
Pathogenesis of Type 1 Diabetes: Established Facts and New Insights

 , and
, and
Abstract
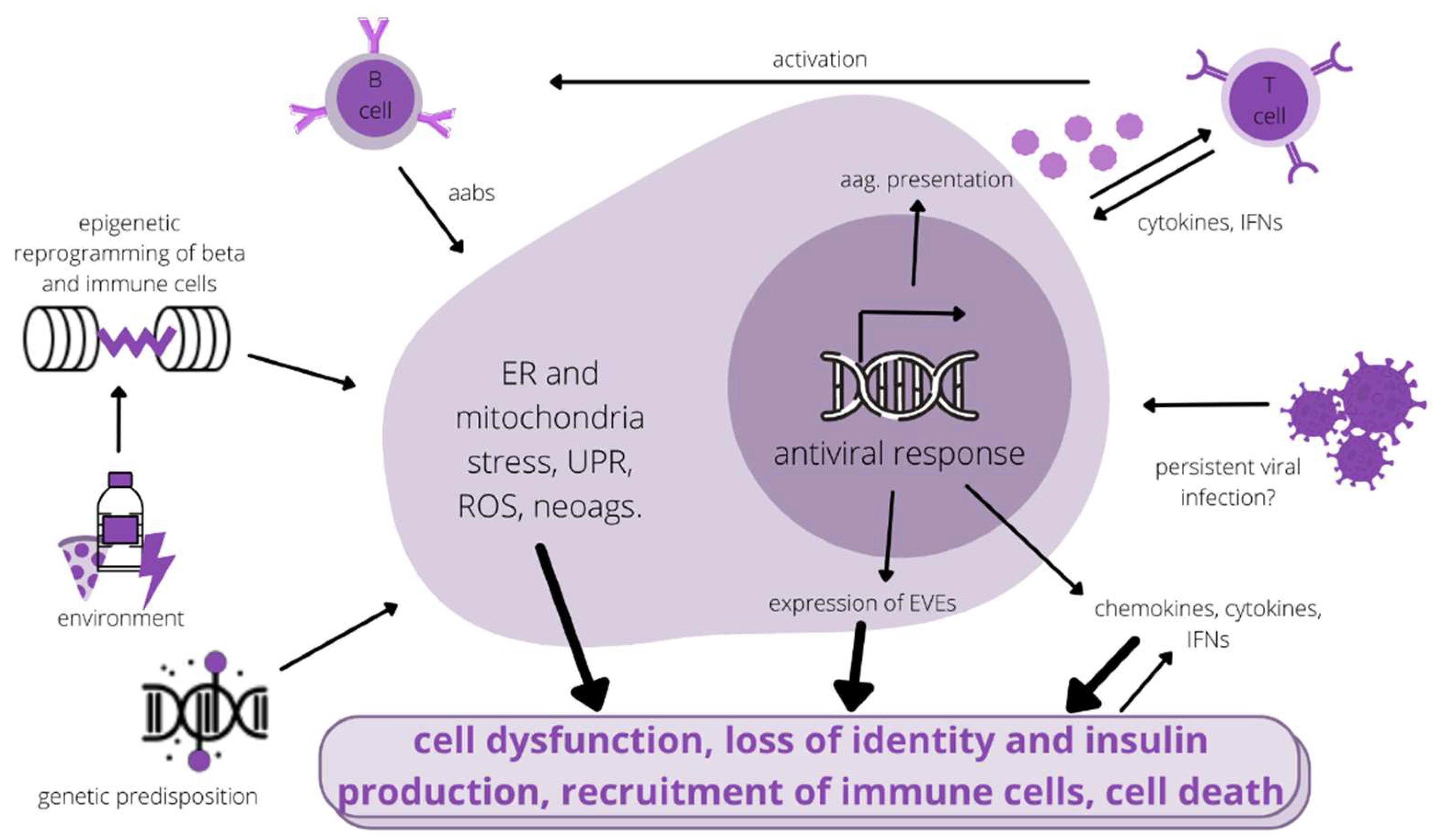
:1. Introduction
2. Pathology of β-Cells
3. Genetic Predisposition
4. Environmental Risk Factors in Connection to Genetic Predisposition
4.1. Viral Infections
4.2. Endogenized Viral Elements in the Genome
4.3. Gut Biome
5. Epigenetic Factors in T1D Pathology
6. Extracellular Vesicles and Non-Coding RNAs
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Draznin, B.; Aroda, V.R.; Bakris, G.; Benson, G.; Brown, F.M.; Freeman, R.; Green, J.; Huang, E.; Isaacs, D.; Kahan, S.; et al. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S17–S38. [Google Scholar] [CrossRef]
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Fard, H.H.; Ghojazadeh, M. Prevalence and Incidence of Type 1 Diabetes in the World: A Systematic Review and Meta-Analysis. Health Promot. Perspect. 2020, 10, 98–115. [Google Scholar] [CrossRef]
- Dayan, C.M.; Korah, M.; Tatovic, D.; Bundy, B.N.; Herold, K.C. Changing the Landscape for Type 1 Diabetes: The First Step to Prevention. Lancet 2019, 394, 1286–1296. [Google Scholar] [CrossRef]
- Beik, P.; Ciesielska, M.; Kucza, M.; Kurczewska, A.; Kuzminska, J.; Maćkowiak, B.; Niechciał, E. Prevention of Type 1 Diabetes: Past Experiences and Future Opportunities. J. Clin. Med. 2020, 9, 2805. [Google Scholar] [CrossRef]
- Frommer, L.; Kahaly, G.J. Type 1 Diabetes and Autoimmune Thyroid Disease—The Genetic Link. Front. Endocrinol. 2021, 12, 618213. [Google Scholar] [CrossRef]
- Cai, Y.; Yan, J.; Gu, Y.; Chen, H.; Chen, Y.; Xu, X.; Zhang, M.; Yu, L.; Zheng, X.; Yang, T. Autoimmune Thyroid Disease Correlates to Islet Autoimmunity on Zinc Transporter 8 Autoantibody. Endocr. Connect. 2021, 10, 534–542. [Google Scholar] [CrossRef]
- Prieto, J.; Singh, K.B.; Nnadozie, M.C.; Abdal, M.; Shrestha, N.; Abe, R.A.M.; Masroor, A.; Khorochkov, A.; Mohammed, L. New Evidence in the Pathogenesis of Celiac Disease and Type 1 Diabetes Mellitus: A Systematic Review. Cureus 2021, 13, e16721. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 Diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Li, Y.; Sun, F.; Yue, T.-T.; Wang, F.-X.; Yang, C.-L.; Luo, J.-H.; Rong, S.-J.; Xiong, F.; Zhang, S.; Wang, C.-Y. Revisiting the Antigen-Presenting Function of Beta Cells in T1D Pathogenesis. Front. Immunol. 2021, 12, 6907838. [Google Scholar] [CrossRef]
- Ahmed, S.; Cerosaletti, K.; James, E.; Long, S.A.; Mannering, S.; Speake, C.; Nakayama, M.; Tree, T.; Roep, B.O.; Herold, K.C.; et al. Standardizing T-Cell Biomarkers in Type 1 Diabetes: Challenges and Recent Advances. Diabetes 2019, 68, 1366–1379. [Google Scholar] [CrossRef]
- Lernmark, Å. Etiology of Autoimmune Islet Disease: Timing Is Everything. Diabetes 2021, 70, 1431–1439. [Google Scholar] [CrossRef]
- Colli, M.L.; Szymczak, F.; Eizirik, D.L. Molecular Footprints of the Immune Assault on Pancreatic Beta Cells in Type 1 Diabetes. Front. Endocrinol. 2020, 11, 568446. [Google Scholar] [CrossRef]
- Mallone, R.; Eizirik, D.L. Presumption of Innocence in Type 1 Diabetes: Why Are Beta Cells Such Vulnerable Autoimmune Targets in Type 1 Diabetes? Diabetologia 2020, 63, 1999–2006. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T.; Richardson, S.J.; Pugliese, A. Pancreas Pathology During the Natural History of Type 1 Diabetes. Curr. Diabetes Rep. 2018, 18, 124. [Google Scholar] [CrossRef] [Green Version]
- Carré, A.; Richardson, S.J.; Larger, E.; Mallone, R. Presumption of Guilt for T Cells in Type 1 Diabetes: Lead Culprits or Partners in Crime Depending on Age of Onset? Diabetologia 2020, 64, 15–25. [Google Scholar] [CrossRef]
- Colli, M.L.; Ramos-Rodríguez, M.; Nakayasu, E.S.; Alvelos, M.I.; Lopes, M.; Hill, J.L.E.; Turatsinze, J.V.; Coomans de Brachène, A.; Russell, M.A.; Raurell-Vila, H.; et al. An Integrated Multi-Omics Approach Identifies the Landscape of Interferon-α-Mediated Responses of Human Pancreatic Beta Cells. Nat. Commun. 2020, 11, 2584. [Google Scholar] [CrossRef]
