
Characterization of Altered Molecular Pathways in the Entorhinal Cortex of Alzheimer’s Disease Patients and In Silico Prediction of Potential Repurposable Drugs

 , , , , and
, , , , and
Abstract
:1. Introduction
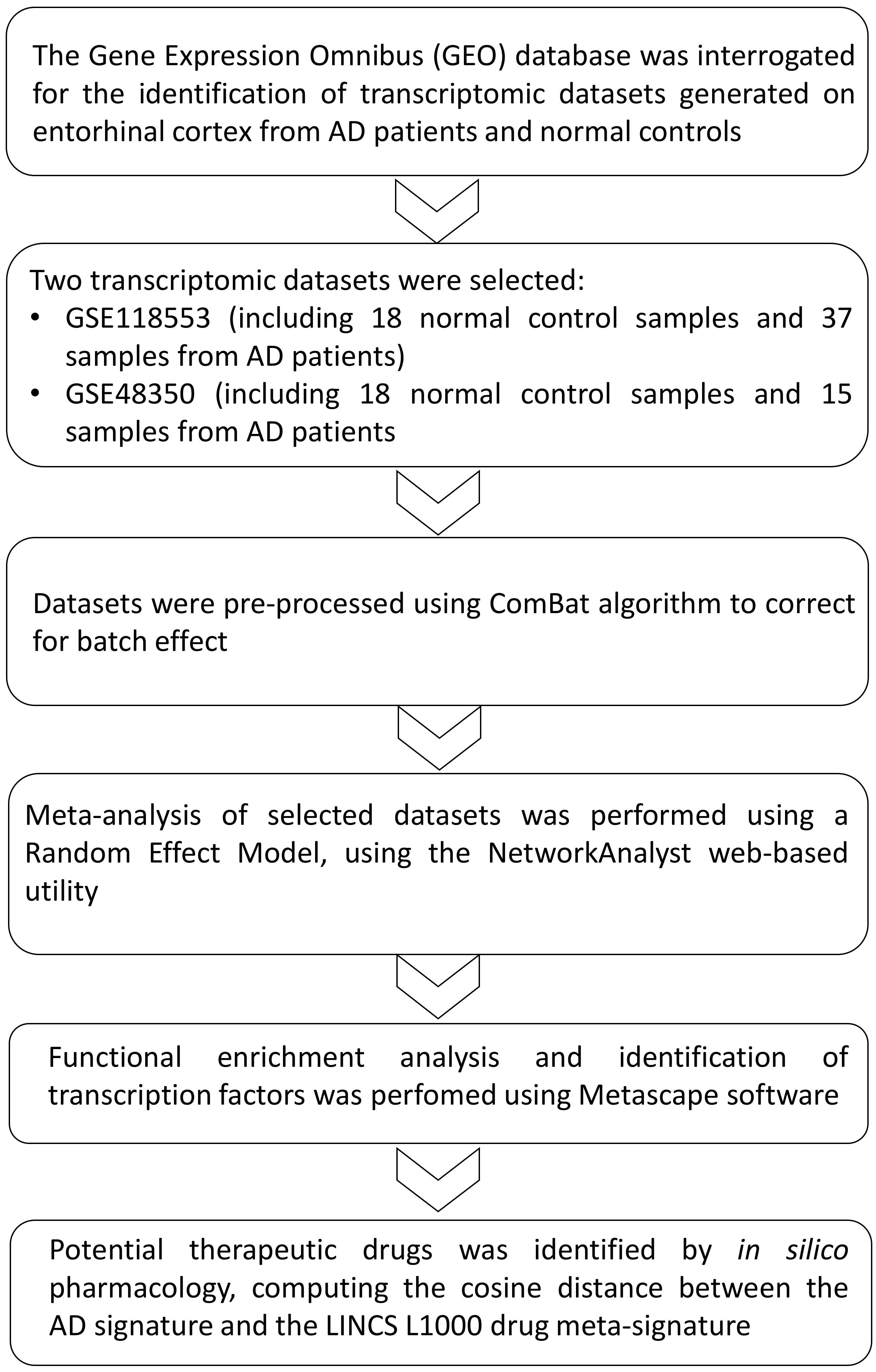
2. Materials and Methods
2.1. Dataset Selection
2.2. In Silico Pharmacology
2.3. Statistical Analysis
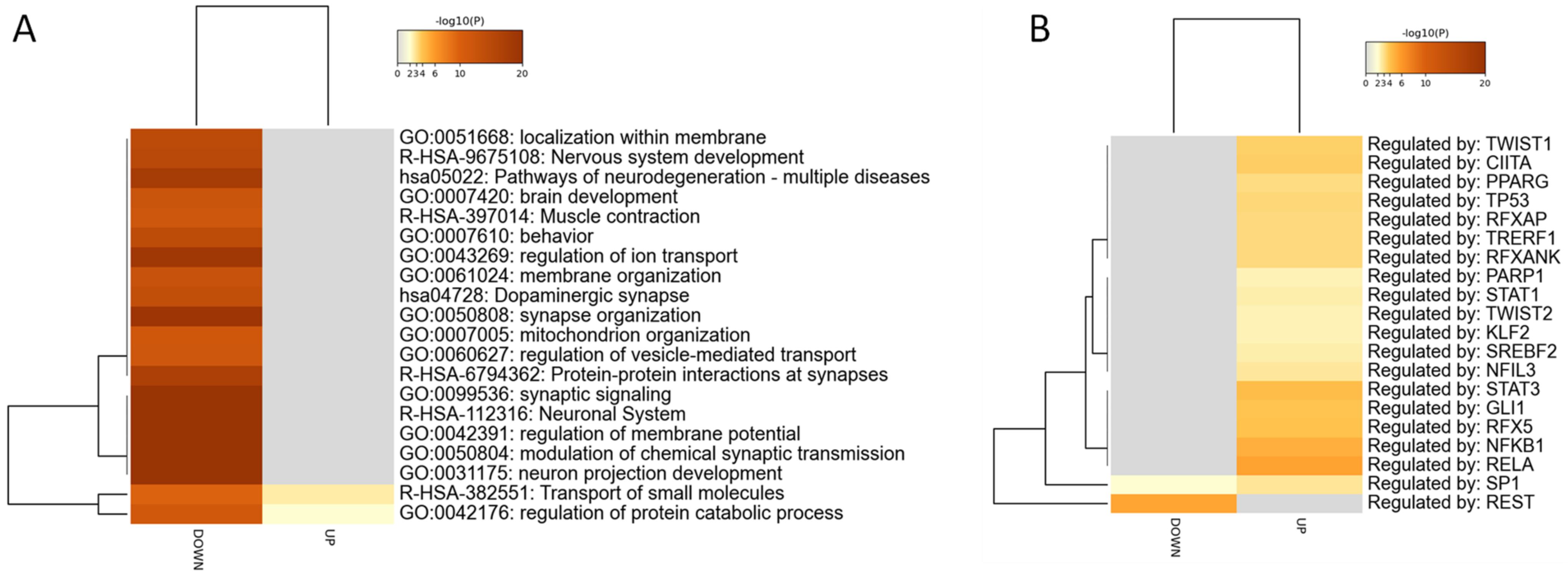
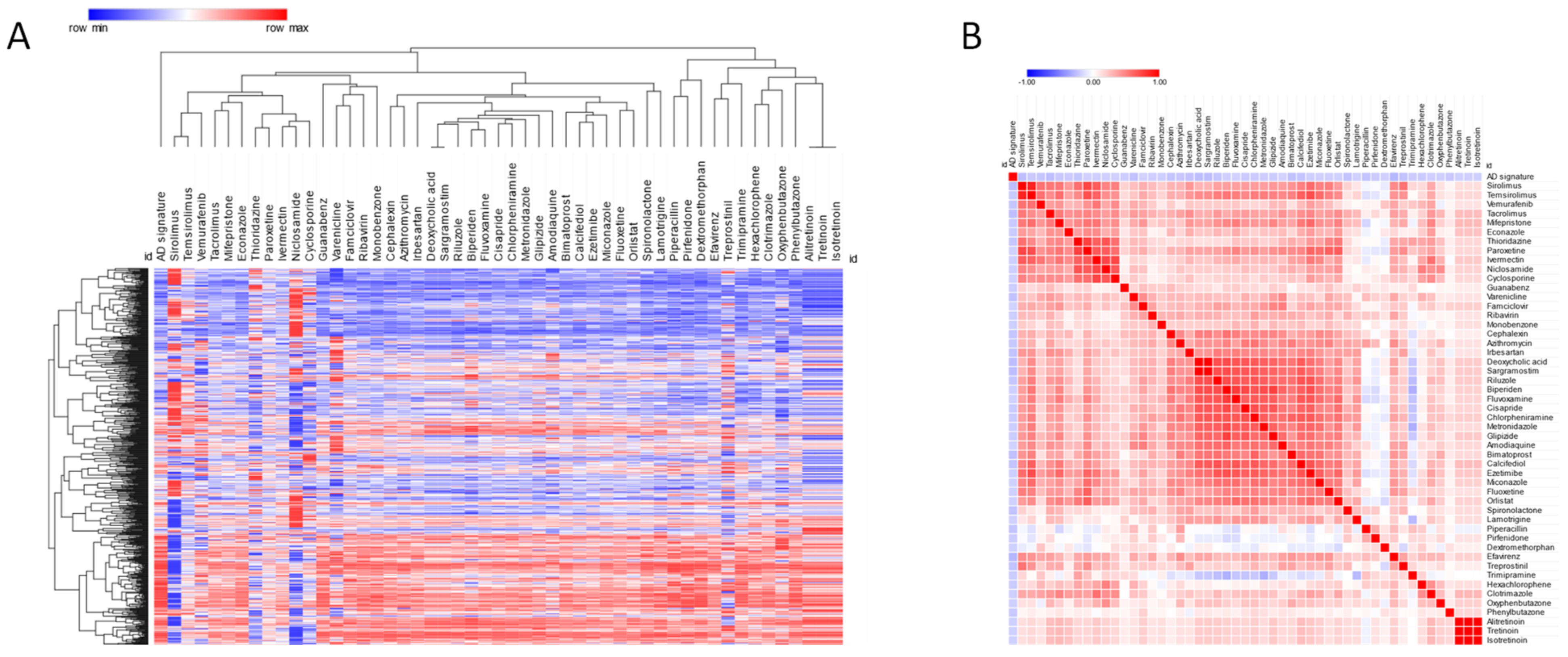
3. Results
3.1. Identification of the AD Gene Expression Profile in EC
3.2. Prediction of Novel Chemotherapeutics for AD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cavalli, E.; Battaglia, G.; Basile, M.S.; Bruno, V.; Petralia, M.C.; Lombardo, S.D.; Pennisi, M.; Kalfin, R.; Tancheva, L.; Fagone, P.; et al. Exploratory Analysis of iPSCS-Derived Neuronal Cells as Predictors of Diagnosis and Treatment of Alzheimer Disease. Brain Sci. 2020, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desikan, R.S.; McEvoy, L.K.; Thompson, W.K.; Holland, D.; Brewer, J.B.; Aisen, P.S.; Sperling, R.A.; Dale, A.M. Amyloid-β-associated clinical decline occurs only in the presence of elevated P-tau. Arch. Neurol. 2012, 69, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garcidueñas, A.L.; Duyckaerts, C. Alzheimer disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 325–337. [Google Scholar]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- McDaid, J.; Mustaly-Kalimi, S.; Stutzmann, G.E. Ca2+ Dyshomeostasis Disrupts Neuronal and Synaptic Function in Alzheimer’s Disease. Cells 2020, 9, 2655. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The dual role of glutamatergic neurotransmission in Alzheimer’s disease: From pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of alzheimer disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, D.J.; Lourida, I.; Hannon, E.; Littlejohns, T.J.; Langa, K.M.; Hyppönen, E.; Kuźma, E. Association of Lifestyle and Genetic Risk with Incidence of Dementia. JAMA-J. Am. Med. Assoc. 2019, 322, 430–437. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; De Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Jack, C.R.; Wiste, H.J.; Schwarz, C.G.; Lowe, V.J.; Senjem, M.L.; Vemuri, P.