
Identification of a Novel SHANK2 Pathogenic Variant in a Patient with a Neurodevelopmental Disorder

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample and DNA Extraction
2.2. Exome Sequencing
2.3. Filtering and Variant Prioritization
2.4. Sanger Sequencing
2.5. Mutation Nomenclature
3. Results
3.1. Clinical Description
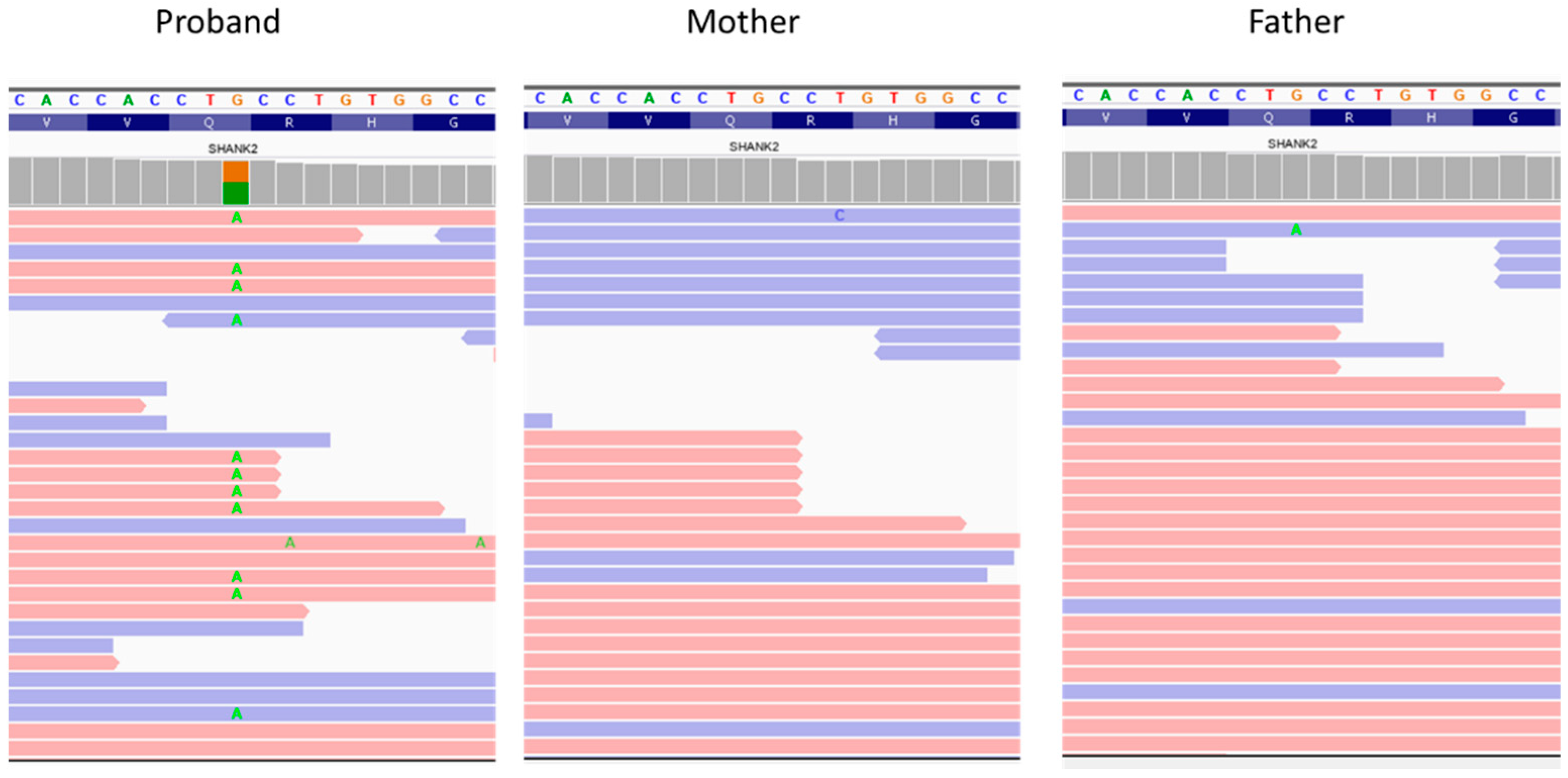
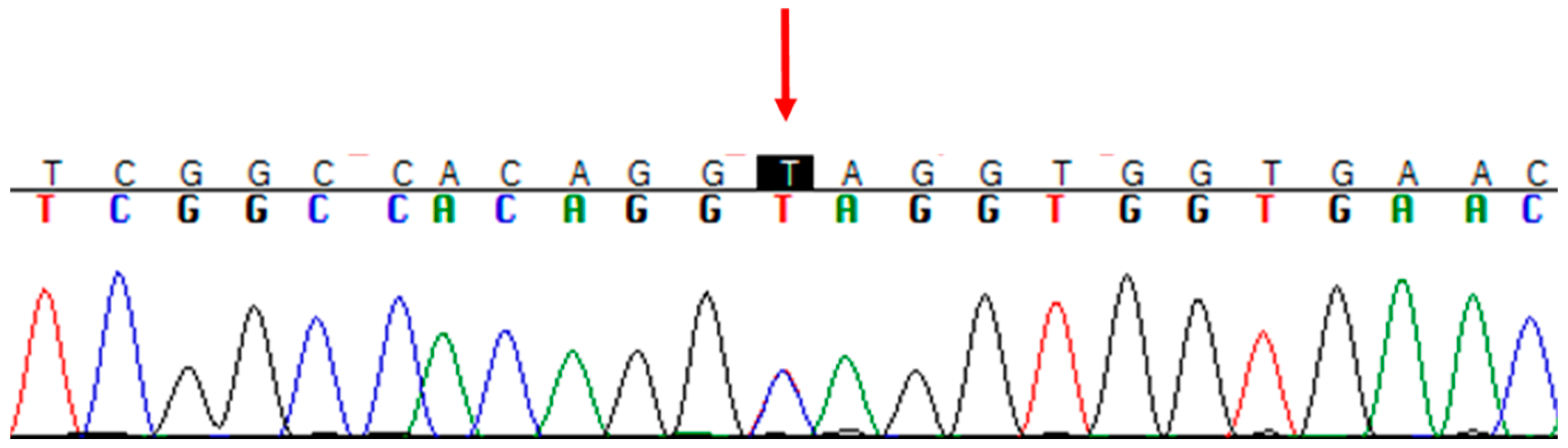
3.2. Genetic Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyle, C.A.; Boulet, S.; Schieve, L.A.; Cohen, R.A.; Blumberg, S.J.; Yeargin-Allsopp, M.; Visser, S.; Kogan, M.D. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics 2011, 127, 1034–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.N.; Coe, B.P.; Dickel, D.E.; Hoekzema, K.; Nelson, B.J.; Zody, M.C.; Kronenberg, Z.N.; Hormozdiari, F.; Raja, A.; Pennacchio, L.A.; et al. Genomic patterns of de novo mutation in simplex autism. Cell 2017, 171, 710–722. [Google Scholar] [CrossRef] [Green Version]
- Samocha, K.E.; Robinson, E.B.; Sanders, S.J.; Stevens, C.; Sabo, A.; McGrath, L.M.; Kosmicki, J.A.; Rehnstrom, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- The Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Sheng, M.; Sala, C. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfnger, E.D. ProSAP/Shank proteins—A family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Ehlers, M.D. Modeling autism by SHANK gene mutations in mice. Neuron 2013, 78, 8–27. [Google Scholar] [CrossRef] [Green Version]
- Berkel, S.; Tang, W.; Treviño, M.; Vogt, M.; Obenhaus, H.A.; Gass, P.; Scherer, S.W.; Sprengel, R.; Schratt, G.; Rappold, G.A. Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 2012, 21, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischmeijer, A.; Magini, P.; Giorda, R.; Gnoli, M.; Ciccone, R.; Cecconi, L.; Franzoni, E.; Mazzanti, L.; Romeo, G.; Zuffardi, O.; et al. Olfactory Receptor-Related Duplicons Mediate a Microdeletion at 11q13.2q13.4 Associated with a Syndromic Phenotype. Mol. Syndromol. 2011, 1, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Leblond, C.S.; Heinrich, J.; Delorme, R.; Proepper, C.; Betancur, C.; Huguet, G.; Konyukh, M.; Chaste, P.; Ey, E.; Rastam, M.; et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012, 8, e1002521. [Google Scholar] [CrossRef] [Green Version]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [Green Version]
