in cells with high energy demands. DJ-1 exerts an antioxidative stress function through scavenging reactive oxygen species, regulation of transcription and signal transduction pathways, and acting as a molecular chaperone and enzyme [69]. Mutations within the PARK7 gene substantially affect the survival of cells in oxidative environments, potentially leading to PD [70,71].
4. X-Linked Dystonia-Parkinsonism
4.1. Genotype-Phenotype
X-linked dystonia-parkinsonism (XDP), also referred to as Lubag, is a movement disorder initially described in Filipino males, caused by the insertion of a SINE-VNTR-Alu (SVA)-type retrotransposon in intron 32 of the TAF1 gene [72,73]. The prevalence is 0.34 per 100,000 in the Philippines, with a high prevalence on the Island of Panay of 5.24 per 100,000 [74]. It initially presents with dystonia, and predominantly involves the craniocervical region that can become generalized at a later stage [72,75]. It may also present with parkinsonism, or this can develop later in the disease course [75]. Therefore, it can show longitudinal evolution from a hyperkinetic to a hypokinetic movement disorder. Although it primarily affects males, manifesting female carriers have been reported. The median age at onset is 40 years from a recent MDSGene review [72].
4.2. Pathophysiology
Recent evidence suggests that probands with XDP have reduced expression of the canonical TAF1 transcript [73]. De novo assembly of multiple neuronal lineages derived from pluripotent stem cells showed reduced expression due to alternative splicing and intron retention close to the SVA [73]. CRISPR/Cas 9 excision of the SVA was able to rescue TAF1 expression, providing evidence of abnormal transcription mediated by the SVA in the pathophysiology of XDP [73]. Further evidence suggests that a hexanucleotide repeat within the SVA modifies disease expressivity, with the number of repeats showing an inverse correlation with the age at onset [76].
5. Complex or Atypical Forms
5.1. ATP13A2
5.1.1. Genotype-Phenotype
Biallelic mutations in ATP13A2 have been found to cause a complex form of parkinsonism known as Kufor-Rakeb syndrome (KRS), characterized by juvenile onset parkinsonism, cognitive impairment, and a supranuclear gaze palsy. ATP13A2 mutations can also cause a range of phenotypes, including neuronal ceroid lipofuscinosis, hereditary spastic paraplegia, and juvenile amyotrophic lateral sclerosis.
Recently, perhaps the first postmortem study of KRS was reported [77]. This showed accumulation of lipofuscin in the neurons and glia, but an absence of Lewy body pathology as well as alpha-synuclein, TDP43, tau, and beta amyloid pathology. This provides evidence for a pathological link with neuronal lipofuscinosis rather than the typical findings in PD [77].
5.1.2. Pathophysiology
ATP13A2 mutations impair lysosomal and mitochondrial function. The mechanism may involve impaired lysosomal polyamine transport resulting in lysosome-dependent cell death [78].
5.2. DCTN1
5.2.1. Genotype-Phenotype
DCTN1-associated Parkinson-plus disorder, also called Perry syndrome, is a rare autosomal dominant disorder characterized by rapidly progressive parkinsonism, depression and mood changes, weight loss, and progressive respiratory changes, chiefly tachypnoea and nocturnal hypoventilation [79].
The disease is linked to mutations in exon 2 of the DCTN1 gene. The mean age at onset of disease is 48 years (range: 35–61) and the mean duration to death is 5 years since diagnosis, from either respiratory failure, sudden unexplained death, or suicide [80]. DCTN1 mutations have been associated with additional phenotypes, including distal spinal and bulbar muscular atrophy and amyotrophic lateral sclerosis.
5.2.2. Pathophysiology
5.3. DNAJC6
5.3.1. Genotype-Phenotype
Biallelic mutations in DNAJC6 cause juvenile-onset, atypical parkinsonism with onset during childhood and a very rapid disease progression with loss of ambulation within 10 years from onset [82,83]. Patients are poorly responsive to levodopa therapy and have additional manifestations such as developmental delay, intellectual disability, seizures, and other movement disorders (e.g., dystonia, spasticity, myoclonus). A minority of patients have early-onset parkinsonism, with symptom onset in the third to fourth decade of life and an absence of additional features [84]. These patients generally have a slower rate of disease progression and a favorable response to levodopa therapy.
5.3.2. Pathophysiology
DNAJC6 encodes for auxilin 1, a brain-specific form of auxilin and a co-chaperone protein involved in the clathrin-mediated synaptic vesicle endocytosis. Auxilin deficiency has been found in animal models to result in impaired synaptic vesicle endocytosis, and thus negatively impacts synaptic neurotransmission, homeostasis, and signaling [85]. However, the exact mechanism by which auxilin deficiency leads to dopaminergic neurodegeneration and atypical neurological symptoms remains unclear.
5.4. FBXO7
5.4.1. Genotype-Phenotype
Mutations in FBXO7 cause autosomal recessive, juvenile/early-onset parkinsonian-pyramidal syndrome (also called PARK15). Missense, splice site, and nonsense mutations have been reported. The median age at onset was 17 years, with a range of 10 to 52 years. The typical presenting symptoms were bradykinesia and tremor, and patients affected by this atypical parkinsonism frequently show pyramidal signs, dysarthria, and dyskinesia. Psychiatric manifestations, such as visual hallucination, agitation, aggression, disinhibition, and impulsive control disorder, are prominent in these patients as a complication of dopaminergic therapy [86,87,88,89].
5.4.2. Pathophysiology
5.5. PLA2G6
5.5.1. Genotype-Phenotype
Mutations in PLA2G6 have been linked to a variety of neurological disorders, including infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation 2B, and Karak syndrome. PLA2G6 mutations may also result in another phenotype—autosomal recessive, adult-onset dystonia-parkinsonism (also called PARK14) [91].
Patients with PLA2G6-related parkinsonism first show symptoms in their childhood or early adulthood, with an age at onset ranging from 8 to 36. In addition to parkinsonism, the majority have dystonia [92,93]. Neuropsychiatric presentations such as depression, psychosis, and cognitive decline are common. There is a good response to levodopa therapy. Magnetic resonance imaging of the brain in most patients showed an absence of iron deposition, and if iron was present, it was found in the substantia nigra or globus pallidus, or both [94].
5.5.2. Pathophysiology
PLA2G6, a phospholipase 2, catalyzes the hydrolysis of the sn-2 acyl-ester bonds in phospholipids to form arachidonic acid and other fatty acids. This is involved in the phospholipid remodeling, apoptosis, and prostaglandin and leukotriene synthesis. The exact mechanism of PLA2G6 in neurodegenerative diseases remains obscure, however defective phospholipases have been implicated in the pathogenesis of neurodegenerative conditions with iron dyshomeostasis.
5.6. SYNJ1
5.6.1. Genotype-Phenotype
Mutations in SYNJ1 are linked to autosomal recessive, early-onset Parkinson disease-20 (PARK20). Individuals affected by SYNJ1-associated parkinsonism generally show symptoms in the third decade of life, and manifest parkinsonism (tremor, bradykinesia) with a poor response to levodopa treatment, as well as additional atypical signs such as dystonia, seizures, cognitive impairment, and developmental delay [95].
5.6.2. Pathophysiology
Synaptojanin-1 plays a crucial role in synaptic vesicle dynamics, including endocytosis and recycling. SJ1-knockout mice display endocytic defects and a remarkable accumulation of clathrin-coated intermediates [96]. Fasano et al. further showed that SYNJ1 is critically involved in early endosome function, and that a loss of SYNJ1 leads to impaired recycling of the transferrin receptor to the plasma membrane, highlighting the important role that the autophagy-lysosome pathway plays in PD pathogenesis [92].
6. Recently Described Parkinson’s Disease Genes
6.1. CHCHD2
6.1.1. Genotype-Phenotype
Mutations in the CHCHD2 gene were linked to an autosomal dominant, late-onset form of PD (PARK22) in the Japanese population in 2015 by Funayama et al., who reported two missense mutations (p.T61I, p.R145Q) and a splice-site mutation (c.300+5G>A) in the CHCHD2 gene [93]. Both missense mutations were also reported in the Chinese population [97,98], although were not found in a study on a large cohort of PD patients of western European ancestry [99]. Instead, three rare variants (p.A32T, p.P34L, and p.I80V) in the CHCHD2 gene were found in the western European cohort, occurring in highly conserved residues [99]. A homozygous missense mutation (p.A71P) has also been reported in a 26-year-old Caucasian woman with recessive early-onset PD [100]. Patients affected by CHCHD2-associated PD typically present with typical parkinsonian features, with a significant response to levodopa.
