
De Novo Transcriptome Analysis Reveals Putative Genes Involved in Anthraquinone Biosynthesis in Rubia yunnanensis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. RNA Isolation and cDNA Library Preparation for Illumina Sequencing
2.3. De Novo Assembly and Sequence Analysis
2.4. Functional Annotation of Transcripts
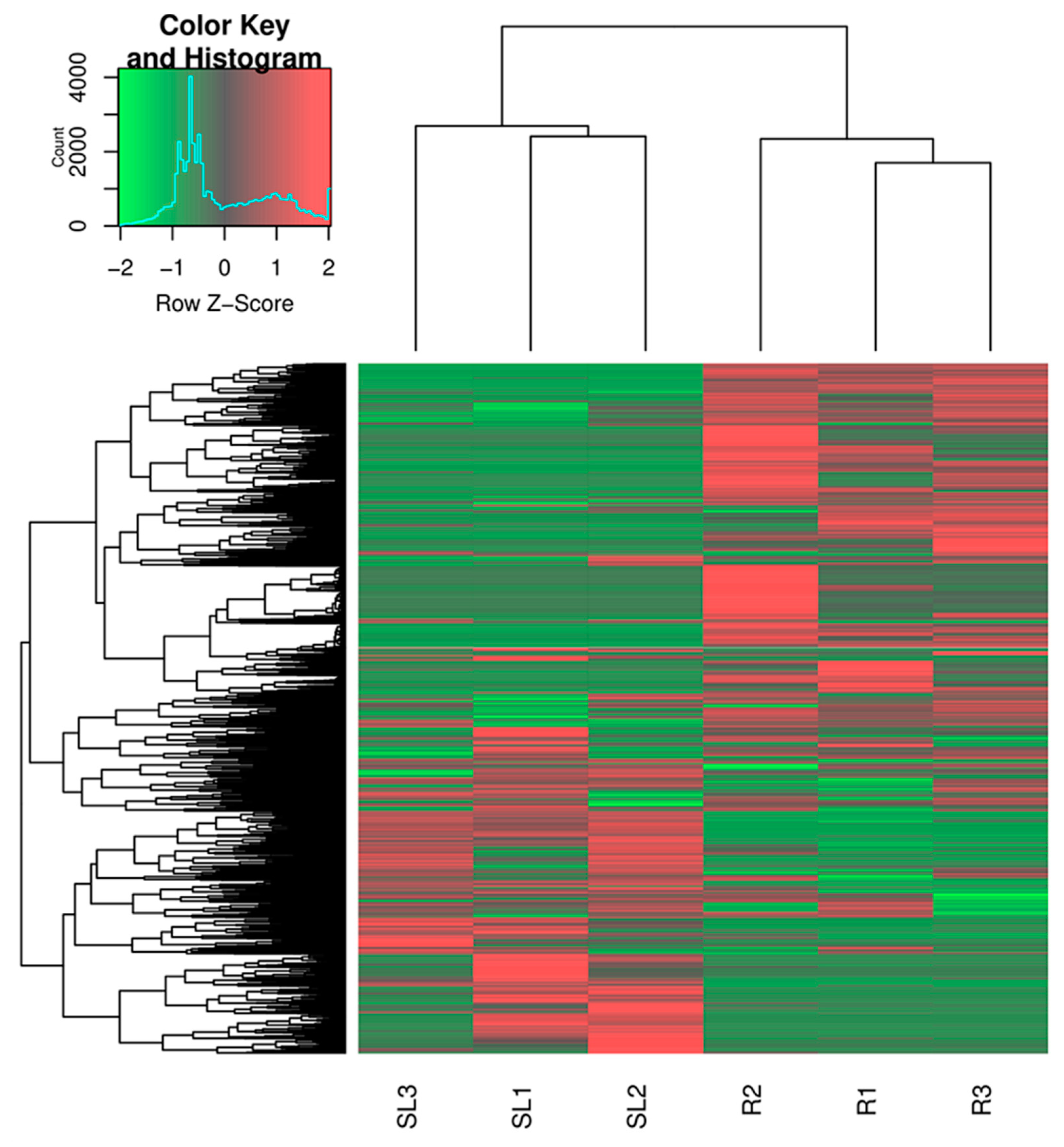
2.5. Identification of Differentially Expressed Genes (DEGs)
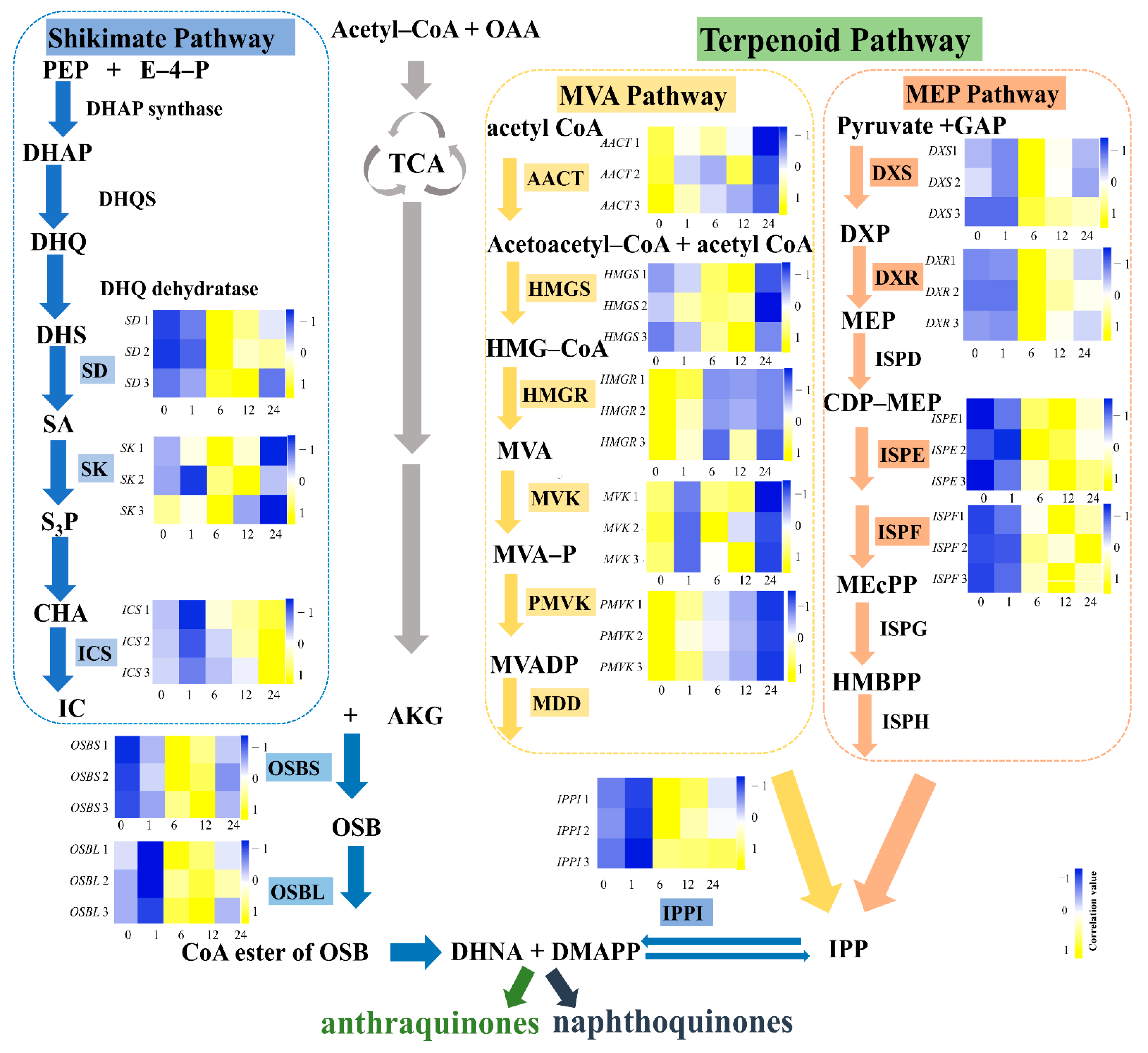
2.6. Analysis of Putative Genes Involved in Anthraquinone Biosynthesis in the R. yunnanensis Transcriptome
2.7. RT-qPCR Validation of the Gene Expression Level Quantified Using Illumina Sequencing
2.8. RT-qPCR Analysis for 15 Genes Involved in Anthraquinone Biosynthesis
2.9. Quantitative Analysis of Six Anthraquinones and Two Naphthoquinones in the Hairy Roots of R. yunnanensis
2.10. Statistical Analysis
3. Results
3.1. Illumina Sequencing and De Novo Sequence Assembly
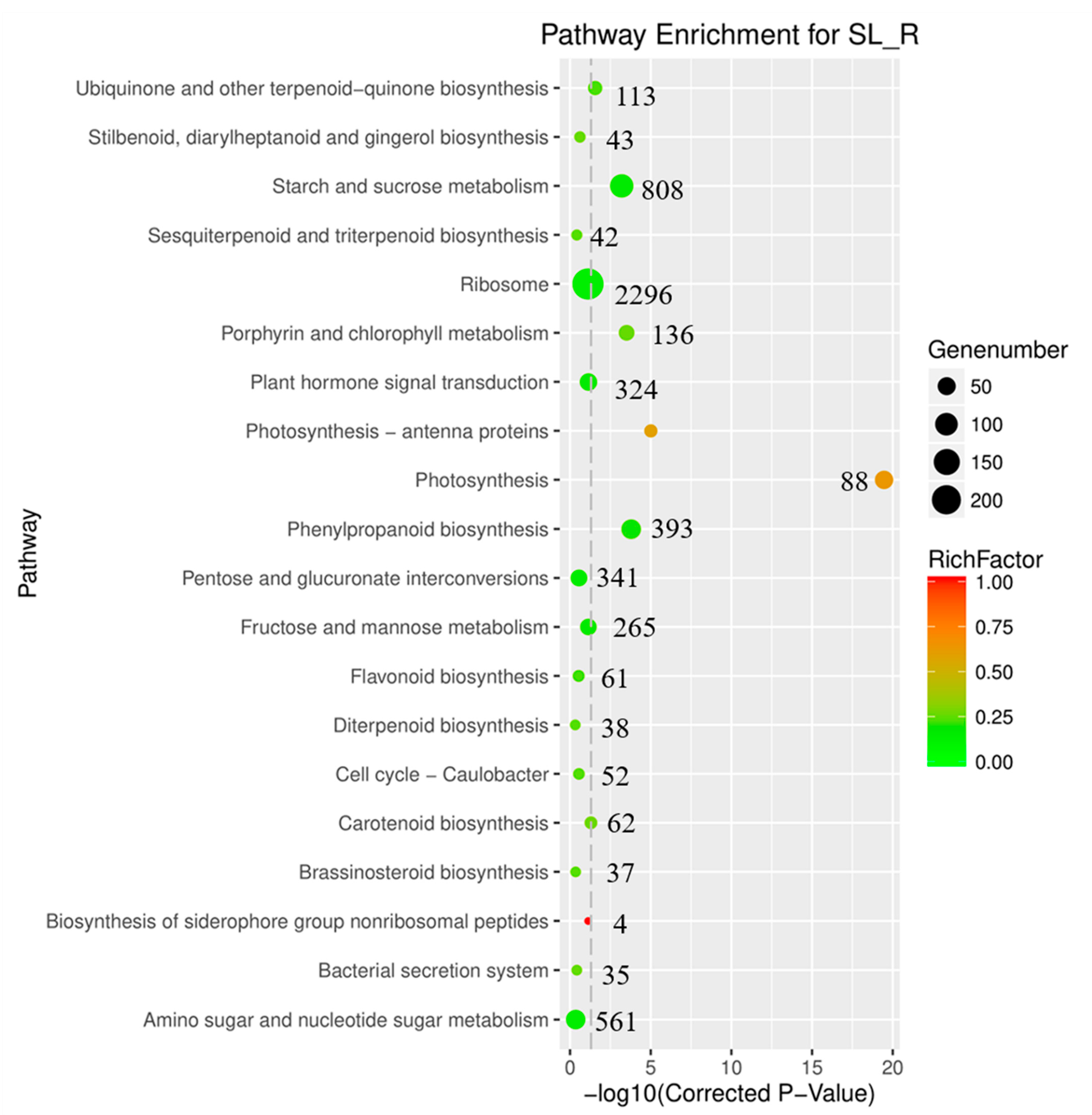
3.2. Function Annotation Analysis
3.3. Identification of Genes Involved in Anthraquinone Biosynthesis
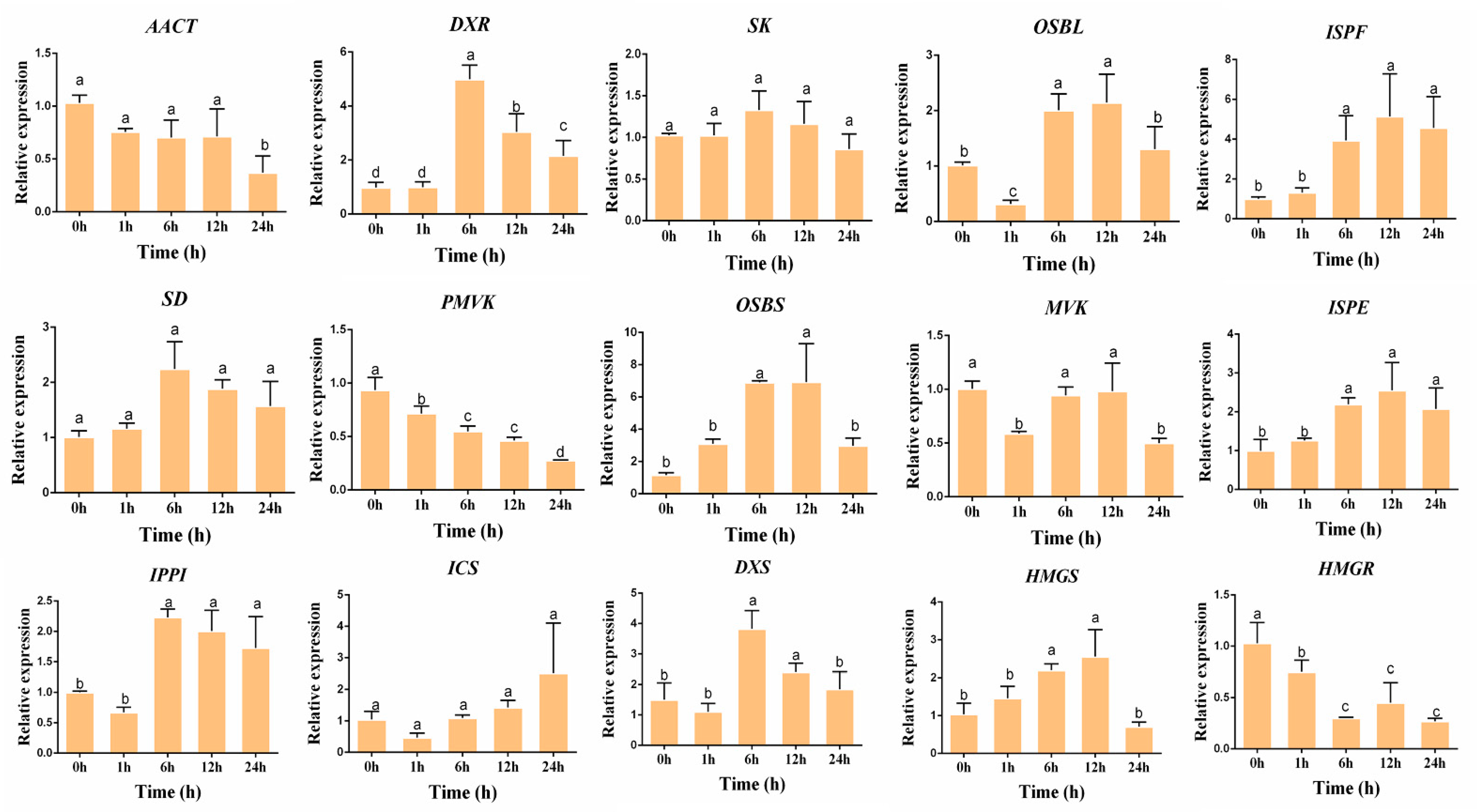
3.4. Validation of the Gene Expression Level
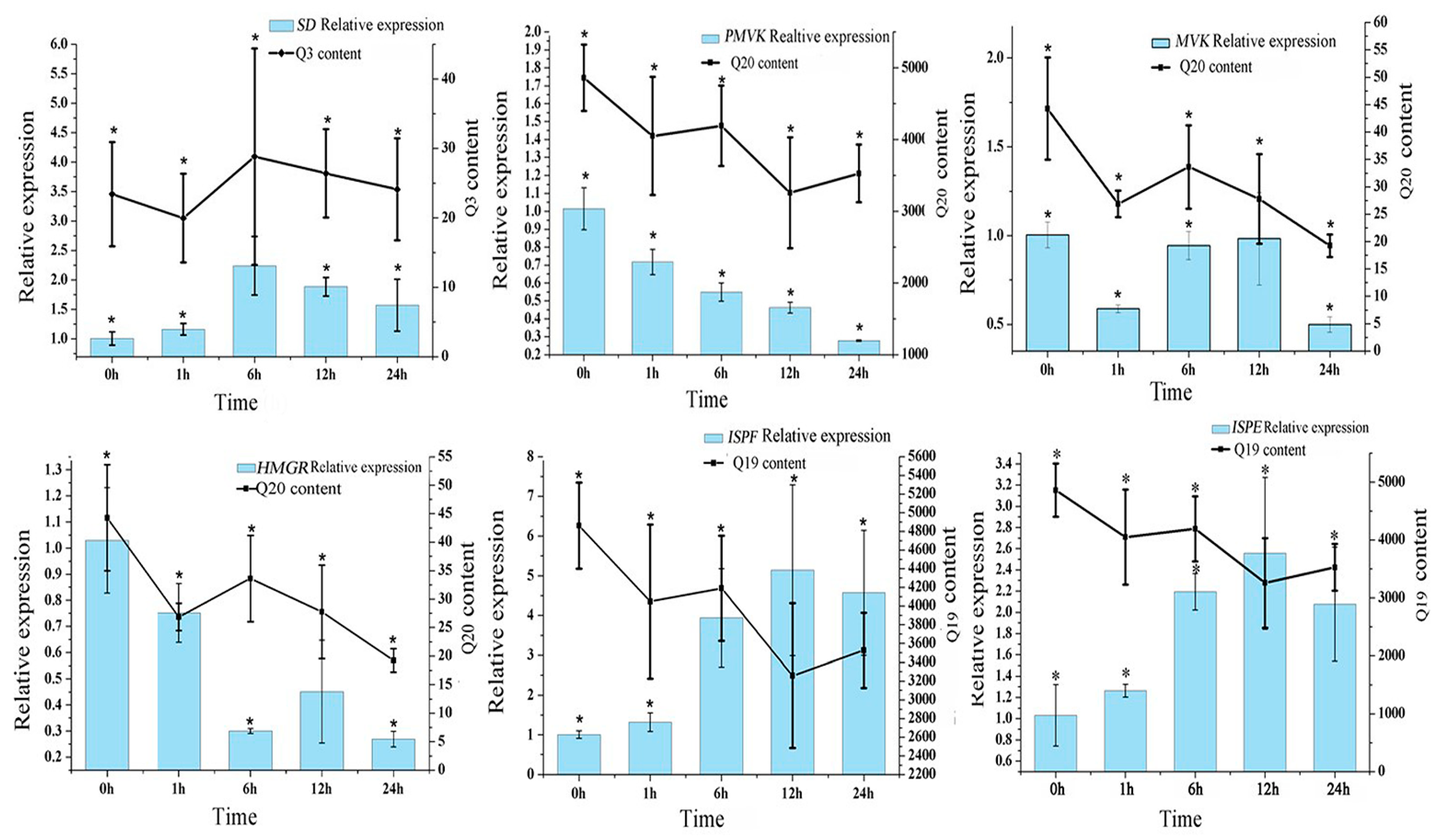
3.5. Gene Expressions Levels and Anthraquinone Contents in the Hairy Roots of R. yunnanensis under MeJA Treatment
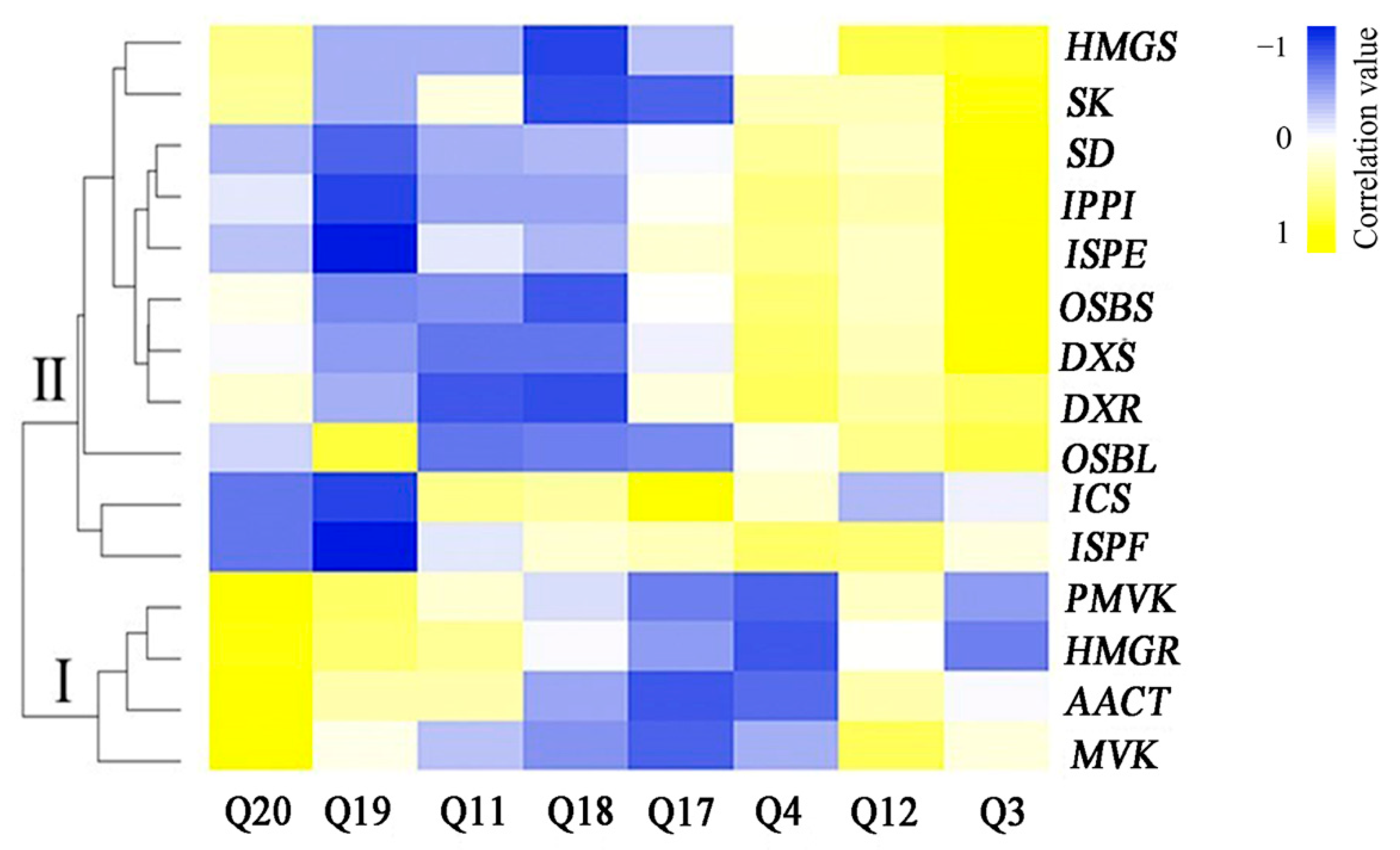
3.6. Correlation Analysis between the Expression Levels of Genes Involved in Anthraquinone Biosynthesis and Anthraquinone Contents
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chinese Flora Editorial Board; Chinese Academy of Science. Flora of China; Science Press: Beijing, China, 1999; pp. 303–305. [Google Scholar]
- Lan, M. Dian Nan Ben Cao; Yunnan People’s Publishing House: Kunming, China, 1978; pp. 349–351. [Google Scholar]