- Szymczak, F.; Colli, M.L.; Mamula, M.J.; Evans-Molina, C.; Eizirik, D.L. Gene Expression Signatures of Target Tissues in Type 1 Diabetes, Lupus Erythematosus, Multiple Sclerosis, and Rheumatoid Arthritis. Sci. Adv. 2021, 7, eabd7600. [Google Scholar] [CrossRef]
- Benninger, R.K.P.; Dorrell, C.; Hodson, D.J.; Rutter, G.A. The Impact of Pancreatic Beta Cell Heterogeneity on Type 1 Diabetes Pathogenesis. Curr. Diabetes Rep. 2018, 18, 112. [Google Scholar] [CrossRef] [Green Version]
- Vilas-Boas, E.A.; Carlein, C.; Nalbach, L.; Almeida, D.C.; Ampofo, E.; Carpinelli, A.R.; Roma, L.P.; Ortis, F. Early Cytokine-Induced Transient NOX2 Activity Is ER Stress-Dependent and Impacts β-Cell Function and Survival. Antioxidants 2021, 10, 1305. [Google Scholar] [CrossRef]
- Krischer, J.P.; Liu, X.; Lernmark, Å.; Hagopian, W.A.; Rewers, M.J.; She, J.-X.; Toppari, J.; Ziegler, A.G.; Akolkar, B. Characteristics of Children Diagnosed with Type 1 Diabetes before vs after 6 Years of Age in the TEDDY Cohort Study. Diabetologia 2021, 64, 2247–2257. [Google Scholar] [CrossRef]
- Panzer, J.K.; Hiller, H.; Cohrs, C.M.; Almaça, J.; Enos, S.J.; Beery, M.; Cechin, S.; Drotar, D.M.; Weitz, J.R.; Santini, J.; et al. Pancreas Tissue Slices from Organ Donors Enable in Situ Analysis of Type 1 Diabetes Pathogenesis. JCI Insight 2020, 5, e134525. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [Green Version]
- Evans-Molina, C.; Sims, E.K.; Dimeglio, L.A.; Ismail, H.M.; Steck, A.K.; Palmer, J.P.; Krischer, J.P.; Geyer, S.; Xu, P.; Sosenko, J.M. β Cell Dysfunction Exists More than 5 Years before Type 1 Diabetes Diagnosis. JCI Insight 2018, 3, e120877. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-Cells in Type 1 and Type 2 Diabetes Mellitus: Different Pathways to Failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Erdem, N.; Montero, E.; Roep, B.O. Breaking and Restoring Immune Tolerance to Pancreatic Beta-Cells in Type 1 Diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2021, 28, 397–403. [Google Scholar] [CrossRef]
- Vig, S.; Lambooij, J.M.; Zaldumbide, A.; Guigas, B. Endoplasmic Reticulum-Mitochondria Crosstalk and Beta-Cell Destruction in Type 1 Diabetes. Front. Immunol. 2021, 12, 669492. [Google Scholar] [CrossRef]
- Kim, Y.K.; Sussel, L.; Davidson, H.W. Inherent Beta Cell Dysfunction Contributes to Autoimmune Susceptibility. Biomolecules 2021, 11, 512. [Google Scholar] [CrossRef]
- Muralidharan, C.; Linnemann, A.K. Beta-Cell Autophagy in the Pathogenesis of Type 1 Diabetes. Am. J. Physiol.-Endocrinol. Metab. 2021, 321, E410–E416. [Google Scholar] [CrossRef]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 Diabetes Mellitus as a Disease of the β-Cell (Do Not Blame the Immune System?). Nat. Rev. Endocrinol. 2020, 17, 150–161. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T.; Johnson, J.D.; Overbergh, L.; Dunne, J.L. Neoepitopes in Type 1 Diabetes: Etiological Insights, Biomarkers and Therapeutic Targets. Front. Immunol. 2021, 12, 667989. [Google Scholar] [CrossRef]
- Carre, A.; Mallone, R. Making Insulin and Staying Out of Autoimmune Trouble: The Beta-Cell Conundrum. Front. Immunol. 2021, 12, 639682. [Google Scholar] [CrossRef]
- Balakrishnan, S.; Kumar, P.; Prabhakar, B.S. Post-Translational Modifications Contribute to Neoepitopes in Type-1 Diabetes: Challenges for Inducing Antigen-Specific Tolerance. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2020, 1868, 140478. [Google Scholar] [CrossRef]
- Oshima, M.; Knoch, K.P.; Diedisheim, M.; Petzold, A.; Cattan, P.; Bugliani, M.; Marchetti, P.; Choudhary, P.; Huang, G.C.; Bornstein, S.R.; et al. Virus-like Infection Induces Human β Cell Dedifferentiation. JCI Insight 2018, 3, e97732. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kepple, J.D.; Shalev, A.; Hunter, C.S. LDB1-Mediated Transcriptional Complexes Are Sensitive to Islet Stress. Islets 2022, 14, 58–68. [Google Scholar] [CrossRef]
- Baker, R.L.; Jamison, B.L.; Haskins, K. Hybrid Insulin Peptides Are Neo-Epitopes for CD4 T Cells in Autoimmune Diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 195–200. [Google Scholar] [CrossRef]