6.1.2. Pathophysiology
CHCHD2 contains a mitochondrial-targeting sequence at the N-terminus and localizes to the mitochondrial intermembrane space. Its close homologue CHCHD10 is enriched at crista junctions of the mitochondria and is believed to be involved in oxidative phosphorylation or in maintenance of crista morphology [101]. The loss of CHCHD2 in flies leads to mitochondrial and neural phenotypes associated with PD pathology and causes chronic oxidative stress and thus age-dependent neurodegeneration in the dopaminergic neurons [102].
6.2. LRP10
6.2.1. Genotype-Phenotype
Through genome-wide linkage analysis of an Italian family with autosomal dominant PD, Quadri and colleagues implicated the LRP10 gene on chromosome 14 as a possible causative disease gene [31]. This was verified through analysis of a larger cohort of patients, where rare, potential mutations in LRP10 were found to be associated with PD and dementia with Lewy bodies [31]. These findings were unable to be replicated in a study by Tesson et al., whose co-segregation analysis did not support a causal role for LRP10 in PD [103]. Since then, several additional variants in the LRP10 have been identified in patients with PD, progressive supranuclear palsy, frontotemporal dementia, and amyotrophic lateral sclerosis, although the correlation of LRP10 variants with the development of α-synucleinopathies and other neurodegenerative diseases has been debated [104,105,106].
6.2.2. Pathophysiology
LRP10 is a single-pass transmembrane protein and a member of a subfamily of LDL receptors. Grochowska et al. discovered that LRP10 expression was high in non-neuronal cells but undetectable in neurons, and that it was present in the trans-Golgi network, plasma membrane, retromer, and early endosomes in astrocytes [107]. They suggested that LRP10-mediated pathogenicity involves the interaction of LRP10 and SORL1 in vesicle tracking pathways, as they were shown to co-localize and interact, and that disturbed vesicle trafficking and loss of LRP10 function were crucial in the pathogenesis of neurodegenerative diseases [107].
6.3. TMEM230
6.3.1. Genotype-Phenotype
The link with PD was first proposed in 2016 by Deng et al., who investigated a large Canadian Mennonite pedigree with autosomal dominant, typical PD, and discovered a p.R141L mutation in TMEM230 which reportedly fully co-segregated with disease [34]. The same pedigree was investigated by Vilarino-Guell and colleagues, who identified a heterozygous missense variant in DNAJC13 (p.N855S) which did not fully co-segregate with disease [108]. Whilst TMEM230 variants have been identified in further studies on PD patient groups, other follow-up genetic studies have failed to detect PD-associated TMEM230 variants, and whether evidence exists for ‘proof of pathogenecity’ has been debated [109,110].
6.3.2. Pathophysiology
TMEM230 is a transmembrane protein with ubiquitous expression. It is a trafficking protein of secretory and recycling vesicles, including neuronal synaptic vesicles. Expression of mutant TMEM230 was found to lead to increased α-synuclein levels [34]. Loss of function of TMEM230 impairs secretory autophagy, Golgi-derived vesicle secretion, and retromer trafficking [111].
6.4. UQCRC1
6.4.1. Genotype-Phenotype
An association between UQCRC1 mutations and familial PD was first reported by Lin et al. in 2020, who identified a novel heterozygous mutation (p.Y314S) in the UQCRC1 gene which co-segregated with disease in a Taiwanese family with autosomal dominant parkinsonism with polyneuropathy [112]. An additional variant in UQCRC1 (p.I311L) also co-segregated with disease [112]. In a subsequent study, no common variant was found to be significantly associated with PD in the European population [113].
6.4.2. Pathophysiology
UQCRC1 is a core component of complex III in the respiratory chain. In Drosophila and mouse models, URCRC1 p.Y314S knock-in organisms showed dopaminergic neuronal loss, age-dependent locomotor deficits, and peripheral neuropathy [112]. Disruption of the Uqcrc1 gene in mice causes embryonic lethality [114], and deficiency of Uqcrc1 in Drosophila increases the cytochrome c in the cytoplasmic fraction and activates the caspase cascade, thus causing a reduction of dopaminergic neurons and neurodegeneration [115].
6.5. VPS13C
6.5.1. Genotype-Phenotype
Lesage et al. first reported five truncating mutations in VPS13C in three unrelated PD patients [35]. These probands were either homozygous or compound heterozygous and had a distinct phenotype of EOPD which progressed rapidly and showed a good but transient initial response to levodopa treatment. Additional variants in VPS13C have been identified in further reports on autosomal recessive, early-onset forms of parkinsonism, although not in late-onset PD [116].
6.5.2. Pathophysiology
VPS13C is part of the family of conserved VPS13 proteins and behaves similarly to VPS35 (see above). VPS13C is a phospholipid transporter and localizes to the contact sites between the endoplasmic reticulum (ER) and late endosome [117]. VPS13 proteins are thought to mediate endoplasmic reticulum-phagy at late endosomes [117].
7. Rare, Atypical, and Unconfirmed Forms
There are many genes that can cause parkinsonian phenotypes, and comprehensive lists can be found elsewhere, with over 70 different genes causing early-onset parkinsonism or parkinsonism as part of a complex neurological disorder [118]. Clinicians should be especially vigilant for treatable causes such as Wilson’s disease [118]. Mutations in GCH1 can cause dopa-responsive dystonia and PD and should also be considered. POLG mutations can cause movement disorders including parkinsonism and dystonia. Mutations in PTRHD1 can cause autosomal recessive PD with intellectual impairment but are rare [119]. RAB39B mutations can cause X-linked intellectual impairment and parkinsonism with classic Lewy body pathology on autopsy studies [120]. Several additional reported genes have not been independently replicated and perhaps require further validation before being considered PD genes, such as DNAJC13, EIF4G1, GIGYF2, HTRA2, and UCHL1 [121].
8. Risk Variants versus Monogenic Forms
When discussing genetic risk in PD, one should differentiate risk variants from causative monogenic ones. Risk variants are relatively common, each with an individual small effect size, yet collectively they significantly increase disease risk. A recent large meta-analysis of genome-wide association studies (GWAS) identified 90 such genome-wide risk alleles that collectively account for 16–36% of PD heritability [122]. A causative monogenic variant, on the other hand, is a rare variant with a large effect size, that is considered the causative culprit of the disease. Complicating this oversimplified dichotomic differentiation is the fact that autosomal dominant forms of monogenic PD have incomplete age-dependent penetrance to a variable extent, which may be affected by the causative gene and the specific pathogenic variant as well as the patient’s ethnicity. Moreover, a complex interplay between monogenic causative variants and risk variants may affect disease penetrance, as exemplified by a recent study which showed that disease penetrance of the LRRK2 variant p.G2019S is modified by a polygenic risk score [45].
9. GBA Variants
A notable issue is the one related to pathogenic GBA (or GBA1) variants, which constitute the most common genetic risk factor for PD. These variants are found in approximately 8.5% of PD patients [123]. However, this number varies significantly across different ethnic groups, ranging between 2.3% and 12% in populations of non-Ashkenazi Jewish origin to 10–31% in Ashkenazi Jews [124]. GBA variants were more common in patients with early-onset disease (<50 years), more rapid development of dementia, and a more aggressive motor course [125,126]. Pathogenic variants in this gene have a low, age-dependent penetrance in PD, which is highly variable across different reports, ranging between 8% and 30% by age 80 years [127,128,129,130]. In a recent study, the authors used a kin-cohort design to evaluate the penetrance of pathogenic GBA variants in a cohort of unselected PD patients, showing that the risk to develop disease by age 60, 70, and 80 years was 10%, 16%, and 19%, respectively [131]. This study also found a trend towards a greater PD penetrance for severe pathogenic variants compared to mild pathogenic variants in the GBA gene, although this difference did not reach statistical significance [131].
Adding to the complexity of GBA-associated PD, a recent study demonstrated an association between PD polygenic risk score and both penetrance and age at onset in individuals carrying a disease-associated GBA variant [132]. Another study examined PD clustering in eight families of non-Parkinsonian GBA-p.N370S homozygote Gaucher patients, showing that all PD cases in these families stemmed from only one of the proband’s parents, further highlighting the potential role of genetic modifiers in PD risk among carriers of GBA variants [133].
Furthermore, a recent study showed that both pathogenic (i.e., associated with Gaucher disease) and non-pathogenic (i.e., not associated with Gaucher) variants in GBA are common in PD, with a more aggressive course in terms of dementia and motor progression [126].
In summary, GBA variants are a common risk factor for PD. They should be clearly differentiated as such from monogenic causes for PD, to avoid ambiguity and terminological and conceptual perplexity when discussing PD risk with patients and clinicians.