- Liou, M.J.; Wu, T.S. Triterpenoids from Rubia yunnanensis. J. Nat. Prod. 2002, 65, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Su, Y.; Huo, Y.; Mi, J.; Wang, X.; Wang, Z.; Liu, Y.; Zhang, H. Identification of antihyperlipidemic constituents from the roots of Rubia yunnanensis Diels. J. Ethnopharmacol. 2014, 155, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.Z.; Fan, J.T.; Xu, J.J.; Li, Y.; Tan, N.H. Apoptosis induction and G2/M arrest of 2-methyl-1,3,6-trihydroxy-9,10-anthraquinone from Rubia yunnanensis in human cervical cancer HeLa cells. Pharmazie 2013, 68, 293–299. [Google Scholar] [PubMed]
- Liu, M.J.; Teng, C.M.; Wu, T.S. Constituents from Rubia ustulata Diels and R. yunnanensis Diels and their antiplatelet aggregation activity. J. Chin. Chem. Soc. Taip. 2002, 49, 1025–1030. [Google Scholar] [CrossRef]
- Fan, J.T.; Kuang, B.; Zeng, G.Z.; Zhao, S.M.; Ji, C.J.; Zhang, Y.M.; Tan, N.H. Biologically active arborinane-type triterpenoids and anthraquinones from Rubia yunnanensis. J. Nat. Prod. 2011, 74, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Zhang, X.J.; Zhang, Z.H.; Wang, Z.; Tan, N.H. Qualitative and quantitative analyses of quinones in multi-origin Rubia species by ultra-performance liquid chromatography-tandem mass spectrometry combined with chemometrics. J. Pharm. Biomed. Anal. 2020, 189, 113471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.M.; Kuang, B.; Zeng, G.Z.; Wang, Z.; Wang, J.; Chen, X.Q.; Tan, N.H. Nematicidal quinone derivatives from three Rubia plants. Tetrahedron 2018, 74, 2115–2220. [Google Scholar] [CrossRef]
- Bonadonna, G.; Monfardini, S.; De Lena, M.; Fossati-Bellani, F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br. Med. J. 1969, 3, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessler, N.N.; Egorova, A.S.; Belozerskaya, T.A. Fungal anthraquinones. Appl. Biochem. Microbiol. 2013, 49, 85–99. [Google Scholar] [CrossRef]
- Pankewitz, F.; Zöllmer, A.; Hilker, M.; Gräser, Y. Presence of Wolbachia in insect eggs containing antimicrobially active anthraquinones. Microb. Ecol. 2007, 54, 713–721. [Google Scholar] [CrossRef]
- Malik, E.M.; Müller, C.E. Anthraquinones as pharmacological tools and drugs. Med. Res. Rev. 2016, 36, 705–748. [Google Scholar] [CrossRef] [PubMed]