- Wang, Y.; Sosinowski, T.; Novikov, A.; Crawford, F.; White, J.; Jin, N.; Liu, Z.; Zou, J.; Neau, D.; Davidson, H.W.; et al. How C-terminal Additions to Insulin B-Chain Fragments Create Superagonists for T Cells in Mouse and Human Type 1 Diabetes. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Reed, B.; Crawford, F.; Hill, R.C.; Jin, N.; White, J.; Krovi, S.H.; Marrack, P.; Hansen, K.; Kappler, J.W. Lysosomal Cathepsin Creates Chimeric Epitopes for Diabetogenic CD4 T Cells via Transpeptidation. J. Exp. Med. 2020, 218, e20192135. [Google Scholar] [CrossRef]
- Reed, B.K.; Kappler, J.W. Hidden in Plain View: Discovery of Chimeric Diabetogenic CD4 T Cell Neo-Epitopes. Front. Immunol. 2021, 12, 669986. [Google Scholar] [CrossRef]
- Mannering, S.I.; Rubin, A.F.; Wang, R.; Bhattacharjee, P. Identifying New Hybrid Insulin Peptides (HIPs) in Type 1 Diabetes. Front. Immunol. 2021, 12, 2–7. [Google Scholar] [CrossRef]
- Benkahla, M.A.; Sabouri, S.; Kiosses, W.B.; Rajendran, S.; Quesada-masachs, E.; von Herrath, M.G. HLA Class I Hyper-Expression Unmasks Beta Cells but Not Alphaα Cells to the Immune System in Pre-Diabetes. J. Autoimmun. 2021, 119, 102628. [Google Scholar] [CrossRef]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. Alpha Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigi, L.; Brusco, N.; Grieco, G.E.; Licata, G.; Krogvold, L.; Marselli, L.; Gysemans, C.; Overbergh, L.; Marchetti, P.; Mathieu, C.; et al. Pancreatic Alpha-Cells Contribute Together With Beta-Cells to CXCL10 Expression in Type 1 Diabetes. Front. Endocrinol. 2020, 11, 630. [Google Scholar] [CrossRef] [PubMed]
- Wendt, A.; Eliasson, L. Pancreatic α-Cells—The Unsung Heroes in Islet Function. Semin. Cell Dev. Biol. 2020, 103, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells from Donors with Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Rodríguez, M.; Raurell-vila, H.; Colli, M.L.; Alvelos, M.I.; Subirana-Granés, M.; Juan-mateu, J.; Norris, R.; Turatsinze, J.; Nakayasu, E.S.; Webb-Robertson, B.-J.M.; et al. The Impact of Pro-Inflammatory Cytokines on the Beta-Cell Regulatory Landscape Provides Insights into the Genetics of Type 1 Diabetes. Nat. Genet. 2019, 51, 1588–1595. [Google Scholar] [CrossRef]
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of Type 1 Diabetes. Pediatr. Diabetes 2018, 19, 346–353. [Google Scholar] [CrossRef]
- Pociot, F.; Lernmark, Å. Genetic Risk Factors for Type 1 Diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Vandewalle, J.; Van der Auwera, B.J.; Amin, H.; Quartier, E.; Desouter, A.K.; Tenoutasse, S.; Gillard, P.; De Block, C.; Keymeulen, B.; Gorus, F.K.; et al. Genetic Variation at ERBB3/IKZF4 and Sexual Dimorphism in Epitope Spreading in Single Autoantibody-Positive Relatives. Diabetologia 2021, 64, 2511–2516. [Google Scholar] [CrossRef]
- Diedisheim, M.; Carcarino, E.; Vandiedonck, C.; Roussel, R.; Gautier, J.; Venteclef, N. Regulation of Inflammation in Diabetes: From Genetics to Epigenomics Evidence. Mol. Metab. 2020, 41, 101041. [Google Scholar] [CrossRef]
- Pang, H.; Luo, S.; Huang, G.; Xia, Y.; Xie, Z.; Zhou, Z. Advances in Knowledge of Candidate Genes Acting at the Beta-Cell Level in the Pathogenesis of T1DM. Front. Endocrinol. 2020, 11, 119. [Google Scholar] [CrossRef]
- Huang, Z.; Qi, G.; Miller, J.S.; Zheng, S.G. CD226: An Emerging Role in Immunologic Diseases. Front. Cell Dev. Biol. 2020, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.R.; Yeh, W.I.; Longfield, J.R.; Gallagher, J.; Infante, C.M.; Wellford, S.; Posgai, A.L.; Atkinson, M.A.; Campbell-Thompson, M.; Lieberman, S.M.; et al. CD226 Deletion Reduces Type 1 Diabetes in the NOD Mouse by Impairing Thymocyte Development and Peripheral T Cell Activation. Front. Immunol. 2020, 11, 2180. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The Diabetes Pandemic and Associated Infections: Suggestions for Clinical Microbiology. Rev. Med. Microbiol. 2019, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Messina, G.; Giovenzana, A.; Petrelli, A. New Evidence of Exocrine Pancreatopathy in Pre-Symptomatic and Symptomatic Type 1 Diabetes. Curr. Diabetes Rep. 2019, 19, 92. [Google Scholar] [CrossRef]