10. Genetic Testing in Parkinson’s Disease
Genetic workup is not routinely performed as part of PD evaluation, and movement disorder specialists only very occasionally suggest genetic testing to PD patients. This is due to a combination of factors related to cost, lack of physician’s perceived impact on patient’s management, and physician’s discomfort regarding test selection and its results or their impact on the patients and their family members [134]. The field of genetic testing in PD is rapidly evolving during recent years, due to the better availability of next-generation sequencing (NGS)-based molecular tests and the initiation of genetic diagnosis-based interventional clinical trials.
10.1. Who Should Be Offered Genetic Testing in Parkinson’s Disease?
Traditionally, a monogenic cause would most probably be suspected, and therefore a genetic test considered, in patients with early-onset PD before age 50 years, and particularly before age 40 years. Furthermore, although polygenic risk and multifactorial inheritance would probably explain most cases with familial clustering of PD, a striking familial history, either of autosomal dominant or autosomal recessive pattern, is yet another clue for a possible monogenic cause that may suggest that a genetic test should be considered. Ethnic origin may also affect the decision to perform genetic testing, for example in patients of Ashkenazi Jewish or African Berber origin. As opposed to this traditional case-by-case approach, as molecular testing is becoming more available, a recently suggested permissive approach supports a more widespread use of genetic testing in PD to improve patient care, to allow inclusion of patients in molecular diagnosis-based clinical trials, and to benefit therapeutic insights and strategies for the larger PD population, including patients with idiopathic disease [135]. This notion can tremendously benefit PD patients both individually and collectively. However, it should be backed up by thorough knowledge of the different evolving aspects of genetic testing in PD, and by an individually tailored explanation to patients and potential carriers in their family prior to testing as well as when returning them the test results, regarding the test and the potential implications of its results for them and for their family members.
10.2. The Implications of a Genetic Diagnosis in Parkinson’s Disease
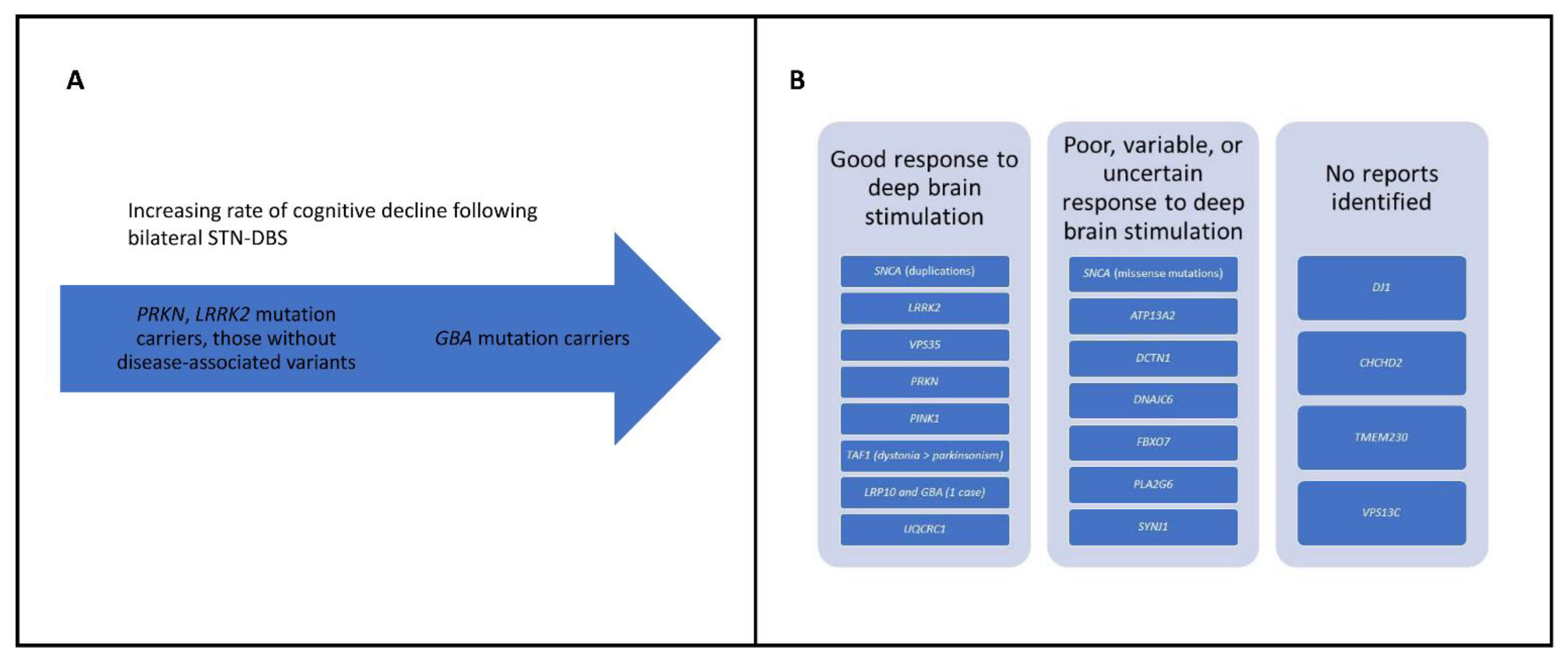
A genetic diagnosis may have significant implications for PD patients, both for expected disease course and response to therapeutic interventions. As mentioned, several monogenic forms are expected to respond well to levodopa medication (e.g., PRKN), whereas others are poorly responsive (e.g., DNAJC6) (Table 2). Additionally, a recent study found that the rate of cognitive decline for GBA mutation carriers after bilateral subthalamic nucleus deep brain stimulation (STN-DBS) is higher than that of carriers of PRKN and LRRK2 mutations and those without identified disease-associated pathogenic variants [136] (Figure 1A). These findings were further corroborated by a new study which suggests that STN-DBS is associated with a greater rate of cognitive decline in GBA mutation carriers [137]. A recommendation that arose from this study is that PD patients should be screened for GBA pathogenic variants prior to DBS surgery, and that carriers of such variants should be counseled on the greater risk of cognitive decline [137].
For SNCA-PD, the response to DBS may also differ according to the type of mutation (Figure 1B). A recent report of four patients with SNCA mutations showed a good response in the three patients with duplications and a poor response in the patient with a missense mutation (p.A53E) [138] (Table 1).
In addition to implications for DBS, the emerging importance of a genetic diagnosis in PD is also related to new gene-based targeted approaches that are being developed in recent years [3], since a specific molecular genetic diagnosis may allow inclusion in interventional clinical trials that target a genetically determined subgroup of PD patients. Moreover, a genetic diagnosis for additional family members at risk of developing PD allows for a more accurate estimation of recurrence risk and informs genetic counseling and family planning. Moreover, some patients are greatly distressed just by the uncertainty regarding the cause for their condition and a genetic diagnosis may bring them great relief.
10.3. Challenges in Genetic Testing
The challenges in genetic testing in PD are related to the patient, the choice of genetic test, and the test results. Patients may be reluctant to perform genetic testing due to different reasons, including a lack of perceived benefit, concern regarding the implications of the test results for them or their family members, or cost. Genetic counseling prior to performing a genetic test is non-directive, meaning that patients or their relatives cannot be directed to have a genetic test, however it should include a thorough, individually tailored explanation regarding the reason why a genetic test is offered, the test itself, its advantages and limitations, and the potential implications of the test results for the patient and their family members. This type of pre-test discussion with the patient is necessary to address the patient’s concerns and to ensure that they are given all the required information to make a knowledge-based decision on whether to proceed with genetic testing or not.
Many types of genetic tests are available in clinical and research settings, ranging from focused testing for a single gene or a specific variant, through variant panels and gene panels, to exome or genome sequencing. Due to the increase in availability and decrease in cost of NGS-based tests, the traditional approach of testing one gene at a time was largely replaced in recent years with broader tests, such as gene panels and exome or genome sequencing, except when a known pathogenic variant has been previously found in the patient’s family, or in uncommon cases where a very high suspicion is raised for a specific gene. When choosing to use a gene panel, one should consider the considerable variability in gene content of different panels. A recent study evaluated the types of clinical genetic tests that are used in PD, revealing notable differences in gene panel size, ranging from 5 to 62 genes. That study showed that five genes were included in all panels (SNCA, PRKN, PINK1, PARK7 (DJ1), and LRRK2), while VPS35 and GBA were only variably included, and that the differences between panels were mainly the result of the variable inclusion of genes associated with atypical parkinsonism and dystonia disorders, or genes with an uncertain association with PD [139]. The selected gene panel should ideally include all established genes for PD with both sequence and deletion/duplication analysis. In cases where the patient presents a combined or an atypical phenotype, a broader approach should be considered, either by using a more comprehensive gene panel or by a genomic analysis with exome or genome sequencing, depending on the specific clinical indicators. Notable limitations that should be taken into consideration are the ones associated with the GBA gene, for which a related pseudogene and structural variations may complicate the detection of pathogenic variants. A novel approach is to use long-read sequencing to assess this gene, with the GridION nanopore sequencing platform recently used in a New Zealand cohort of patients [139]. Another factor to consider is the cost of genetic tests, which might not be covered by the patient’s insurance and therefore may inevitably affect decisions in the molecular workup in some cases. In summary, the decision regarding which genetic test should be used depends on case-specific factors and requires to consider the different types of tests available, their advantages and limitations, and their suitability for each individual patient.