- Caro, Y.; Anamale, L.; Fouillaud, M.; Laurent, P.; Petit, T.; Dufosse, L. Natural hydroxyanthraquinoid pigments as potent food grade colorants: An overview. Nat. Prod. Bioprospect. 2012, 2, 174–193. [Google Scholar] [CrossRef]
- Dufossé, L. Anthraquinones, the Dr Jekyll and Mr Hyde of the food pigment family. Food Res. Int. 2014, 65, 132–136. [Google Scholar] [CrossRef]
- Han, Y.S.; van der Heijden, R.; Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant. Cell Tiss. Org. 2001, 67, 201–220. [Google Scholar] [CrossRef]
- Liang, W.; Sun, J.C.; Guo, F.X.; Zhang, X.M.; Xu, B.; Chen, Y.; Li, X. Research progress on synthesis of anthraquinones based on shikimic acid/o-succinylbenzoic acid pathway. Chin. Trad. Herb. Drugs 2020, 51, 1939–1950. [Google Scholar]
- Han, Y.S.; van der Heijden, R.; Lefeber, A.W.M.; Eekelens, C.; Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of Cinchona ‘Robusta’ proceeds via the methylerythritol 4-phosphate pathway. Phytochemistry 2002, 59, 45–55. [Google Scholar] [CrossRef]
- Ramos-Valdivia, A.C.; van der Heijden, R.; Verpoorte, R. Isopentenyl diphosphate isomerase: A core enzyme in isoprenoid biosynthesis. A review of its biochemistry and function. Nat. Prod. Rep. 1997, 14, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.Y.; Kuang, T.D.; Miao, Y.Y.; Xu, Y.Q.; Wang, Z.; Dong, L.D.; Tan, N.H. Discovery and characterization of four glycosyltransferases involved in anthraquinone glycosides biosynthesis in Rubia yunnanensis. Org. Chem. Front. 2020, 7, 2442–2448. [Google Scholar] [CrossRef]
- Rama Reddy, N.R.; Mehta, R.H.; Soni, P.H.; Makasana, J.; Gajbhiye, N.A.; Ponnuchamy, M.; Kumar, J. Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS ONE 2015, 10, e0129422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, W.P.; Zhang, Y.; Song, J.; Zhao, L.J.; Wang, Z.Z. De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar]