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Gunter, J.L.; et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 2018, 141, 1517–1528. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, J.; Liu, J.; Wang, Y.; Li, K. Magnetic resonance imaging measurement of entorhinal cortex in the diagnosis and differential diagnosis of mild cognitive impairment and alzheimer’s disease. Brain Sci. 2021, 11, 1129. [Google Scholar] [CrossRef]
- Olajide, O.J.; Suvanto, M.E.; Chapman, C.A. Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer’s disease. Biol. Open 2021, 10, 56796. [Google Scholar] [CrossRef]
- Ding, B.; Xi, Y.; Gao, M.; Li, Z.; Xu, C.; Fan, S.; He, W. Gene expression profiles of entorhinal cortex in Alzheimer’s disease. Am. J. Alzheimers. Dis. Other Demen. 2014, 29, 526–532. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Miyashita, A.; Hatsuta, H.; Kikuchi, M.; Nakaya, A.; Saito, Y.; Tsukie, T.; Hara, N.; Ogishima, S.; Kitamura, N.; Akazawa, K.; et al. Genes associated with the progression of neurofibrillary tangles in alzheimer’s disease. Transl. Psychiatry 2014, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Hodges, A.K.; Curtis, C.; Lee, S.H.; Troakes, C.; Dobson, R.J.B.; Newhouse, S.J. Transcriptomic analysis of probable asymptomatic and symptomatic alzheimer brains. Brain. Behav. Immun. 2019, 80, 644–656. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Coleman, P.D.; Cribbs, D.H.; Rogers, J.; Gillen, D.L.; Cotman, C.W. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1653–1661. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Campo, G.; Corsico, F.; Presti, M.; Bramanti, P.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; et al. Modulation of Tetraspanin 32 (TSPAN32) Expression in T Cell-Mediated Immune Responses and in Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4323. [Google Scholar] [CrossRef] [Green Version]
- Basile, M.S.; Mazzon, E.; Mangano, K.; Pennisi, M.; Petralia, M.C.; Lombardo, S.D.; Nicoletti, F.; Fagone, P.; Cavalli, E. Impaired Expression of Tetraspanin 32 (TSPAN32) in Memory T Cells of Patients with Multiple Sclerosis. Brain Sci. 2020, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Mangano, K.; Di Marco, R.; Reyes-Castillo, Z.; Muñoz-Valle, J.F.; Nicoletti, F. Altered expression of tspan32 during b cell activation and systemic lupus erythematosus. Genes 2021, 12, 931. [Google Scholar] [CrossRef]