11. Role for Heterozygosity in Autosomal Recessive Parkinson Genes
The possibility that monoallelic pathogenic variants in autosomal recessive PD genes constitute a risk factor for PD is controversial, and conflicting evidence regarding this issue has been reported.
11.1. PRKN Heterozygotes
A recent population-based study analyzed data of 164 confirmed heterozygous PRKN mutation carriers and 2582 controls from South Tyrol in Northern Italy. This study showed a significantly higher number of carriers than controls with a reported akinesia-related phenotype based on a validated PD screening questionnaire [140]. Another study evaluated PRKN as a risk factor for PD in three large independent case-control cohorts and revealed a 1.55-fold risk increase in heterozygous carriers, who also had a younger age of disease onset [141]. However, ~70% of potentially monoallelic cases were not assessed for a second PRKN mutation. To further address this, the authors conducted a meta-analysis of available cohorts and studies of individuals from European ancestry, demonstrating a significant 1.65-fold increase in PD risk in monoallelic PRKN mutation carriers. Nevertheless, when excluding from the analysis studies which did not search for biallelic carriers and those that focused on early-onset PD, no association between monoallelic PRKN mutation and disease risk was found, highlighting the importance of confounding factors that might bias this association [141]. In a recent study, full sequencing and CNV analysis of PRKN in 2809 PD patients and 3629 controls revealed no association between all types of heterozygous PRKN variants and PD risk [142].
11.2. PINK1 Heterozygotes
Several studies have previously suggested that heterozygous PINK1 variants may act as a risk factor for late-onset PD. Of note, one study in a large German family suggested that heterozygous PINK1 mutations may increase the risk for the development of at least subtle motor and non-motor signs of PD [143]. Puschmann et al. investigated the functional effects of the heterozygous PINK1 p.G411S variant and concluded that it acts as a risk factor for PD, which confers its effect by a partial dominant-negative mechanism [144]. A recent comprehensive analysis contradicted these studies. By harnessing combined data from several large datasets totaling 13,708 cases and 362,850 control individuals, this investigation found no evidence of association between heterozygous PINK1 mutations and PD risk [145], further highlighting the complexity and controversy in this field.
11.3. Conclusion on Heterozygous Carriers
The evidence for the role of heterozygous carriers is conflicting—some studies which were based largely on findings in specific cases or families suggested a possible association, while newer studies that utilized large datasets mostly refuted this possibility.
A hidden trans-acting pathogenic variant on the other allele of the gene may at least partly explain these contradictory findings. This may occur in cases where the chosen methodology could not identify these variants, for example when a deletion/duplication analysis was not performed or when the second allele harbored a disease-associated non-coding, structural, or mosaic variant which the molecular testing strategy that was used could not reveal. In these cases, an apparent association between a monoallelic variant and disease may be erroneously concluded. This scenario, however, would not explain cases of families with a clear autosomal dominant inheritance pattern across several generations. Another possible explanation for the conflicting evidence may be that monoallelic deleterious variants in autosomal recessive Parkinson-related genes confer an increased disease risk to some extent as part of a multifactorial inheritance, where each individual, family, or ethnic group are affected by a certain genetic background and/or environmental factors. In this potential scenario, while a monoallelic pathogenic variant may indeed increase the risk for PD, the threshold for disease expression may vary substantially between different individuals, families, or ethnic groups, depending on other genetic variants and environmental factors. This might be missed when analyzing very large, grouped datasets or data that are limited to specific ethnic groups. Other potential factors that might contribute to those contradictory findings may stem from data collection-related biases, such as a recall bias or cases of subtle signs of parkinsonism in reportedly healthy individuals which are considered in the analysis as unaffected controls.
12. Dual LRRK2 and GBA Mutation Carriers
It would be anticipated that having a mutation in both LRRK2 and GBA would have an added deleterious effect, as suggested by laboratory studies [146,147]. However, a recent longitudinal study of a large PD sample measuring progression using the Montreal Cognitive Assessment and Movement Disorders Society—Unified Parkinson Disease Rating Scale–Part III, showed that patients with both the p.G2019S mutation and GBA-PD had a slower rate of decline than those with GBA-PD alone, which was no different from LRRK2-G2019S alone [148]. Similarly, a retrospective observational study of Ashkenazi Jewish patients revealed that patients with mutations in LRRK2 and GBA (described by the authors as “GBA-LRRK2-PD”) were less frequently affected by dementia, probable REM-behavior sleep disorder, and psychosis, compared to other groups (GBA-PD, LRRK2-PD, mutation-negative PD) [149]. This raises the possibility of a protective effect of having the LRRK2 p.G2019S mutation in GBA mutation carriers [149].
13. Conclusions
There have been major advances in research into monogenic PD in recent years. There have been multiple PD gene discoveries, although we highlight the importance of independent validation of these findings. There have been greater insights into genotype–phenotype relationships, and laboratory studies have translated the genetic discoveries into an improved understanding of the pathophysiological mechanism underlying PD.
It has become apparent that there are major ethnic and regional differences in the distribution of mutations in PD genes. There has been further evidence on the role of heterozygous carriers in autosomal recessive PD genes, and the effect of having mutations in both LRRK2 and GBA in the same individual. Additionally, there is a suggestion that the underlying monogenic cause may influence the disease course as well as the response to levodopa and DBS.
Advances in genomic technology provide individuals with PD with greater access to genetic testing through both clinical and research pathways. Global efforts will play a key role in exploiting this genomic data. Worldwide studies can pool many patients to identify rare genetic causes of PD and can also be used to attempt to replicate important genetic discoveries. Furthermore, they offer greater representation of underrepresented populations from different ethnic groups and geographic regions. There are several major global projects to identify new disease genes in PD, including established initiatives such as the International Parkinson Disease Genomics Consortium [150] and newer initiatives such as the Global Parkinson’s disease genetics program (GP2) [151].
PD currently remains an incurable disorder but advances in our understanding of the genetics of PD may inform our understanding of the pathophysiology and thus help with efforts to develop targeted therapies.