- Zhao, D.K.; Shen, Y.; Shi, Y.N.; Shi, X.Q.; Qiao, Q.; Zi, S.H.; Zhao, E.Q.; Yu, D.Q.; Kennelly, E.J. Probing the transcriptome of Aconitum carmichaelii reveals the candidate genes associated with the biosynthesis of the toxic aconitine-type C19-diterpenoid alkaloids. Phytochemistry 2018, 152, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Choudhri, P.; Rani, M.; Sangwan, R.S.; Kumar, R.; Kumar, A.; Chhokar, V. De novo sequencing, assembly, and characterisation of Aloe vera transcriptome and analysis of expression profiles of genes related to saponin and anthraquinone metabolism. BMC Genomics 2018, 19, 427. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.W.; Gao, S.; Wang, H.; Lei, T.X.; Shen, J.W.; Gao, J.; Chen, S.L.; Yin, J.; Liu, J.Q. De novo sequencing transcriptome of endemic Gentiana straminea (Gentianaceae) to identify genes involved in the biosynthesis of active ingredients. Gene 2016, 575, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.Z.; Yoo, M.J.; Koh, J.; Liu, L.H.; Chen, Y.Z.; Acikgoz, D.G.; Wang, Q.M.; Chen, S.X. Molecular reprogramming of Arabidopsis in response to perturbation of jasmonate signaling. J. Proteome Res. 2014, 13, 5751–5766. [Google Scholar] [CrossRef] [PubMed]
- Kochan, E.; Balcerczak, E.; Lipert, A.; Szymańska, G.; Szymczyk, P. Methyl jasmonate as a control factor of the synthase squalene gene promoter and ginsenoside production in American ginseng hairy root cultured in shake flasks and a nutrient sprinkle bioreactor. Ind. Crops Prod. 2018, 115, 182–193. [Google Scholar] [CrossRef]
- Wang, C.H.; Zheng, L.P.; Tian, H.; Wang, J.W. Synergistic effects of ultraviolet-B and methyl jasmonate on tanshinone biosynthesis in Salvia miltiorrhiza hairy roots. J. Photochem. Photobiol. B 2016, 159, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, M.C.; Alfieri, M.; Malafronte, N.; De Tommasi, N.; Leone, A. Increasing the synthesis of bioactive abietane diterpenes in Salvia sclarea hairy roots by elicited transcriptional reprogramming. Plant. Cell Rep. 2017, 36, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Li, R.S.; Zhu, J.H.; Guo, D.; Li, H.L.; Wang, Y.; Ding, X.P.; Mei, W.L.; Chen, Z.B.; Dai, H.F.; Peng, S.Q. Genome-wide identification and expression analysis of terpene synthase gene family in Aquilaria sinensis. Plant. Physiol. Biochem. 