- Cavalli, E.; Mazzon, E.; Mammana, S.; Basile, M.S.; Lombardo, S.D.; Mangano, K.; Bramanti, P.; Nicoletti, F.; Fagone, P.; Petralia, M.C. Overexpression of macrophage migration inhibitory factor and its homologue d-dopachrome tautomerase as negative prognostic factor in neuroblastoma. Brain Sci. 2019, 9, 284. [Google Scholar] [CrossRef] [Green Version]
- Presti, M.; Mazzon, E.; Basile, M.S.; Petralia, M.C.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886. [Google Scholar] [CrossRef] [Green Version]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; Baranzini, S.E. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife 2017, 6, 26726. [Google Scholar] [CrossRef] [PubMed]
- Keenan, A.B.; Jenkins, S.L.; Jagodnik, K.M.; Koplev, S.; He, E.; Torre, D.; Wang, Z.; Dohlman, A.B.; Silverstein, M.C.; Lachmann, A.; et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Meng, F.; Xi, Y.; Huang, J.; Ayers, P.W. A curated diverse molecular database of blood-brain barrier permeability with chemical descriptors. Sci. Data 2021, 8, 1–11. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Citron, B.A.; Dennis, J.S.; Zeitlin, R.S.; Echeverria, V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 2499–2504. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Zukin, R.S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 2018, 48, 193–200. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent Advances in Drug Repositioning for the Discovery of New Anticancer Drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning—what we need to know. Drug Discov. Today 2013, 18, 110–115. [Google Scholar] [CrossRef]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Wong, S.T.C. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Jadamba, E.; Shin, M. A Systematic Framework for Drug Repositioning from Integrated Omics and Drug Phenotype Profiles Using Pathway-Drug Network. Biomed. Res. Int. 2016, 2016, 7147039. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.M.; Lam, M.; Mast, N.; Moon, J.; Li, Y.; Maxfield, E.; Pikuleva, I.A. CYP46A1 Activation by Efavirenz Leads to Behavioral Improvement without Significant Changes in Amyloid Plaque Load in the Brain of 5XFAD Mice. Neurotherapeutics 2019, 16, 710–724. [Google Scholar] [CrossRef]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimer’s Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Chen, G.; He, W.; Xiao, M.; Yan, L.J. Activation of mTOR: A culprit of Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2015, 11, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of alzheimer’s disease. PLoS ONE 2010, 5, 9979. [Google Scholar] [CrossRef] [Green Version]
- Kendziorra, K.; Wolf, H.; Meyer, P.M.; Barthel, H.; Hesse, S.; Becker, G.A.; Luthardt, J.; Schildan, A.; Patt, M.; Sorger, D.; et al. Decreased cerebral α4β2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer’s disease assessed with positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 515–525. [Google Scholar] [CrossRef]
- Zhang, L.J.; Xiao, Y.; Qi, X.L.; Shan, K.R.; Pei, J.J.; Kuang, S.X.; Liu, F.; Guan, Z.Z. Cholinesterase activity and mRNA level of nicotinic acetylcholine receptors (α4 and β2 subunits) in blood of elderly Chinese diagnosed as Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 849–858. [Google Scholar] [CrossRef]
- Rollema, H.; Hajós, M.; Seymour, P.A.; Kozak, R.; Majchrzak, M.J.; Guanowsky, V.; Horner, W.E.; Chapin, D.S.; Hoffmann, W.E.; Johnson, D.E.; et al. Preclinical pharmacology of the α4β2 nAChR partial agonist varenicline related to effects on reward, mood and cognition. Biochem. Pharmacol. 2009, 78, 813–824. [Google Scholar] [CrossRef]
- Smith, R.C.; Lindenmayer, J.P.; Davis, J.M.; Cornwell, J.; Noth, K.; Gupta, S.; Sershen, H.; Lajtha, A. Cognitive and antismoking effects of varenicline in patients with schizophrenia or schizoaffective disorder. Schizophr. Res. 2009, 110, 149–155. [Google Scholar] [CrossRef]
- Kim, S.Y.; Choi, S.H.; Rollema, H.; Schwam, E.M.; McRae, T.; Dubrava, S.; Jacobsen, J. Phase II Crossover Trial of Varenicline in Mild-to-Moderate Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2014, 37, 232–245. [Google Scholar] [CrossRef]