- Onengut-Gumuscu, S.; Paila, U.; Chen, W.M.; Ratan, A.; Zhu, Z.; Steck, A.K.; Frohnert, B.I.; Waugh, K.C.; Webb-Robertson, B.J.M.; Norris, J.M.; et al. Novel Genetic Risk Factors Influence Progression of Islet Autoimmunity to Type 1 Diabetes. Sci. Rep. 2020, 10, 19193. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From Mechanism to Autoimmune Therapy. Int. Immunopharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Scoville, D.W.; Jetten, A.M. GLIS3: A Critical Transcription Factor in Islet β-Cell Generation. Cells 2021, 10, 3471. [Google Scholar] [CrossRef]
- Mine, K.; Yoshikai, Y.; Takahashi, H.; Mori, H.; Anzai, K.; Nagafuchi, S. Genetic Susceptibility of the Host in Virus-Induced Diabetes. Microorganisms 2020, 8, 1133. [Google Scholar] [CrossRef]
- Mori, H.; Takahashi, H.; Mine, K.; Higashimoto, K.; Inoue, K.; Kojima, M.; Kuroki, S.; Eguchi, T.; Ono, Y.; Inuzuka, S.; et al. TYK2 Promoter Variant Is Associated with Impaired Insulin Secretion and Lower Insulin Resistance in Japanese Type 2 Diabetes Patients. Genes 2021, 12, 400. [Google Scholar] [CrossRef]
- Mordes, J.P.; Cort, L.; Liu, Z.; Eberwine, R.; Blankenhorn, E.P.; Pierce, B.G. T Cell Receptor Genotype and Ubash3a Determine Susceptibility to Rat Autoimmune Diabetes. Genes 2021, 12, 852. [Google Scholar] [CrossRef]
- Kaur, S.; Mirza, A.H.; Overgaard, A.J.; Pociot, F.; Størling, J. A Dual Systems Genetics Approach Identifies Common Genes, Networks, and Pathways for Type 1 and 2 Diabetes in Human Islets. Front. Genet. 2021, 12, 630109. [Google Scholar] [CrossRef] [PubMed]
- Howlader, M.; Sultana, M.I.; Akter, F.; Hossain, M. Adiponectin Gene Polymorphisms Associated with Diabetes Mellitus: A Descriptive Review. Heliyon 2021, 7, e07851. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.C.; Inshaw, J.R.J.; Onengut-Gumuscu, S.; Chen, W.-M.; Santa Cruz, D.F.; Yang, H.; Cutler, A.J.; Crouch, D.J.M.; Farber, E.; Bridges, S.L., Jr.; et al. Fine-Mapping, Trans-Ancestral and Genomic Analyses Identify Causal Variants, Cells, Genes and Drug Targets for Type 1 Diabetes. Nat. Genet. 2021, 53, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Inshaw, J.R.J.; Sidore, C.; Cucca, F.; Stefana, M.I.; Crouch, D.J.M.; Mccarthy, M.I.; Mahajan, A.; Todd, J.A. Analysis of Overlapping Genetic Association in Type 1 and Type 2 Diabetes. Diabetologia 2021, 64, 1342–1347. [Google Scholar] [CrossRef]
- Nyaga, D.M.; Vickers, M.H.; Jefferies, C.; Fadason, T.; O’Sullivan, J.M. Untangling the Genetic Link between Type 1 and Type 2 Diabetes Using Functional Genomics. Sci. Rep. 2021, 11, 13871. [Google Scholar] [CrossRef]
- Sticht, J.; Álvaro-benito, M.; Konigorski, S. Type 1 Diabetes and the HLA Region: Genetic Association Besides Classical HLA Class II Genes. Front. Genet. 2021, 12, 683946. [Google Scholar] [CrossRef]
- Nygård, L.; Laine, A.-P.; Kiviniemi, M.; Toppari, J.; Härkönen, T.; Knip, M.; Veijola, R.; Lempainen, J.; Ilonen, J. Tri-SNP Polymorphism in the Intron of HLA-DRA1 Affects Type 1 Diabetes Susceptibility in the Finnish Population. Hum. Immunol. 2021, 82, 912–916. [Google Scholar] [CrossRef]
- Zhao, L.P.; Papadopoulos, G.K.; Lybrand, T.P.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Larsson, H.E.; Ludvigsson, J.; Marcus, C.; Persson, M.; et al. The KAG Motif of HLA-DRB1 (Β71, Β74, Β86) Predicts Seroconversion and Development of Type 1 Diabetes. eBioMedicine 2021, 69, 103431. [Google Scholar] [CrossRef]
- Jiang, Z.; Ren, W.; Liang, H.; Yan, J.; Yang, D.; Luo, S.; Zheng, X.; Lin, G.-W.; Xian, Y.; Xu, W.; et al. HLA Class I Genes Modulate Disease Risk and Age at Onset Together with DR-DQ in Chinese Patients with Insulin-Requiring Type 1 Diabetes. Diabetologia 2021, 64, 2026–2036. [Google Scholar] [CrossRef]
- Pang, H.; Xia, Y.; Luo, S.; Huang, G.; Li, X.; Xie, Z.; Zhou, Z. Emerging Roles of Rare and Low-Frequency Genetic Variants in Type 1 Diabetes Mellitus. J. Med. Genet. 2021, 58, 289–296. [Google Scholar] [CrossRef]