2021, 164, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.Y.; Hu, Y.Y.; Yi, S.Y.; Zhang, X.J.; Tan, N.H. Establishment of hairy root culture of Rubia yunnanensis Diels: Production of Rubiaceae-type cyclopeptides and quinones. J. Biotechnol. 2021, 341, 21–29. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.B.; Zhang, Y.; Li, S.K.; Li, Y.; Ye, J.; Yu, C.; Zhou, L.; et al. SOAPnuke: A mapreduce acceleration supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.; Steven, L. Fast gapped-read alignment with Bowtie2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichinger, D.; Bacher, A.; Zenk, M.H.; Eisenerich, W. Quantitative assessment of metabolic flux by 13C NMR analysis. Biosynthesis of anthraquinones in Rubia tinctorum. J. Am. Chem. Soc. 1999, 121, 7469–7475. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2011, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.Y.; Lin, Q.W.; Zhang, X.J.; Wang, J.; Miao, Y.Y.; Tan, N.H. Selection and validation of appropriate reference genes for quantitative RT-PCR analysis in Rubia yunnanensis Diels based on transcriptome data. Biomed. Res. Int. 2020, 2020, 5824841. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.T.; Murthy, H.N.; Park, S.Y. Methyl jasmonate induced oxidative stress and accumulation of secondary metabolites in plant cell and organ cultures. Int. J. Mol. Sci. 2020, 21, 716. [Google Scholar] [CrossRef] [Green Version]
- Perassolo, M.; Cardillo, A.B.; Mugas, M.L.; Núñez Montoya, S.C.; Giulietti, A.M.; Rodríguez Talou, J. Enhancement of anthraquinone production and release by combination of culture medium selection and methyl jasmonate elicitation in hairy root cultures of Rubia tinctorum. Ind. Crops Prod. 2017, 105, 124–132. [Google Scholar] [CrossRef]