- Pereira, A.C.; Lambert, H.K.; Grossman, Y.S.; Dumitriu, D.; Waldman, R.; Jannetty, S.K.; Calakos, K.; Janssen, W.G.; McEwen, B.S.; Morrison, J.H. Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc. Natl. Acad. Sci. USA 2014, 111, 18733–18738. [Google Scholar] [CrossRef] [Green Version]
- Hunsberger, H.C.; Weitzner, D.S.; Rudy, C.C.; Hickman, J.E.; Libell, E.M.; Speer, R.R.; Gerhardt, G.A.; Reed, M.N. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J. Neurochem. 2015, 135, 381–394. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| EntrezID | Name | GSE48350_FC | GSE118553_FC | GSE48350_Adj Pval | GSE118553_Adj Pval | CombinedES | Adj Pval |
|---|---|---|---|---|---|---|---|
| 285268 | ZNF621 | 2.4746 | 0.92173 | 1.34 × 10−8 | 3.23 × 10−8 | 2.2507 | 6.12 × 10−5 |
| 121260 | SLC15A4 | 0.23632 | 0.46129 | 0.15057 | 1.87 × 10−9 | 1.5736 | 0.038196 |
| 79819 | WDR78 | 0.26701 | 0.43115 | 0.16414 | 6.5 × 10−8 | 1.4171 | 0.013998 |
| 91947 | ARRDC4 | 1.0171 | 1.4368 | 0.17458 | 2.45 × 10−8 | 1.4171 | 0.042945 |
| 51574 | LARP7 | 0.20582 | 0.34122 | 0.17458 | 7.05 × 10−8 | 1.3661 | 0.035172 |
| 677 | ZFP36L1 | 0.5541 | 0.89219 | 0.17458 | 5.46 × 10−8 | 1.3635 | 0.045701 |
| 1842 | ECM2 | 1.3153 | 1.3997 | 0.16414 | 3.42 × 10−7 | 1.3563 | 0.004831 |
| 25937 | WWTR1 | 0.95877 | 1.2911 | 0.17458 | 1.78 × 10−7 | 1.3539 | 0.015527 |
| 79887 | PLBD1 | 0.5105 | 0.59817 | 0.11938 | 1.64 × 10−6 | 1.3531 | 6.12 × 10−5 |
| 84532 | ACSS1 | 0.32735 | 0.45851 | 0.14265 | 1.18 × 10−6 | 1.3436 | 0.000416 |
| 3769 | KCNJ13 | 0.18731 | 0.36619 | 0.17458 | 3.72 × 10−7 | 1.3225 | 0.011159 |
| 1903 | S1PR3 | 0.50774 | 0.75184 | 0.17458 | 2.56 × 10−7 | 1.3125 | 0.023432 |
| 828 | CAPS | 1.2364 | 1.2386 | 0.17458 | 2.52 × 10−7 | 1.2953 | 0.033873 |
| 3176 | HNMT | 0.32837 | 0.37213 | 0.081897 | 1.38 × 10−5 | 1.2911 | 1.69 × 10−5 |
| 6542 | SLC7A2 | 0.88797 | 1.1676 | 0.17458 | 3.23 × 10−7 | 1.2669 | 0.043421 |
| 169792 | GLIS3 | 0.79637 | 0.8679 | 0.16731 | 7.38 × 10−6 | 1.238 | 0.000211 |
| 137075 | CLDN23 | 0.14904 | 0.33434 | 0.17458 | 3.76 × 10−6 | 1.2175 | 0.003207 |
| 730112 | FAM166B | 0.27099 | 0.39649 | 0.17458 | 5.21 × 10−6 | 1.2045 | 0.00246 |
| 83538 | TTC25 | 0.31451 | 0.58902 | 0.17458 | 3.44 × 10−6 | 1.1801 | 0.013422 |