- Qu, H.Q.; Qu, J.; Bradfield, J.; Marchand, L.; Glessner, J.; Chang, X.; March, M.; Li, J.; Connolly, J.J.; Roizen, J.D.; et al. Genetic Architecture of Type 1 Diabetes with Low Genetic Risk Score Informed by 41 Unreported Loci. Commun. Biol. 2021, 4, 908. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.J.; Dennis, J.M.; Sharp, S.A.; Kaur, A.; Misra, S.; Walkey, H.C.; Johnston, D.G.; Oliver, N.S.; Hagopian, W.A.; Weedon, M.N.; et al. DR15-DQ6 Remains Dominantly Protective against Type 1 Diabetes throughout the First Five Decades of Life. Diabetologia 2021, 64, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, J.; James, E.; Chow, I.-T.; Reijonen, H.; Kwok, W.W. Increased Islet Antigen-Specific Regulatory and Effector CD4+ T Cells in Healthy Subjects with the Type 1 Diabetes Protective Haplotype. Sci. Immunol. 2020, 5, eaax8767. [Google Scholar] [CrossRef]
- Schweiger, D.S.; Goricar, K.; Hovnik, T.; Mendez, A.; Bratina, N.; Brecelj, J.; Vidan-Jeras, B.; Battelino, T.; Dolzan, V. Dual Role of PTPN22 but Not NLRP3 Inflammasome Polymorphisms in Type 1 Diabetes and Celiac Disease in Children. Front. Pediatr. 2019, 7, 63. [Google Scholar] [CrossRef]
- Grant, S.F.A.; Wells, A.D.; Rich, S.S. Next Steps in the Identification of Gene Targets for Type 1 Diabetes. Diabetologia 2020, 63, 2260–2269. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.H.; Fontaine, D.A.; Lodh, S.; Blumer, J.T.; Roopra, A.; Davis, D.B. TCF19 Impacts a Network of Inflammatory and DNA Damage Response Genes in the Pancreatic β-Cell. Metabolites 2021, 11, 513. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Morgan, N.G. Enteroviral Infections in the Pathogenesis of Type 1 Diabetes: New Insights for Therapeutic Intervention. Curr. Opin. Pharmacol. 2018, 43, 11–19. [Google Scholar] [CrossRef]
- Pedersen, K.; Haupt-Jorgensen, M.; Krogvold, L.; Kaur, S.; Gerling, I.C.; Pociot, F.; Dahl-Jørgensen, K.; Buschard, K.; Dk, K.P. Genetic Predisposition in the 2′-5′ A Pathway in the Development of Type 1 Diabetes: Potential Contribution to Dysregulation of Innate Antiviral Immunity. Diabetologia 2021, 64, 1805–1815. [Google Scholar] [CrossRef]
- Enczmann, J.; Balz, V.; Hoffmann, M.; Kummer, S.; Reinauer, C.; Döing, C.; Förtsch, K.; Welters, A.; Mayatepek, E.; Meissner, T.; et al. Next Generation Sequencing Identifies the HLA-DQA1*03:03 Allele in the Type 1 Diabetes Risk-Associated HLA-DQ8 Serotype. Genes 2021, 12, 1879. [Google Scholar] [CrossRef]
- Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both Which Cause T1D. Microorganisms 2020, 8, 1017. [Google Scholar] [CrossRef]
- Tremblay, J.; Hamet, P. Environmental and Genetic Contributions to Diabetes. Metab. Clin. Exp. 2019, 100S, 153952. [Google Scholar] [CrossRef] [PubMed]
- Taha-Khalde, A.; Haim, A.; Karakis, I.; Shashar, S.; Biederko, R.; Shtein, A.; Hershkowits, E.; Novack, L. Air Pollution and Meteorological Conditions during Gestation and Type 1 Diabetes in Offspring. Environ. Int. 2021, 154, 106546. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Portincasa, P. Relationships between Emissions of Toxic Airborne Molecules and Type 1 Diabetes Incidence in Children: An Ecologic Study. World J. Diabetes 2021, 12, 673–684. [Google Scholar] [CrossRef]
- Montresor-López, J.A.; Reading, S.R.; Yanosky, J.D.; Mittleman, M.A.; Bell, R.A.; Crume, T.L.; Dabelea, D.; Dolan, L.; D’Agostino, R.B.; Marcovina, S.M.; et al. The Relationship between Traffic-Related Air Pollution Exposures and Allostatic Load Score among Youth with Type 1 Diabetes in the SEARCH Cohort. Environ. Res. 2021, 197, 111075. [Google Scholar] [CrossRef] [PubMed]
- Nurminen, N.; Cerrone, D.; Lehtonen, J.; Parajuli, A.; Roslund, M.; Lönnrot, M.; Ilonen, J.; Toppari, J.; Veijola, R.; Knip, M.; et al. Land Cover of Early-Life Environment Modulates the Risk of Type 1 Diabetes. Diabetes Care 2021, 44, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Camaya, I.; Mok, T.Y.; Lund, M.; To, J.; Braidy, N.; Robinson, M.W.; Santos, J.; O’Brien, B.; Donnelly, S. The Parasite-Derived Peptide FhHDM-1 Activates the PI3K/Akt Pathway to Prevent Cytokine-Induced Apoptosis of β-Cells. J. Mol. Med. 2021, 99, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Marietta, E.; Horwath, I.; Meyer, S.; Khaleghi-Rostamkolaei, S.; Norman, E.; Luckey, D.; Balakrishnan, B.; Mangalam, A.; Choung, R.S.; Taneja, V.; et al. Administration of Human Derived Upper Gut Commensal Prevotella Histicola Delays the Onset of Type 1 Diabetes in NOD Mice. BMC Microbiol. 2022, 22, 8. [Google Scholar] [CrossRef]