- Marcou, C.A.; Studinski Jones, A.L.; Murphree, M.I.; Kirmani, S.; Hoppman, N.L. De novo 11q deletion including SHANK2 in a patient with global developmental delay. Am. J. Med. Genet. A 2017, 173, 801–805. [Google Scholar] [CrossRef]
- Bowling, K.M.; Thompson, M.L.; Amaral, M.D.; Finnila, C.R.; Hiatt, S.M.; Engel, K.L.; Cochran, J.N.; Brothers, K.B.; East, K.M.; Gray, D.E.; et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.Z.; Zhang, J.; Li, Z.; Lin, X.; Li, J.; Wang, S.; Yang, C.; Wu, Q.; Ye, A.Y.; Wang, M.; et al. Targeted resequencing of 358 candidate genes for autism spectrum disorder in a Chinese cohort reveals diagnostic potential and genotype-phenotype correlations. Hum. Mutat. 2019, 40, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Caumes, R.; Smol, T.; Thuillier, C.; Balerdi, M.; Lestienne-Roche, C.; Manouvrier-Hanu, S.; Ghoumid, J. Phenotypic spectrum of SHANK2-related neurodevelopmental disorder. Eur. J. Med. Genet. 2020, 63, 104072. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, J.; Qiao, Y.; Calli, K.; Martell, S.; Race, S.; Chijiwa, C.; Glodjo, A.; Jones, S.; Rajcan-Separovic, E.; Scherer, S.; et al. Contribution of Multiple Inherited Variants to Autism Spectrum Disorder (ASD) in a Family with 3 Affected Siblings. Genes 2021, 12, 1053. [Google Scholar] [CrossRef]
- Eltokhi, A.; Gonzalez-Lozano, M.A.; Oettl, L.L.; Rozov, A.; Pitzer, C.; Röth, R.; Berkel, S.; Hüser, M.; Harten, A.; Kelsch, W.; et al. Imbalanced post- and extrasynaptic SHANK2A functions during development affect social behavior in SHANK2-mediated neuropsychiatric disorders. Mol. Psychiatry 2021, 26, 6482–6504. [Google Scholar] [CrossRef]
- Han, K.A.; Yoon, T.H.; Shin, J.; Um, J.W.; Ko, J. Differentially altered social dominance- and cooperative-like behaviors in Shank2- and Shank3-mutant mice. Mol. Autism 2020, 11, 87. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.; Chen, J.; Li, X.; Yi, Q.; Shi, Y. Identification of SHANK2 Pathogenic Variants in a Chinese Uygur Population with Schizophrenia. J. Mol. Neurosci. 2021, 71, 1–8. [Google Scholar] [CrossRef]
- Lee, Y.S.; Yu, N.K.; Chun, J.; Yang, J.E.; Lim, C.S.; Kim, H.; Park, G.; Lee, J.A.; Lee, K.; Kaang, B.K.; et al. Identification of a novel Shank2 transcriptional variant in Shank2 knockout mouse model of autism spectrum disorder. Mol. Brain 2020, 13, 54. [Google Scholar] [CrossRef]