| 58487 | CREBZF | 0.15424 | 0.33269 | 0.17458 | 2.51 × 10−6 | 1.1742 | 0.024247 |
| EntrezID | Name | GSE48350_FC | GSE118553_FC | GSE48350_Adj Pval | GSE118553_Adj Pval | CombinedES | Adj Pval |
|---|---|---|---|---|---|---|---|
| 10361 | NPM2 | −0.3746 | −0.67012 | 0.1148 | 1.12 × 10−8 | −1.5404 | 0.009671 |
| 2596 | GAP43 | −0.61551 | −0.91532 | 0.17458 | 1.28 × 10−8 | −1.4592 | 0.039307 |
| 246176 | GAS2L2 | −0.22421 | −0.24684 | 0.002432 | 3.17 × 10−5 | −1.443 | 1.21 × 10−6 |
| 1917 | EEF1A2 | −0.45676 | −0.73972 | 0.17458 | 3.71 × 10−8 | −1.4062 | 0.032214 |
| 22859 | ADGRL1 | −0.34859 | −0.52543 | 0.15557 | 2.14 × 10−7 | −1.389 | 0.004429 |
| 10423 | CDIPT | −0.19717 | −0.29224 | 0.15057 | 6.77 × 10−7 | −1.3519 | 0.001392 |
| 4004 | LMO1 | −0.68291 | −0.51695 | 0.004138 | 0.000147 | −1.3449 | 6.58 × 10−6 |
| 534 | ATP6V1G2 | −0.4706 | −0.6834 | 0.17458 | 4.98 × 10−7 | −1.2865 | 0.017316 |
| 51686 | OAZ3 | −0.56799 | −0.40996 | 0.004184 | 0.000515 | −1.2743 | 1.81 × 10−5 |
| 55643 | BTBD2 | −0.36764 | −0.50131 | 0.17458 | 2.1 × 10−6 | −1.2587 | 0.002795 |
| 9853 | RUSC2 | −0.21069 | −0.386 | 0.17458 | 4.36 × 10−7 | −1.2458 | 0.045965 |
| 27132 | CPNE7 | −0.43312 | −0.66894 | 0.17458 | 8.82 × 10−7 | −1.2335 | 0.026278 |
| 9556 | ATP5MPL | −0.30239 | −0.39911 | 0.17458 | 8.17 × 10−6 | −1.2175 | 0.000416 |
| 80146 | UXS1 | −0.2609 | −0.4633 | 0.17458 | 9.22 × 10−7 | −1.2141 | 0.036783 |
| 55530 | SVOP | −0.74564 | −1.128 | 0.17458 | 2.11 × 10−6 | −1.1977 | 0.0181 |
| 6835 | SURF2 | −0.34906 | −0.28302 | 0.017189 | 0.000789 | −1.1975 | 6.12 × 10−5 |
| 55294 | FBXW7 | −0.34269 | −0.47962 | 0.17458 | 3.19 × 10−6 | −1.1863 | 0.012874 |
| 226 | ALDOA | −0.30882 | −0.44703 | 0.17458 | 2.51 × 10−6 | −1.182 | 0.019997 |
| 9143 | SYNGR3 | −0.52952 | −0.78748 | 0.17458 | 3.18 × 10−6 | −1.1782 | 0.015749 |
| 7280 | TUBB2A | −0.33387 | −0.52954 | 0.17458 | 2.34 × 10−6 | −1.1746 | 0.026663 |
| Drug | Cosine Similarity | p_Value | FDR |
|---|---|---|---|
| Efavirenz | −0.28 | 0 | 0.01 |
| Tacrolimus | −0.25 | 0 | 0.01 |
| Sirolimus | −0.25 | 0 | 0.01 |
| Deoxycholic acid | −0.25 | 0 | 0.01 |
| Sargramostim | −0.25 | 0 | 0.01 |
| Bimatoprost | −0.24 | 0 | 0.01 |
| Varenicline | −0.24 | 0 | 0.01 |