Author Contributions
All authors made substantial contributions to the conception and design, drafting of the work and revision, approved the submitted version, and agree to be personally accountable for the authors’ own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and documented in the literature (F.J., A.F. and K.R.K.). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
K.R.K. receives funding to study dystonia from the Paul Ainsworth Family Foundation and receives a Working Group Co-Lead Award from the Michael J. Fox Foundation, Aligning Science Across Parkinson’s (ASAP) initiative, which is unrelated to the current paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kumar, K.R.; Lohmann, K.; Klein, C. Genetics of Parkinson disease and other movement disorders. Curr. Opin. Neurol. 2012, 25, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The cell biology of Parkinson’s disease. J. Cell Biol. 2021, 220, e202012095. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, K.; Rudakou, U.; Gan-Or, Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and Parkin. Neuropharmacology 2022, 202, 108822. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grunewald, A.; Almuammar, S.; Konig, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDS Gene systematic review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Kia, D.A.; Pihlstrom, L.; Gan-Or, Z.; Lesage, S.; Gibbs, J.R.; Ding, J.; Alcalay, R.N.; Hassin-Baer, S.; Pittman, A.M.; et al. Insufficient evidence for pathogenicity of SNCA His50Gln (H50Q) in Parkinson’s disease. Neurobiol. Aging 2018, 64, 159.e5–159.e8. [Google Scholar] [CrossRef]
- Liu, H.; Koros, C.; Strohaker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibanez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J.; SNCA Multiplication Investigators of the GEoPD Consortium. A Meta-Analysis of alpha-Synuclein Multiplication in Familial Parkinsonism. Front. Neurol. 2018, 9, 1021. [Google Scholar] [CrossRef]
- Lesage, S.; Houot, M.; Mangone, G.; Tesson, C.; Bertrand, H.; Forlani, S.; Anheim, M.; Brefel-Courbon, C.; Broussolle, E.; Thobois, S.; et al. Genetic and Phenotypic Basis of Autosomal Dominant Parkinson’s Disease in a Large Multi-Center Cohort. Front. Neurol. 2020, 11, 682. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Over, L.; Bruggemann, N.; Lohmann, K. Therapies for Genetic Forms of Parkinson’s Disease: Systematic Literature Review. J. Neuromuscul. Dis. 2021, 8, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Leaver, K.; Viser, A.; Kopell, B.H.; Ortega, R.A.; Miravite, J.; Okun, M.S.; Elango, S.; Raymond, D.; Bressman, S.B.; Saunders-Pullman, R.; et al. Clinical profiles and outcomes of deep brain stimulation in G2019S LRRK2 Parkinson disease. J. Neurosurg. 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Lunati, A.; Houot, M.; Romdhan, S.B.; Clot, F.; Tesson, C.; Mangone, G.; Toullec, B.L.; Courtin, T.; Larcher, K.; et al. Characterization of recessive Parkinson’s disease in a large multicenter study. Ann. Neurol. 2020, 88, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Buchanan, C.M.; Mulroy, E.; Simpson, M.; Reid, H.A.; Drake, K.M.; Merriman, M.E.; Phipps-Green, A.; Cadzow, M.; Merriman, T.R.; et al. Potential PINK1 Founder Effect in Polynesia Causing Early-Onset Parkinson’s Disease. Mov. Disord. 2021, 36, 2199–2200. [Google Scholar] [CrossRef]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhaes, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687. [Google Scholar] [CrossRef]
- Evidente, V.G.H. X-Linked Dystonia-Parkinsonism. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bruggemann, N.; Domingo, A.; Rasche, D.; Moll, C.K.E.; Rosales, R.L.; Jamora, R.D.G.; Hanssen, H.; Munchau, A.; Graf, J.; Weissbach, A.; et al. Association of Pallidal Neurostimulation and Outcome Predictors With X-linked Dystonia Parkinsonism. JAMA Neurol. 2019, 76, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Wittke, C.; Petkovic, S.; Dobricic, V.; Schaake, S.; Group, M.D.-e.P.S.; Respondek, G.; Weissbach, A.; Madoev, H.; Trinh, J.; Vollstedt, E.J.; et al. Genotype-Phenotype Relations for the Atypical Parkinsonism Genes: MDS Gene Systematic Review. Mov. Disord. 2021, 36, 1499–1510. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef]
- Williams, D.R.; Hadeed, A.; al-Din, A.S.; Wreikat, A.L.; Lees, A.J. Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov. Disord. 2005, 20, 1264–1271. [Google Scholar] [CrossRef]
- Wang, D.; Gao, H.; Li, Y.; Jiang, S.; Yang, X. ATP13A2 Gene Variants in Patients with Parkinson’s Disease in Xinjiang. BioMed Res. Int. 2020, 2020, 6954820. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.F.; Rodriguez, R.D.; Bonifati, V.; Nitrini, R.; Pasqualucci, C.A.; Gelpi, E.; Barbosa, E.R. Neuropathologic Findings in a Patient with Juvenile-Onset Levodopa-Responsive Parkinsonism Due to ATP13A2 Mutation. Neurology 2021, 97, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Dickson, D.W.; Nabeshima, K.; Schmeichel, A.M.; Wszolek, Z.K.; Yamada, T.; Benarroch, E.E. Neurodegeneration involving putative respiratory neurons in Perry syndrome. Acta Neuropathol. 2008, 115, 263–268. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Mishima, T.; Fujioka, S. Perry Disease: Concept of a New Disease and Clinical Diagnostic Criteria. J. Mov. Disord. 2021, 14, 1–9. [Google Scholar] [CrossRef]
- Kurian, M.A.; Abela, L. DNAJC6 Parkinson Disease. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Correa-Vela, M.; Lupo, V.; Montpeyo, M.; Sancho, P.; Marce-Grau, A.; Hernandez-Vara, J.; Darling, A.; Jenkins, A.; Fernandez-Rodriguez, S.; Tello, C.; et al. Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect. Ann. Clin. Transl. Neurol. 2020, 7, 1436–1442. [Google Scholar] [CrossRef]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Ikeda, A.; Nishioka, K.; Meng, H.; Takanashi, M.; Hasegawa, I.; Inoshita, T.; Shiba-Fukushima, K.; Li, Y.; Yoshino, H.; Mori, A.; et al. Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum. Mol. Genet. 2019, 28, 3895–3911. [Google Scholar] [CrossRef]
- Liao, T.W.; Wang, C.C.; Chung, W.H.; Su, S.C.; Chin, S.H.; Fung, H.C.; Wu, Y.R. Role of LRP10 in Parkinson’s disease in a Taiwanese cohort. Parkinsonism Relat. Disord. 2021, 89, 79–83. [Google Scholar] [CrossRef]
- Quadri, M.; Mandemakers, W.; Grochowska, M.M.; Masius, R.; Geut, H.; Fabrizio, E.; Breedveld, G.J.; Kuipers, D.; Minneboo, M.; Vergouw, L.J.M.; et al. LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: A genome-wide linkage and sequencing study. Lancet Neurol. 2018, 17, 597–608. [Google Scholar] [CrossRef]
- Manini, A.; Straniero, L.; Monfrini, E.; Percetti, M.; Vizziello, M.; Franco, G.; Rimoldi, V.; Zecchinelli, A.; Pezzoli, G.; Corti, S.; et al. Screening of LRP10 mutations in Parkinson’s disease patients from Italy. Parkinsonism Relat. Disord. 2021, 89, 17–21. [Google Scholar] [CrossRef]
- Neri, M.; Braccia, A.; Panteghini, C.; Garavaglia, B.; Gualandi, F.; Cavallo, M.A.; Scerrati, A.; Ferlini, A.; Sensi, M. Parkinson’s disease-dementia in trans LRP10 and GBA variants: Response to deep brain stimulation. Parkinsonism Relat. Disord. 2021, 92, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Shi, Y.; Yang, Y.; Ahmeti, K.B.; Miller, N.; Huang, C.; Cheng, L.; Zhai, H.; Deng, S.; Nuytemans, K.; et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 2016, 48, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serratos, I.N.; Hernandez-Perez, E.; Campos, C.; Aschner, M.; Santamaria, A. An Update on the Critical Role of alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Tissue to Cellular and Molecular Levels. Mol. Neurobiol. 2022, 59, 620–642. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, P.A.; Ohnmacht, J.; Santos, B.F.R.; Antony, P.M.; Massart, F.; Cruciani, G.; Dording, C.M.; Pavelka, L.; Casadei, N.; Kwon, Y.J.; et al. Gene-corrected p.A30P SNCA patient-derived isogenic neurons rescue neuronal branching and function. Sci. Rep. 2021, 11, 21946. [Google Scholar] [CrossRef] [PubMed]