- Luo, H.M.; Zhu, Y.J.; Song, J.Y.; Xu, L.J.; Sun, C.; Zhang, X.; Xu, Y.H.; Sun, W.; Xu, H.B.; Wang, B.; et al. Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol. Plant. 2014, 152, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Piao, X.C.; Jiang, J.; Jiang, X.L.; Yin, C.R.; Lian, M.L. A high production of flavonoids and anthraquinones via adventitious root culture of Oplopanax elatus and evaluating antioxidant activity. Plant. Cell Tiss. Org. 2019, 137, 173–179. [Google Scholar] [CrossRef]
- Han, Y.S.; Roytrakul, S.; Verberne, M.C.; van der Heijden, R.; Linthorst, H.J.; Verpoorte, R. Cloning of a cDNA encoding 1-deoxy-D-xylulose 5-phosphate synthase from Morinda citrifolia and analysis of its expression in relation to anthraquinone accumulation. Plant. Sci. 2003, 164, 911–917. [Google Scholar] [CrossRef]
- Stalman, M.; Koskamp, A.M.; Luderer, R.; Vernooy, J.H.; Wind, J.C.; Wullems, G.J.; Croes, A.F. Regulation of anthraquinone biosynthesis in cell cultures of Morinda citrifolia. J. Plant. Physiol. 2003, 160, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Veremeichik, G.N.; Bulgakov, V.P.; Shkryl, Y.N.; Siliantieva, S.A.; Makhazen, D.S.; Tchernoded, G.K.; Mischenko, N.P.; Fedoreyev, S.A.; Vasileva, E.A. Activation of anthraquinone biosynthesis in long-cultured callus culture of Rubia cordifolia transformed with the rolA plant oncogene. J. Biotechnol. 2019, 306, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Anthony, J.R.; Anthony, L.C.; Nowroozi, F.; Kwon, G.; Newman, J.D.; Keasling, J.D. Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab. Eng. 2009, 11, 13–19. [Google Scholar] [CrossRef]
- Yang, J.; Xian, M.; Su, S.Z.; Zhao, G.; Nie, Q.J.; Jiang, X.L.; Zheng, Y.N.; Liu, W. Enhancing production of bio-isoprene using hybrid MVA pathway and isoprene synthase in E. coli. PLoS ONE 2012, 7, e33509. [Google Scholar] [CrossRef]
- Yuan, Y.; Yu, J.; Jiang, C.; Li, M.H.; Lin, S.F.; Wang, X.M.; Huang, L.Q. Functional diversity of genes for the biosynthesis of paeoniflorin and its derivatives in Paeonia. Int. J. Mol. Sci. 2013, 14, 18502–18519. [Google Scholar] [CrossRef]