- Dedrick, S.; Sundaresh, B.; Huang, Q.; Brady, C.; Yoo, T.; Cronin, C.; Rudnicki, C.; Flood, M.; Momeni, B.; Ludvigsson, J.; et al. The Role of Gut Microbiota and Environmental Factors in Type 1 Diabetes Pathogenesis. Front. Endocrinol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Fuhri Snethlage, C.M.; Nieuwdorp, M.; van Raalte, D.H.; Rampanelli, E.; Verchere, B.C.; Hanssen, N.M.J. Auto-Immunity and the Gut Microbiome in Type 1 Diabetes: Lessons from Rodent and Human Studies. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101544. [Google Scholar] [CrossRef]
- Hamilton-Williams, E.E.; Lorca, G.L.; Norris, J.M.; Dunne, J.L. A Triple Threat? The Role of Diet, Nutrition, and the Microbiota in T1D Pathogenesis. Front. Nutr. 2021, 8, 600756. [Google Scholar] [CrossRef]
- Kalbermatter, C.; Fernandez Trigo, N.; Christensen, S.; Ganal-Vonarburg, S.C. Maternal Microbiota, Early Life Colonization and Breast Milk Drive Immune Development in the Newborn. Front. Immunol. 2021, 12, 683022. [Google Scholar] [CrossRef] [PubMed]
- Khamis, T.; Abdelalim, A.F.; Saeed, A.A.; Edress, N.M.; Nafea, A.; Ebian, H.F.; Algendy, R.; Hendawy, D.M.; Arisha, A.H.; Abdallah, S.H. Breast Milk MSCs Upregulated β-Cells PDX1, Ngn3, and PCNA Expression via Remodeling ER Stress /Inflammatory /Apoptotic Signaling Pathways in Type 1 Diabetic Rats. Eur. J. Pharmacol. 2021, 905, 174188. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.R.; Foskett, D.B.; Maxwell, A.J.; Ward, E.J.; Faulkner, C.L.; Luo, J.Y.X.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms 2021, 9, 1519. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, S.; Krogvold, L.; Edwin, B.; Buanes, T.; Korsgren, O.; Laiho, J.E.; Oikarinen, M.; Ludvigsson, J.; Skog, O.; Anagandula, M.; et al. Characterisation of Enterovirus RNA Detected in the Pancreas and Other Specimens of Live Patients with Newly Diagnosed Type 1 Diabetes in the DiViD Study. Diabetologia 2021, 64, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Geravandi, S.; Richardson, S.; Pugliese, A.; Maedler, K. Localization of Enteroviral RNA within the Pancreas in Donors with T1D and T1D-Associated Autoantibodies. Cell Rep. Med. 2021, 2, 100371. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, A.; Nekoua, M.P.; Michaux, H.; Sane, F.; Halouani, A.; Engelmann, I.; Alidijinou, E.K.; Martens, H.; Jaidane, H.; Geenen, V.; et al. Effect of Coxsackievirus B4 Infection on the Thymus: Elucidating Its Role in the Pathogenesis of Type 1 Diabetes. Microorganisms 2021, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective Virome Analyses in Young Children at Increased Genetic Risk for Type 1 Diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Blum, S.I.; Tse, H.M. Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development. Microorganisms 2020, 8, 993. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Bertin, A.; Sane, F.; Gimeno, J.-P.; Fournier, I.; Salzet, M.; Engelmann, I.; Alidjinou, E.K.; Hober, D. Persistence of Coxsackievirus B4 in Pancreatic β Cells Disturbs Insulin Maturation, Pattern of Cellular Proteins, and DNA Methylation. Microorganisms 2021, 9, 1125. [Google Scholar] [CrossRef]
- Akhbari, P.; Richardson, S.J.; Morgan, N.G. Type 1 Diabetes: Interferons and the Aftermath of Pancreatic Beta-Cell Enteroviral Infection. Microorganisms 2020, 8, 1419. [Google Scholar] [CrossRef]