- Lutz, A.K.; Pérez Arévalo, A.; Ioannidis, V.; Stirmlinger, N.; Demestre, M.; Delorme, R.; Bourgeron, T.; Boeckers, T.M. SHANK2 Mutations Result in Dysregulation of the ERK1/2 Pathway in Human Induced Pluripotent Stem Cells-Derived Neurons and Shank2(−/−) Mice. Front. Mol. Neurosci. 2021, 14, 773571. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, K.; Zhang, W.B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Schmeisser, M.J.; Schoen, M.; Boeckers, T.M. Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 2011, 21, 594–603. [Google Scholar] [CrossRef]
- Alsufiani, H.M.; Alkhanbashi, A.S.; Laswad, N.A.B.; Bakhadher, K.K.; Alghamdi, S.A.; Tayeb, H.O.; Tarazi, F.I. Zinc deficiency and supplementation in autism spectrum disorder and Phelan-McDermid syndrome. J. Neurosci. Res. 2022, 100, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Flieger, W.; Flieger, M.; Forma, A.; Sitarz, E.; Skórzyńska-Dziduszko, K.; Grochowski, C.; Maciejewski, R.; Karakuła-Juchnowicz, H. Autism spectrum disorder: Trace elements imbalances and the pathogenesis and severity of autistic symptoms. Neurosci. Biobehav. Rev. 2021, 129, 117–132. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Present Study | Caumes et al., 2020 | Zhou et al., 2019 | Guo et al., 2018 | Bowling et al., 2017 | Marcou et al., 2016 | Leblond et al., 2014 | Leblond et al., 2012 | Wischmeijer et al., 2011 | Pinto et al., 2010 | Berkel et al., 2010 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pat.1 | Pat.1 | Pat.2 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.1 | Pat.2 | Pat.3 | |
| AGE | 9 | 6 | 6 | 4 | NA | NA | 12 | NA | 11 | 8 | NA | NA | NA | NA |
| GENDER | F | M | F | M | M | NA | F | M | M | F | M | F | M | M |
| VARIANT | c.334C>T | c.1322del | c.132dup | c.2540_2541del | c.87C>G | c.1896dup | 11q13.2 to 11q13.4 | del_11q13.3q13.4 (all SHANK2 exons) | loss of exon 5-16 | del_11q13.3q13.4 (all SHANK2 exons) | del exon 5-16 | del exon 7 | del exon 7-6 | c.2521C>T |
| p.(Gln112*) | p.(Ile441Thsfs*8) | p.(Asp45Argfs*3) | p.(Ser847*) | p.(Tyr29*) | p.(Asp633Argfs*3) | p.(?) | p.(?) | p.(?) | p.(?) | p.(?) | p.(?) | p.(?) | p.(Arg841*) | |
| PATTERNS OF INHERITANCE | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo |
| SPEECH DELAY | + | + | + | + | + | + | + | + | + | + | + | + | + | NA |
| AUTISM/ASD | − | ASD | AUTISM | ASD | ASD | NA | NA | AUTISM | AUTISM | AUTISM | AUTISM | AUTISM | AUTISM | AUTISM |
| ID | − | Mild | Moderate | NA | + | Moderate | Moderate/ Severe | Severe | Moderate | Moderate/Severe | Mild | Moderate | Mild | Moderate |
| MOTOR DELAY | − | − | − | − | − | NA | + | + | + | + | NA | + | NA | NA |