- Saunders-Pullman, R.; Raymond, D.; Elango, S. LRRK2 Parkinson Disease. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kestenbaum, M.; Alcalay, R.N. Clinical Features of LRRK2 Carriers with Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 31–48. [Google Scholar] [CrossRef]
- Waro, B.J.; Aasly, J.O. Exploring cancer in LRRK2 mutation carriers and idiopathic Parkinson’s disease. Brain Behav. 2018, 8, e00858. [Google Scholar] [CrossRef] [Green Version]
- Agalliu, I.; San Luciano, M.; Mirelman, A.; Giladi, N.; Waro, B.; Aasly, J.; Inzelberg, R.; Hassin-Baer, S.; Friedman, E.; Ruiz-Martinez, J.; et al. Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: A pooled analysis. JAMA Neurol. 2015, 72, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Agalliu, I.; Ortega, R.A.; Luciano, M.S.; Mirelman, A.; Pont-Sunyer, C.; Brockmann, K.; Vilas, D.; Tolosa, E.; Berg, D.; Waro, B.; et al. Cancer outcomes among Parkinson’s disease patients with leucine rich repeat kinase 2 mutations, idiopathic Parkinson’s disease patients, and nonaffected controls. Mov. Disord. 2019, 34, 1392–1398. [Google Scholar] [CrossRef]
- Macias-Garcia, D.; Perinan, M.T.; Munoz-Delgado, L.; Jesus, S.; Jimenez-Jaraba, M.V.; Buiza-Rueda, D.; Bonilla-Toribio, M.; Adarmes-Gomez, A.; Carrillo, F.; Gomez-Garre, P.; et al. Increased Stroke Risk in Patients with Parkinson’s Disease with LRRK2 Mutations. Mov. Disord. 2022, 37, 225–227. [Google Scholar] [CrossRef]
- San Luciano, M.; Tanner, C.M.; Meng, C.; Marras, C.; Goldman, S.M.; Lang, A.E.; Tolosa, E.; Schule, B.; Langston, J.W.; Brice, A.; et al. Nonsteroidal Anti-inflammatory Use and LRRK2 Parkinson’s Disease Penetrance. Mov. Disord. 2020, 35, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Blauwendraat, C.; Makarious, M.B.; Bandres-Ciga, S.; Leonard, H.L.; Gibbs, J.R.; Hernandez, D.G.; Scholz, S.W.; Faghri, F.; International Parkinson’s Disease Genomics Consortium; et al. Penetrance of Parkinson’s Disease in LRRK2 p.G2019S Carriers Is Modified by a Polygenic Risk Score. Mov. Disord. 2020, 35, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Durr, A.; Tazir, M.; Lohmann, E.; Leutenegger, A.L.; Janin, S.; Pollak, P.; Brice, A.; French Parkinson’s Disease Genetics Study Group. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N. Engl. J. Med. 2006, 354, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Tan, A.H.; Ahmad-Annuar, A.; Klein, C.; Tan, L.C.S.; Rosales, R.L.; Bhidayasiri, R.; Wu, Y.R.; Shang, H.F.; Evans, A.H.; et al. Parkinson’s disease in the Western Pacific Region. Lancet Neurol. 2019, 18, 865–879. [Google Scholar] [CrossRef]
- Xie, C.L.; Pan, J.L.; Wang, W.W.; Zhang, Y.; Zhang, S.F.; Gan, J.; Liu, Z.G. The association between the LRRK2 G2385R variant and the risk of Parkinson’s disease: A meta-analysis based on 23 case-control studies. Neurol. Sci. 2014, 35, 1495–1504. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Q.; Yi, M.; Zhou, X.; Guo, J.; Xu, Q.; Tang, B.; Yan, X. Genetic Analysis of LRRK2 R1628P in Parkinson’s Disease in Asian Populations. Parkinsons Dis. 2017, 2017, 8093124. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Reed, X.; Kia, D.A.; Gan-Or, Z.; Lesage, S.; Pihlstrom, L.; Guerreiro, R.; Gibbs, J.R.; Sabir, M.; Ahmed, S.; et al. Frequency of Loss of Function Variants in LRRK2 in Parkinson Disease. JAMA Neurol. 2018, 75, 1416–1422. [Google Scholar] [CrossRef]
- Bryant, N.; Malpeli, N.; Ziaee, J.; Blauwendraat, C.; Liu, Z.; Consortium, A.P.; West, A.B. Identification of LRRK2 missense variants in the accelerating medicines partnership Parkinson’s disease cohort. Hum. Mol. Genet. 2021, 30, 454–466. [Google Scholar] [CrossRef]
- Berwick, D.C.; Heaton, G.R.; Azeggagh, S.; Harvey, K. LRRK2 Biology from structure to dysfunction: Research progresses, but the themes remain the same. Mol. Neurodegener. 2019, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, M.; Funayama, M.; Li, Y.; Kashihara, K.; Murakami, Y.; Ishizu, N.; Toyoda, C.; Noguchi, K.; Hashimoto, T.; Nakano, N.; et al. VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov. Disord. 2012, 27, 1413–1417. [Google Scholar] [CrossRef]
- Kumar, K.R.; Weissbach, A.; Heldmann, M.; Kasten, M.; Tunc, S.; Sue, C.M.; Svetel, M.; Kostic, V.S.; Segura-Aguilar, J.; Ramirez, A.; et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch. Neurol. 2012, 69, 1360–1364. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.A.; Morrison, B.E. Contributions of VPS35 Mutations to Parkinson’s Disease. Neuroscience 2019, 401, 1–10. [Google Scholar] [CrossRef]
- Ishiguro, M.; Li, Y.; Yoshino, H.; Daida, K.; Ishiguro, Y.; Oyama, G.; Saiki, S.; Funayama, M.; Hattori, N.; Nishioka, K. Clinical manifestations of Parkinson’s disease harboring VPS35 retromer complex component p.D620N with long-term follow-up. Parkinsonism Relat. Disord. 2021, 84, 139–143. [Google Scholar] [CrossRef]
- Cutillo, G.; Simon, D.K.; Eleuteri, S. VPS35 and the mitochondria: Connecting the dots in Parkinson’s disease pathophysiology. Neurobiol. Dis. 2020, 145, 105056. [Google Scholar] [CrossRef]
- Sassone, J.; Reale, C.; Dati, G.; Regoni, M.; Pellecchia, M.T.; Garavaglia, B. The Role of VPS35 in the Pathobiology of Parkinson’s Disease. Cell. Mol. Neurobiol. 2021, 41, 199–227. [Google Scholar] [CrossRef]
- Lesage, S.; Magali, P.; Lohmann, E.; Lacomblez, L.; Teive, H.; Janin, S.; Cousin, P.Y.; Durr, A.; Brice, A.; French Parkinson Disease Genetics Study Group. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum. Mutat. 2007, 28, 27–32. [Google Scholar] [CrossRef]
- Bruggemann, N.; Klein, C. Parkin Type of Early-Onset Parkinson Disease. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, A.V.; Campbell, P.; Schapira, A.H.V.; Morris, H.R.; Taanman, J.W. The PINK1-Parkin mitophagy signalling pathway is not functional in peripheral blood mononuclear cells. PLoS ONE 2021, 16, e0259903. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Klein, C. PINK1 Type of Young-Onset Parkinson Disease. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Guler, S.; Gul, T.; Guler, S.; Haerle, M.C.; Basak, A.N. Early-Onset Parkinson’s Disease: A Novel Deletion Comprising the DJ-1 and TNFRSF9 Genes. Mov. Disord. 2021, 36, 2973–2976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 994. [Google Scholar] [CrossRef]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [Green Version]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- Pauly, M.G.; Ruiz Lopez, M.; Westenberger, A.; Saranza, G.; Bruggemann, N.; Weissbach, A.; Rosales, R.L.; Diesta, C.C.; Jamora, R.D.G.; Reyes, C.J.; et al. Expanding Data Collection for the MDSGene Database: X-linked Dystonia-Parkinsonism as Use Case Example. Mov. Disord. 2020, 35, 1933–1938. [Google Scholar] [CrossRef]
- Aneichyk, T.; Hendriks, W.T.; Yadav, R.; Shin, D.; Gao, D.; Vaine, C.A.; Collins, R.L.; Domingo, A.; Currall, B.; Stortchevoi, A.; et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172, 897–909.e21. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.V.; Maranon, E.; Demaisip, C.; Peralta, O.; Borres-Icasiano, R.; Arancillo, J.; Rivera, C.; Munoz, E.; Tan, K.; Reyes, M.T. The natural history of sex-linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Parkinsonism Relat. Disord. 2002, 9, 29–38. [Google Scholar] [CrossRef]
- Santiano, R.A.S.; Rosales, R.L. A Cross-Cultural Validation of the Filipino and Hiligaynon Versions of the Parts IIIB (Non-Motor Features) and IV (Activities of Daily Living) of the X-Linked Dystonia-Parkinsonism- MDSP Rating Scale. Clin. Parkinsonism Relat. Disord. 2021, 5, 100100. [Google Scholar] [CrossRef]