- Nyalwidhe, J.O.; Jurczyk, A.; Satish, B.; Redick, S.; Qaisar, N.; Trombly, M.I.; Vangala, P.; Racicot, R.; Bortell, R.; Harlan, D.M.; et al. Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge. Microorganisms 2020, 8, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias Junior, A.G.; Sampaio, N.G.; Rehwinkel, J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019, 27, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netanyah, E.; Calafatti, M.; Arvastsson, J.; Cabrera-Rode, E.; Cilio, C.M.; Sarmiento, L. Extracellular Vesicles Released by Enterovirus-Infected EndoC-ΒH1 Cells Mediate Non-Lytic Viral Spread. Microorganisms 2020, 8, 1753. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.W.M.; Stienstra, R.; Jaeger, M.; van Gool, A.J.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P.; Tack, C.J. Understanding the Increased Risk of Infections in Diabetes: Innate and Adaptive Immune Responses in Type 1 Diabetes. Metab. Clin. Exp. 2021, 121, 154795. [Google Scholar] [CrossRef]
- Burke, R.M.; Tate, J.E.; Jiang, B.; Parashar, U.D. Rotavirus and Type 1 Diabetes—Is There a Connection? A Synthesis of the Evidence. J. Infect. Dis. 2020, 222, 1076–1083. [Google Scholar] [CrossRef]
- Dian, Z.; Sun, Y.; Zhang, G.; Xu, Y.; Fan, X.; Yang, X.; Pan, Q.; Peppelenbosch, M.; Miao, Z. Rotavirus-Related Systemic Diseases: Clinical Manifestation, Evidence and Pathogenesis. Crit. Rev. Microbiol. 2021, 47, 580–595. [Google Scholar] [CrossRef]
- Chekhlabi, N.; Haoudar, A.; Echcharii, N.; Ettair, S.; Dini, N. New-Onset Diabetes with Ketoacidosis Precipitated by COVID-19 in Children: A Report of Two Cases. Case Rep. Pediatr. 2021, 2021, 5545258. [Google Scholar] [CrossRef]
- Venkatesh, N.; Astbury, N.; Thomas, M.C.; Rosado, C.J.; Pappas, E.; Krishnamurthy, B.; MacIsaac, R.J.; Kay, T.W.H.; Thomas, H.E.; O’Neal, D.N. Severe Acute Respiratory Syndrome Coronavirus 2 as a Potential Cause of Type 1 Diabetes Facilitated by Spike Protein Receptor Binding Domain Attachment to Human Islet Cells: An Illustrative Case Study and Experimental Data. Diabet. Med. 2021, 38, e14608. [Google Scholar] [CrossRef]
- Alfishawy, M.; Nassar, M.; Mohamed, M.; Fatthy, M.; Elmessiery, R.M. New-Onset Type 1 Diabetes Mellitus with Diabetic Ketoacidosis and Pancreatitis in a Patient with COVID-19. Sci. Afr. 2021, 13, e00915. [Google Scholar] [CrossRef]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 Infects and Replicates in Cells of the Human Endocrine and Exocrine Pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Wu, C.T.; Lidsky, P.V.; Xiao, Y.; Lee, I.T.; Cheng, R.; Nakayama, T.; Jiang, S.; Demeter, J.; Bevacqua, R.J.; Chang, C.A.; et al. SARS-CoV-2 Infects Human Pancreatic β Cells and Elicits β Cell Impairment. Cell Metab. 2021, 33, 1565–1576.e5. [Google Scholar] [CrossRef] [PubMed]
- Boddu, S.K.; Aurangabadkar, G.; Kuchay, M.S. New Onset Diabetes, Type 1 Diabetes and COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- El-Senousy, W.M.; Abdel-Moneim, A.; Abdel-Latif, M.; El-hefnawy, M.H.; Khalil, R.G. Coxsackievirus B4 as a Causative Agent of Diabetes Mellitus Type 1: Is There a Role of Inefficiently Treated Drinking Water and Sewage in Virus Spreading? Food Environ. Virol. 2018, 10, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.D.; Dey, R.; Scott, C.; Li, Q.; Pang, X.L.; Ashbolt, N.J. Persistence of Infectious Enterovirus within Free-Living Amoebae—A Novel Waterborne Risk Pathway? Water Res. 2018, 144, 204–214. [Google Scholar] [CrossRef]
- Berbudi, A.; Ajendra, J.; Wardani, A.P.F.; Hoerauf, A.; Hübner, M.P. Parasitic Helminths and Their Beneficial Impact on Type 1 and Type 2 Diabetes. Diabetes/Metab. Res. Rev. 2016, 32, 238–250. [Google Scholar] [CrossRef]
- Esposito, S.; Mariotti Zani, E.; Torelli, L.; Scavone, S.; Petraroli, M.; Patianna, V.; Predieri, B.; Iughetti, L.; Principi, N. Childhood Vaccinations and Type 1 Diabetes. Front. Immunol. 2021, 12, 667889. [Google Scholar] [CrossRef]
- Inns, T.; Fleming, K.M.; Iturriza-Gomara, M.; Hungerford, D. Paediatric Rotavirus Vaccination, Coeliac Disease and Type 1 Diabetes in Children: A Population-Based Cohort Study. BMC Med. 2021, 19, 147. [Google Scholar] [CrossRef]
- Yue, T.; Sun, F.; Yang, C.; Wang, F.; Luo, J.; Yang, P.; Xiong, F.; Zhang, S.; Yu, Q.; Wang, C.-Y. The AHR Signaling Attenuates Autoimmune Responses During the Development of Type 1 Diabetes. Front. Immunol. 2020, 11, 1510. [Google Scholar] [CrossRef]