| NEUROLOGICAL SIGNS | Difficulties in mathematical calculation | Sleep and attention disorders | Anxiety | Sleep disorder | Attentional problems Febrile seizures infancy repetitive behavior | Hyperactivity | − | Oral dyspraxia Signs of cerebellar dysfunction Slight hypotonia | − | NA | Slow reaction and limited mimicry | NA | NA | |
| CLINICAL EXAMINATION | Flat profile, thick eyebrows, long eyelashes, bulbous tip and prominent columella, large and spaced teeth, retracted ears | NA | NA | NA | NA | Microcephalic Mild malar hypoplasia, mild retrognathia. Fine hair. Prominent forehead Bilateral epicanthal folds, long palpebral fissures, deep-set eyes. Broad nasal tip, depressed nasal bridge. Small mouth, down-turned corners. Wide-spaced teeth. Long slender fingers, clinodactyly. Long, slender feet, mild pes planus, piezogenic papules | Clinodactyly Deep-set eyes strabismus and ptosis. Large ears Retrognathia Wide nasal bridge Thin upper lip | Prominent chin, hypermetropia and astigmatism | Congenital hip dysplasia, downward slanting palpebral fissures, deep-set eyes, ptosis of the left eyelid, long and fine lashes, broad nasal bridge, tubular nose with round overhanging tip and hypoplastic nares, short philtrum, small and thin upper lip, preauricular tag and small low-set simple ears | Hypermetropia, large and prominent ears, flat feet | NA | NA | NA | |
| MRI | Normal | Normal | Normal | NA | − | NA | Normal | NA | NA | Normal | Normal | NA | NA | NA |
| EEG | Normal | NA | NA | − | − | NA | Normal | NA | Normal | Normal | NA | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doddato, G.; Fabbiani, A.; Scandurra, V.; Canitano, R.; Mencarelli, M.A.; Renieri, A.; Ariani, F. Identification of a Novel SHANK2 Pathogenic Variant in a Patient with a Neurodevelopmental Disorder. Genes 2022, 13, 688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040688
Doddato G, Fabbiani A, Scandurra V, Canitano R, Mencarelli MA, Renieri A, Ariani F. Identification of a Novel SHANK2 Pathogenic Variant in a Patient with a Neurodevelopmental Disorder. Genes. 2022; 13(4):688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040688
Chicago/Turabian StyleDoddato, Gabriella, Alessandra Fabbiani, Valeria Scandurra, Roberto Canitano, Maria Antonietta Mencarelli, Alessandra Renieri, and Francesca Ariani. 2022. "Identification of a Novel SHANK2 Pathogenic Variant in a Patient with a Neurodevelopmental Disorder" Genes 13, no. 4: 688. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040688