- Hawkins, A.R.; Lamb, H.K.; Moore, J.D.; Charles, I.G.; Roberts, C.F. The pre-chorismate (shikimate) and quinate pathways in filamentous fungi: Theoretical and practical aspects. J. Gen. Microbiol. 1993, 139, 2891–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Dudareva, N. The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu. Rev. Plant. Biol. 2012, 63, 73–105. [Google Scholar] [CrossRef] [PubMed]
- Karakas, F.P.; Cingoz, G.S.; Turker, A.U. The effects of oxidative stress on phenolic composition and antioxidant metabolism in callus culture of common daisy. Afr. J. Tradit. Complem. 2016, 13, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Hu, G.S.; Li, N.; Han, Z.F.; Jia, J.M. Effects of lovastatin, clomazone and methyl jasmonate treatment on the accumulation of purpurin and mollugin in cell suspension cultures of Rubia cordifolia. Chin. J. Nat. Med. 2013, 11, 0396–0400. [Google Scholar] [CrossRef]
- Leivar, P.; Monte, E. PIFs: Systems integrators in plant development. Plant. Cell 2014, 26, 56–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, X.; Dong, X.; Li, Q.; Chen, Z.; Liu, L. An update on the function and regulation of methylerythritol phosphate and mevalonate pathways and their evolutionary dynamics. J. Integr. Plant. Biol. 2021, 63, 1211–1226. [Google Scholar] [CrossRef]
- Learned, R.M. Light suppresses 3-hydroxy-3-methylglutaryl coenzyme a reductase gene expression in Arabidopsis thaliana. Plant. Physiol. 1996, 110, 645–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C.; Curtis, N.; Löbs, A.K.; Wheeldon, I. Multiplexed CRISPR activationmof cryptic sugar metabolism enables Yarrowia lipolytica growth on cel-lobiose. Biotechnol. J. 2018, 13, 1700584. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Mochida, K.; Asano, T.; Nakabayashi, R.; Chiba, M.; Udomson, N.; Yamazaki, Y.; Goodenowe, D.B.; Sankawa, U.; Yoshida, T.; et al. Coupling deep transcriptome analysis with untargeted metabolic profiling in Ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant. Cell Physiol. 