| Calcifediol | −0.23 | 0 | 0.01 |
| Piperacillin | −0.23 | 0 | 0.01 |
| Treprostinil | −0.23 | 0 | 0.01 |
| Spironolactone | −0.23 | 0 | 0.01 |
| Cephalexin | −0.23 | 0 | 0.01 |
| Irbesartan | −0.23 | 0 | 0.01 |
| Thioridazine | −0.22 | 0 | 0.01 |
| Riluzole | −0.22 | 0 | 0.01 |
| Vemurafenib | −0.22 | 0 | 0.01 |
| Azithromycin | −0.21 | 0 | 0.01 |
| Phenylbutazone | −0.21 | 0 | 0.01 |
| Mifepristone | −0.21 | 0 | 0.01 |
| Temsirolimus | −0.21 | 0 | 0.01 |
| Cisapride | −0.2 | 0 | 0.01 |
| Guanabenz | −0.2 | 0 | 0.01 |
| Trimipramine | −0.2 | 0 | 0.01 |
| Hexachlorophene | −0.2 | 0 | 0.01 |
| Glipizide | −0.2 | 0 | 0.01 |
| Chlorpheniramine | −0.2 | 0 | 0.01 |
| Econazole | −0.2 | 0 | 0.01 |
| Pirfenidone | −0.2 | 0 | 0.01 |
| Clotrimazole | −0.19 | 0 | 0.01 |
| Monobenzone | −0.19 | 0 | 0.01 |
| Ivermectin | −0.19 | 0 | 0.01 |
| Biperiden | −0.19 | 0 | 0.01 |
| Ribavirin | −0.19 | 0 | 0.01 |
| Metronidazole | −0.19 | 0 | 0.01 |
| Ezetimibe | −0.19 | 0 | 0.01 |
| Niclosamide | −0.19 | 0 | 0.01 |
| Cyclosporine | −0.18 | 0 | 0.01 |
| Famciclovir | −0.18 | 0 | 0.01 |
| Fluoxetine | −0.18 | 0 | 0.01 |
| Dextromethorphan | −0.18 | 0 | 0.01 |
| Alitretinoin | −0.18 | 0 | 0.01 |
| Lamotrigine | −0.18 | 0 | 0.01 |
| Amodiaquine | −0.18 | 0 | 0.01 |
| Paroxetine | −0.18 | 0 | 0.01 |
| Tretinoin | −0.18 | 0 | 0.01 |
| Isotretinoin | −0.18 | 0 | 0.01 |
| Orlistat | −0.18 | 0 | 0.01 |
| Miconazole | −0.18 | 0 | 0.01 |
| Oxyphenbutazone | −0.18 | 0 | 0.01 |
| Fluvoxamine | −0.18 | 0.01 | 0.02 |
| Drug | Cosine Similarity | FDR |
|---|---|---|
| Varenicline | −0.24 | 0.01 |
| Piperacillin | −0.23 | 0.01 |
| Riluzole | −0.22 | 0.01 |
| Thioridazine | −0.22 | 0.01 |
| Phenylbutazone | −0.21 | 0.01 |
| Temsirolimus | −0.21 | 0.01 |
| Chlorpheniramine | −0.2 | 0.01 |
| Cisapride | −0.2 | 0.01 |
| Trimipramine | −0.2 | 0.01 |
| Biperiden | −0.19 | 0.01 |
| Ivermectin | −0.19 | 0.01 |
| Metronidazole | −0.19 | 0.01 |
| Ribavirin | −0.19 | 0.01 |
| Dextromethorphan | −0.18 | 0.01 |
| Fluoxetine | −0.18 | 0.01 |
| Fluvoxamine | −0.18 | 0.02 |
| Lamotrigine | −0.18 | 0.01 |
| Paroxetine | −0.18 | 0.01 |
| Tretinoin | −0.18 | 0.01 |
| Chlorprothixene | −0.17 | 0.01 |
| Clemastine | −0.17 | 0.01 |