- Westenberger, A.; Reyes, C.J.; Saranza, G.; Dobricic, V.; Hanssen, H.; Domingo, A.; Laabs, B.H.; Schaake, S.; Pozojevic, J.; Rakovic, A.; et al. A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Ann. Neurol. 2019, 85, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Nybo, C.J.; Gustavsson, E.K.; Farrer, M.J.; Aasly, J.O. Neuropathological findings in PINK1-associated Parkinson’s disease. Parkinsonism Relat. Disord. 2020, 78, 105–108. [Google Scholar] [CrossRef] [PubMed]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.; McEntagart, M.M.; Isaacs, J.D. DCTN1-related Parkinson-plus disorder (Perry syndrome). Pract. Neurol. 2020, 20, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Dachsel, J.C.; Farrer, M.J.; Dickson, D.W.; Tsuboi, Y.; Wszolek, Z.K. Elucidating the genetics and pathology of Perry syndrome. J. Neurol. Sci. 2010, 289, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dachsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 mutations in Perry syndrome. Nat. Genet. 2009, 41, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Cinnamon, Y.; Ta-Shma, A.; Shaag, A.; Yim, Y.I.; Zenvirt, S.; Jalas, C.; Lesage, S.; Brice, A.; Taraboulos, A.; et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 2012, 7, e36458. [Google Scholar] [CrossRef] [Green Version]
- Koroglu, C.; Baysal, L.; Cetinkaya, M.; Karasoy, H.; Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 2013, 19, 320–324. [Google Scholar] [CrossRef]
- Olgiati, S.; Quadri, M.; Fang, M.; Rood, J.P.; Saute, J.A.; Chien, H.F.; Bouwkamp, C.G.; Graafland, J.; Minneboo, M.; Breedveld, G.J.; et al. DNAJC6 Mutations Associated with Early-Onset Parkinson’s Disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar] [CrossRef]
- Yim, Y.I.; Sun, T.; Wu, L.G.; Raimondi, A.; De Camilli, P.; Eisenberg, E.; Greene, L.E. Endocytosis and clathrin-uncoating defects at synapses of auxilin knockout mice. Proc. Natl. Acad. Sci. USA 2010, 107, 4412–4417. [Google Scholar] [CrossRef] [Green Version]
- Di Fonzo, A.; Dekker, M.C.; Montagna, P.; Baruzzi, A.; Yonova, E.H.; Correia Guedes, L.; Szczerbinska, A.; Zhao, T.; Dubbel-Hulsman, L.O.; Wouters, C.H.; et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 2009, 72, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ding, L.; Li, H.; Lin, Y.; Dai, Y.; Xu, X.; Dong, Q.; Lin, Y.; Long, L. Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-Box only protein 7 gene. Parkinsonism Relat. Disord. 2018, 47, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Yalcin-Cakmakli, G.; Olgiati, S.; Quadri, M.; Breedveld, G.J.; Cortelli, P.; Bonifati, V.; Elibol, B. A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat. Disord. 2014, 20, 1248–1252. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, E.; Coquel, A.S.; Honore, A.; Gurvit, H.; Hanagasi, H.; Emre, M.; Leutenegger, A.L.; Drouet, V.; Sahbatou, M.; Guven, G.; et al. A new F-box protein 7 gene mutation causing typical Parkinson’s disease. Mov. Disord. 2015, 30, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paisan-Ruiz, C.; Bhatia, K.P.; Li, A.; Hernandez, D.; Davis, M.; Wood, N.W.; Hardy, J.; Houlden, H.; Singleton, A.; Schneider, S.A. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 2009, 65, 19–23. [Google Scholar] [CrossRef]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Karkheiran, S.; Shahidi, G.A.; Walker, R.H.; Paisan-Ruiz, C. PLA2G6-associated Dystonia-Parkinsonism: Case Report and Literature Review. Tremor Other Hyperkinet. Mov. 2015, 5, 317. [Google Scholar] [CrossRef]
- Lesage, S.; Mangone, G.; Tesson, C.; Bertrand, H.; Benmahdjoub, M.; Kesraoui, S.; Arezki, M.; Singleton, A.; Corvol, J.C.; Brice, A. Clinical Variability of SYNJ1-Associated Early-Onset Parkinsonism. Front. Neurol. 2021, 12, 648457. [Google Scholar] [CrossRef]
- Cao, M.; Wu, Y.; Ashrafi, G.; McCartney, A.J.; Wheeler, H.; Bushong, E.A.; Boassa, D.; Ellisman, M.H.; Ryan, T.A.; De Camilli, P. Parkinson Sac Domain Mutation in Synaptojanin 1 Impairs Clathrin Uncoating at Synapses and Triggers Dystrophic Changes in Dopaminergic Axons. Neuron 2017, 93, 882–896.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.H.; Mao, C.Y.; Zhang, S.Y.; Yang, J.; Song, B.; Wu, P.; Zuo, C.T.; Liu, Y.T.; Ji, Y.; Yang, Z.H.; et al. CHCHD2 gene mutations in familial and sporadic Parkinson’s disease. Neurobiol. Aging 2016, 38, 217.e9–217.e13. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, Q.; An, R.; Zheng, J.; Tian, S.; Chen, Y.; Xu, Y. Mutational scanning of the CHCHD2 gene in Han Chinese patients with Parkinson’s disease and meta-analysis of the literature. Parkinsonism Relat. Disord. 2016, 29, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Bras, J.M.; Lesage, S.; Schulte, C.; Gibbs, J.R.; Nalls, M.A.; Brice, A.; Wood, N.W.; Morris, H.; Hardy, J.A.; et al. CHCHD2 and Parkinson’s disease. Lancet Neurol. 2015, 14, 678–679. [Google Scholar] [CrossRef]
- Lee, R.G.; Sedghi, M.; Salari, M.; Shearwood, A.J.; Stentenbach, M.; Kariminejad, A.; Goullee, H.; Rackham, O.; Laing, N.G.; Tajsharghi, H.; et al. Early-onset Parkinson disease caused by a mutation in CHCHD2 and mitochondrial dysfunction. Neurol. Genet. 2018, 4, e276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef] [Green Version]
- Tesson, C.; Brefel-Courbon, C.; Corvol, J.C.; Lesage, S.; Brice, A.; French Parkinson’s Disease Genetics Study Group. LRP10 in alpha-synucleinopathies. Lancet Neurol. 2018, 17, 1034. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cen, Z.; Zheng, X.; Pan, Q.; Chen, X.; Zhu, L.; Chen, S.; Wu, H.; Xie, F.; Wang, H.; et al. LRP10 in autosomal-dominant Parkinson’s disease. Mov. Disord. 2019, 34, 912–916. [Google Scholar] [CrossRef]
- Daida, K.; Nishioka, K.; Li, Y.; Yoshino, H.; Kikuchi, A.; Hasegawa, T.; Funayama, M.; Hattori, N. Mutation analysis of LRP10 in Japanese patients with familial Parkinson’s disease, progressive supranuclear palsy, and frontotemporal dementia. Neurobiol. Aging 2019, 84, 235.e11–235.e16. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.; Ou, R.; Gu, X.; Wei, Q.; Cao, B.; Zhang, L.; Hou, Y.; Liu, K.; Chen, X.; et al. Mutation analysis of LRP10 in a large Chinese familial Parkinson disease cohort. Neurobiol. Aging 2021, 99, 99.e1–99.e6. [Google Scholar] [CrossRef] [PubMed]
- Grochowska, M.M.; Carreras Mascaro, A.; Boumeester, V.; Natale, D.; Breedveld, G.J.; Geut, H.; van Cappellen, W.A.; Boon, A.J.W.; Kievit, A.J.A.; Sammler, E.; et al. LRP10 interacts with SORL1 in the intracellular vesicle trafficking pathway in non-neuronal brain cells and localises to Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2021, 142, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Vilarino-Guell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, M.J. Doubts about TMEM230 as a gene for parkinsonism. Nat. Genet. 2019, 51, 367–368. [Google Scholar] [CrossRef]
- Deng, H.X.; Pericak-Vance, M.A.; Siddique, T. Reply to ‘TMEM230 variants in Parkinson’s disease’ and ‘Doubts about TMEM230 as a gene for parkinsonism’. Nat. Genet. 2019, 51, 369–371. [Google Scholar] [CrossRef]
- Kim, M.J.; Deng, H.X.; Wong, Y.C.; Siddique, T.; Krainc, D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 2017, 26, 729–741. [Google Scholar] [CrossRef]
- Lin, C.H.; Tsai, P.I.; Lin, H.Y.; Hattori, N.; Funayama, M.; Jeon, B.; Sato, K.; Abe, K.; Mukai, Y.; Takahashi, Y.; et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain 2020, 143, 3352–3373. [Google Scholar] [CrossRef]
- Senkevich, K.; Bandres-Ciga, S.; Gan-Or, Z.; Krohn, L.; International Parkinson’s Disease Genomics Consortium. Lack of evidence for association of UQCRC1 with Parkinson’s disease in Europeans. Neurobiol. Aging 2021, 101, 297.e1–297.e4. [Google Scholar] [CrossRef]
- Shan, W.; Li, J.; Xu, W.; Li, H.; Zuo, Z. Critical role of UQCRC1 in embryo survival, brain ischemic tolerance and normal cognition in mice. Cell. Mol. Life Sci. 2019, 76, 1381–1396. [Google Scholar] [CrossRef]
- Hung, Y.C.; Huang, K.L.; Chen, P.L.; Li, J.L.; Lu, S.H.; Chang, J.C.; Lin, H.Y.; Lo, W.C.; Huang, S.Y.; Lee, T.T.; et al. UQCRC1 engages cytochrome c for neuronal apoptotic cell death. Cell Rep. 2021, 36, 109729. [Google Scholar] [CrossRef]
- Rudakou, U.; Ruskey, J.A.; Krohn, L.; Laurent, S.B.; Spiegelman, D.; Greenbaum, L.; Yahalom, G.; Desautels, A.; Montplaisir, J.Y.; Fahn, S.; et al. Analysis of common and rare VPS13C variants in late-onset Parkinson disease. Neurol. Genet. 2020, 6, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Mari, M.; Parashar, S.; Liu, D.; Cui, Y.; Reggiori, F.; Novick, P.J.; Ferro-Novick, S. Vps13 is required for the packaging of the ER into autophagosomes during ER-phagy. Proc. Natl. Acad. Sci. USA 2020, 117, 18530–18539. [Google Scholar] [CrossRef] [PubMed]