2013, 54, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Durst, F.; Benveniste, I.; Salaün, J.P.; Werck-Reichhart, D. Function, mechanism and regulation of cytochrome P-450 enzymes in plants. Biochem. Soc. Trans. 1992, 20, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.; Mesarich, C.H.; Saccomanno, B.; Vaisberg, A.; De Wit, P.J.; Cox, R.; Collemare, J. Elucidation of cladofulvin biosynthesis reveals a cytochrome P450 monooxygenase required for anthraquinone dimerization. Proc. Natl. Acad. Sci. USA 2016, 113, 6851–6856. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number of Annotated Transcripts | Ratio of Annotated Transcripts (%) | Number of Annotated Genes | Ratio of Annotated Genes (%) |
|---|---|---|---|---|
| All assembly transcripts | 636,198 | NA | NA | NA |
| All assembly genes | 554,646 | NA | NA | NA |
| NR | 108,753 | 17.09 | 77,188 | 13.92 |
| Swiss-Prot | 98,311 | 15.45 | 76,703 | 13.83 |
| KEGG | 103,529 | 16.27 | 73,112 | 13.18 |
| KOG | 87,215 | 13.71 | 67,466 | 12.16 |
| Pfam | 38,877 | 6.11 | 29,369 | 5.30 |
| GO | 72,573 | 11.41 | 51,102 | 9.21 |
| All annotated transcripts | 140,078 | 22.02 | 107,631 | 19.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Miao, Y.; Chen, L.; Yi, S.; Tan, N. De Novo Transcriptome Analysis Reveals Putative Genes Involved in Anthraquinone Biosynthesis in Rubia yunnanensis. Genes 2022, 13, 521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030521
Zhang R, Miao Y, Chen L, Yi S, Tan N. De Novo Transcriptome Analysis Reveals Putative Genes Involved in Anthraquinone Biosynthesis in Rubia yunnanensis. Genes. 2022; 13(3):521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030521
Chicago/Turabian StyleZhang, Rongfei, Yuanyuan Miao, Lingyun Chen, Shanyong Yi, and Ninghua Tan. 2022. "De Novo Transcriptome Analysis Reveals Putative Genes Involved in Anthraquinone Biosynthesis in Rubia yunnanensis" Genes 13, no. 3: 521. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13030521