| Methocarbamol | −0.17 | 0.01 |
| Sunitinib | −0.17 | 0.01 |
| Tamoxifen | −0.17 | 0.01 |
| Thiethylperazine | −0.17 | 0.01 |
| Granisetron | −0.16 | 0.01 |
| Loperamide | −0.16 | 0.01 |
| Praziquantel | −0.16 | 0.01 |
| Diphenylpyraline | −0.15 | 0.01 |
| Droperidol | −0.15 | 0.01 |
| Pemoline | −0.15 | 0.01 |
| Saquinavir | −0.15 | 0.04 |
| Sertraline | −0.15 | 0.01 |
| Atomoxetine | −0.14 | 0.03 |
| Cytarabine | −0.14 | 0.01 |
| Gefitinib | −0.14 | 0.01 |
| Sulfamethoxazole | −0.14 | 0.02 |
| Pramipexole | −0.13 | 0.01 |
| Naloxone | −0.12 | 0.01 |
| Nortriptyline | −0.12 | 0.04 |
| Pentobarbital | −0.12 | 0.02 |
| Metaxalone | −0.11 | 0.02 |
| Nalbuphine | −0.11 | 0.03 |
| Rivastigmine | −0.11 | 0.01 |
| Terbinafine | −0.11 | 0.02 |
| Cyproheptadine | −0.1 | 0.01 |
| Metoclopramide | −0.1 | 0.01 |
| Naltrexone | −0.1 | 0.03 |
| Prednicarbate | −0.1 | 0.02 |
| Desoximetasone | −0.09 | 0.03 |
| Ethotoin | −0.08 | 0.03 |
| Zonisamide | −0.08 | 0.02 |
| Flucytosine | −0.06 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagone, P.; Mangano, K.; Martino, G.; Quattropani, M.C.; Pennisi, M.; Bella, R.; Fisicaro, F.; Nicoletti, F.; Petralia, M.C. Characterization of Altered Molecular Pathways in the Entorhinal Cortex of Alzheimer’s Disease Patients and In Silico Prediction of Potential Repurposable Drugs. Genes 2022, 13, 703. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040703
Fagone P, Mangano K, Martino G, Quattropani MC, Pennisi M, Bella R, Fisicaro F, Nicoletti F, Petralia MC. Characterization of Altered Molecular Pathways in the Entorhinal Cortex of Alzheimer’s Disease Patients and In Silico Prediction of Potential Repurposable Drugs. Genes. 2022; 13(4):703. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040703
Chicago/Turabian StyleFagone, Paolo, Katia Mangano, Gabriella Martino, Maria Catena Quattropani, Manuela Pennisi, Rita Bella, Francesco Fisicaro, Ferdinando Nicoletti, and Maria Cristina Petralia. 2022. "Characterization of Altered Molecular Pathways in the Entorhinal Cortex of Alzheimer’s Disease Patients and In Silico Prediction of Potential Repurposable Drugs" Genes 13, no. 4: 703. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040703