- Morales-Briceno, H.; Mohammad, S.S.; Post, B.; Fois, A.F.; Dale, R.C.; Tchan, M.; Fung, V.S.C. Clinical and neuroimaging phenotypes of genetic parkinsonism from infancy to adolescence. Brain 2020, 143, 751–770. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, H.; Azcona, L.J.; Aghamollaii, V.; Omrani, M.D.; Garshasbi, M.; Taghavi, S.; Tafakhori, A.; Shahidi, G.A.; Jamshidi, J.; Darvish, H.; et al. PTRHD1 (C2orf79) mutations lead to autosomal-recessive intellectual disability and parkinsonism. Mov. Disord. 2017, 32, 287–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.R.; Sim, J.C.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with alpha-synuclein pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, P.; Rudakou, U.; Yu, E.; Ruskey, J.A.; Asayesh, F.; Laurent, S.B.; Spiegelman, D.; Fahn, S.; Waters, C.; Monchi, O.; et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson’s disease. Neurobiol. Aging 2021, 100, 119.e7–119.e13. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Skrahina, V.; Gaber, H.; Vollstedt, E.J.; Forster, T.M.; Usnich, T.; Curado, F.; Bruggemann, N.; Paul, J.; Bogdanovic, X.; Zulbahar, S.; et al. The Rostock International Parkinson’s Disease (ROPAD) Study: Protocol and Initial Findings. Mov. Disord. 2021, 36, 1005–1010. [Google Scholar] [CrossRef]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Lim, J.L.; Lohmann, K.; Tan, A.H.; Tay, Y.W.; Ibrahim, K.A.; Abdul Aziz, Z.; Mawardi, A.S.; Puvanarajah, S.D.; Lim, T.T.; Looi, I.; et al. Glucocerebrosidase (GBA) gene variants in a multi-ethnic Asian cohort with Parkinson’s disease: Mutational spectrum and clinical features. J. Neural Transm. 2022, 129, 37–48. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Evans, J.; Breen, D.P.; Barker, R.A.; Williams-Gray, C.H. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaiti-Pellie, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Rana, H.Q.; Balwani, M.; Bier, L.; Alcalay, R.N. Age-specific Parkinson disease risk in GBA mutation carriers: Information for genetic counseling. Genet. Med. 2013, 15, 146–149. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Dinur, T.; Becker-Cohen, M.; Revel-Vilk, S.; Zimran, A.; Arkadir, D. Parkinson’s Clustering in Families of Non-Neuronopathic N370S GBA Mutation Carriers Indicates the Presence of Genetic Modifiers. J. Parkinsons Dis. 2021, 11, 615–618. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Kehoe, C.; Shorr, E.; Battista, R.; Hall, A.; Simuni, T.; Marder, K.; Wills, A.M.; Naito, A.; Beck, J.C.; et al. Genetic testing for Parkinson disease: Current practice, knowledge, and attitudes among US and Canadian movement disorders specialists. Genet. Med. 2020, 22, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.; Schulze, J.; Kopil, C.; Hastings, T.; Naito, A.; Wojcieszek, J.; Payne, K.; Alcalay, R.N.; Klein, C.; Saunders-Pullman, R.; et al. Genetic Testing for Parkinson Disease: Are We Ready. Neurol. Clin. Pract. 2021, 11, 69–77. [Google Scholar] [CrossRef]
- Mangone, G.; Bekadar, S.; Cormier-Dequaire, F.; Tahiri, K.; Welaratne, A.; Czernecki, V.; Pineau, F.; Karachi, C.; Castrioto, A.; Durif, F.; et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat. Disord. 2020, 76, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Pal, G.; Mangone, G.; Hill, E.J.; Ouyang, B.; Liu, Y.; Lythe, V.; Ehrlich, D.; Saunders-Pullman, R.; Shanker, V.; Bressman, S.; et al. Parkinson Disease and Subthalamic Nucleus Deep Brain Stimulation: Cognitive Effects in GBA Mutation Carriers. Ann. Neurol. 2022, 91, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.; Oyama, G.; Hattori, N.; Shimo, Y.; Kuusimaki, T.; Kaasinen, V.; Antonini, A.; Kim, D.; Lee, J.I.; Cho, K.R.; et al. Subthalamic deep brain stimulation in Parkinson’s disease with SNCA mutations: Based on the follow-up to 10 years. Brain Behav. 2022, 12, e2503. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.; Schulze, J.; Verbrugge, J.; Beck, J.C.; Marder, K.S.; Saunders-Pullman, R.; Klein, C.; Naito, A.; Alcalay, R.N.; ClinGen Parkinson’s Disease Gene Curation Expert Panel; et al. The commercial genetic testing landscape for Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 92, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Castelo Rueda, M.P.; Raftopoulou, A.; Gogele, M.; Borsche, M.; Emmert, D.; Fuchsberger, C.; Hantikainen, E.M.; Vukovic, V.; Klein, C.; Pramstaller, P.P.; et al. Frequency of Heterozygous Parkin (PRKN) Variants and Penetrance of Parkinson’s Disease Risk Markers in the Population-Based CHRIS Cohort. Front. Neurol. 2021, 12, 706145. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Bustos, B.I.; Hu, J.; Krainc, D.; Joseph, T.; Hehir, J.; Tan, M.; Zhang, W.; Escott-Price, V.; Williams, N.M.; et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. Hum. Mol. Genet. 2021, 30, 78–86. [Google Scholar] [CrossRef]
- Yu, E.; Rudakou, U.; Krohn, L.; Mufti, K.; Ruskey, J.A.; Asayesh, F.; Estiar, M.A.; Spiegelman, D.; Surface, M.; Fahn, S.; et al. Analysis of Heterozygous PRKN Variants and Copy-Number Variations in Parkinson’s Disease. Mov. Disord. 2021, 36, 178–187. [Google Scholar] [CrossRef]
- Eggers, C.; Schmidt, A.; Hagenah, J.; Bruggemann, N.; Klein, J.C.; Tadic, V.; Kertelge, L.; Kasten, M.; Binkofski, F.; Siebner, H.; et al. Progression of subtle motor signs in PINK1 mutation carriers with mild dopaminergic deficit. Neurology 2010, 74, 1798–1805. [Google Scholar] [CrossRef]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Krohn, L.; Grenn, F.P.; Makarious, M.B.; Kim, J.J.; Bandres-Ciga, S.; Roosen, D.A.; Gan-Or, Z.; Nalls, M.A.; Singleton, A.B.; Blauwendraat, C.; et al. Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 2020, 91, 168.e1–168.e5. [Google Scholar] [CrossRef]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.; DeAndrade, M.P.; Novis, H.S.; Lin, S.; Chang, J.; Lengacher, N.; Tomlinson, J.J.; Tansey, M.G.; LaVoie, M.J. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov. Disord. 2020, 35, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.A.; Wang, C.; Raymond, D.; Bryant, N.; Scherzer, C.R.; Thaler, A.; Alcalay, R.N.; West, A.B.; Mirelman, A.; Kuras, Y.; et al. Association of Dual LRRK2 G2019S and GBA Variations with Parkinson Disease Progression. JAMA Netw. Open 2021, 4, e215845. [Google Scholar] [CrossRef] [PubMed]
- Yahalom, G.; Greenbaum, L.; Israeli-Korn, S.; Fay-Karmon, T.; Livneh, V.; Ruskey, J.A.; Ronciere, L.; Alam, A.; Gan-Or, Z.; Hassin-Baer, S. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat. Disord. 2019, 62, 179–184. [Google Scholar] [CrossRef] [PubMed]
- International Parkinson Disease Genomics Consortium. Ten Years of the International Parkinson Disease Genomics Consortium: Progress and Next Steps. J. Parkinsons Dis. 2020, 10, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Global Parkinson’s Genetics Program. GP2: The Global Parkinson’s Genetics Program. Mov. Disord. 2021, 36, 842–851. [Google Scholar] [CrossRef]
Figure 1.
(A) Increasing cognitive decline in GBA carriers versus PRKN, LRRK2, and those without disease-associated variants. (B) Outcome of deep brain stimulation stratified according to Parkinson’s disease monogenic forms.
Figure 1.
(A) Increasing cognitive decline in GBA carriers versus PRKN, LRRK2, and those without disease-associated variants. (B) Outcome of deep brain stimulation stratified according to Parkinson’s disease monogenic forms.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030471
AMA Style
Jia F, Fellner A, Kumar KR. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes. 2022; 13(3):471. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030471
Chicago/Turabian StyleJia, Fangzhi, Avi Fellner, and Kishore Raj Kumar. 2022. "Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing" Genes 13, no. 3: 471. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030471
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.