- Levet, S.; Charvet, B.; Bertin, A.; Deschaumes, A.; Perron, H.; Hober, D. Human Endogenous Retroviruses and Type 1 Diabetes. Curr. Diabetes Rep. 2019, 19, 141. [Google Scholar] [CrossRef] [Green Version]
- Dechaumes, A.; Bertin, A.; Sane, F.; Levet, S.; Varghese, J.; Charvet, B.; Gmyr, V.; Kerr-Conte, J.; Pierquin, J.; Arunkumar, G.; et al. Coxsackievirus-B4 Infection Can Induce the Expression of Human Endogenous Retrovirus W in Primary Cells. Microorganisms 2020, 8, 1335. [Google Scholar] [CrossRef]
- Pircalabioru, G.G.; Corcionivoschi, N.; Gundogdu, O.; Chifiriuc, M.-C.; Marutescu, L.G.; Ispas, B.; Savu, O. Dysbiosis in the Development of Type I Diabetes and Associated Complications: From Mechanisms to Targeted Gut Microbes Manipulation Therapies. Int. J. Mol. Sci. 2021, 22, 2763. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Lednovich, K.; Xu, K.; Gough, S.; Wicksteed, B.; Layden, B.T. FFAR from the Gut Microbiome Crowd: SCFA Receptors in T1D Pathology. Metabolites 2021, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, P.; Metos, J.; Anandh Babu, P.V. Impact of Type 1 Diabetes on the Composition and Functional Potential of Gut Microbiome in Children and Adolescents: Possible Mechanisms, Current Knowledge, and Challenges. Gut Microbes 2021, 13, 1–18. [Google Scholar] [CrossRef]
- Mønsted, M.Ø.; Falck, N.D.; Pedersen, K.; Buschard, K.; Holm, L.J.; Haupt-Jorgensen, M. Intestinal Permeability in Type 1 Diabetes: An Updated Comprehensive Overview. J. Autoimmun. 2021, 122, 102674. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.C.G.; Oliveira, R.P.; Torres, L.; Aguiar, S.L.F.; Pinheiro-Rosa, N.; Lemos, L.; Guimarães, M.A.; Reis, D.; Silveira, T.; Ferreira, Ê.; et al. Abnormalities in the Gut Mucosa of Non-Obese Diabetic Mice Precede the Onset of Type 1 Diabetes. J. Leukoc. Biol. 2019, 106, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Fulci, V.; Stronati, L.; Cucchiara, S.; Laudadio, I.; Carissimi, C. Emerging Roles of Gut Virome in Pediatric Diseases. Int. J. Mol. Sci. 2021, 22, 4127. [Google Scholar] [CrossRef]
- Cinek, O.; Kramna, L.; Ann, M.; Ibekwe, U.; Ahmadov, G.; Mukhtar, B.; Elmahi, E.; Mekki, H.; Lebl, J.; Ahmed, M. Eukaryotic Viruses in the Fecal Virome at the Onset of Type 1 Diabetes: A Study from Four Geographically Distant African and Asian Countries. Pediatr. Diabetes 2021, 22, 558–566. [Google Scholar] [CrossRef]
- Salamon, D.; Sroka-Oleksiak, A.; Gurgul, A.; Arent, Z.; Szopa, M.; Bulanda, M.; Małecki, M.T.; Gosiewski, T. Analysis of the Gut Mycobiome in Adult Patients with Type 1 and Type 2 Diabetes Using Next-Generation Sequencing (NGS) with Increased Sensitivity—Pilot Study. Nutrients 2021, 13, 1066. [Google Scholar] [CrossRef]
- Wernroth, M.L.; Fall, K.; Svennblad, B.; Ludvigsson, J.F.; Sjölander, A.; Almqvist, C.; Fall, T. Early Childhood Antibiotic Treatment for Otitis Media and Other Respiratory Tract Infections Is Associated with Risk of Type 1 Diabetes: A Nationwide Register-Based Study with Sibling Analysis. Diabetes Care 2020, 43, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Paun, A.; Yau, C.; Meshkibaf, S.; Daigneault, M.C.; Marandi, L.; Mortin-Toth, S.; Bar-Or, A.; Allen-Vercoe, E.; Poussier, P.; Danska, J.S. Association of HLA-Dependent Islet Autoimmunity with Systemic Antibody Responses to Intestinal Commensal Bacteria in Children. Sci. Immunol. 2019, 4, eaau8125. [Google Scholar] [CrossRef]
- Al Theyab, A.; Almutairi, T.; Al-suwaidi, A.M.; Bendriss, G.; McVeigh, C.; Chaari, A. Epigenetic Effects of Gut Metabolites: Exploring the Path of Dietary Prevention of Type 1 Diabetes. Front. Nutr. 2020, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, L.M.; Zou, Y.; Zhang, S.; Xiong, F.; Wang, C.Y. Implication of Epigenetic Factors in the Pathogenesis of Type 1 Diabetes. Chin. Med. J. 2021, 134, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and Epigenetic Fine Mapping of Causal Autoimmune Disease Variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Arvastsson, J.; Sarmiento, L.; Cilio, C.M. Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes 2021, 12, 887. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Uzun, Y.; He, B.; Salamati, S.E.; Coffey, J.K.M.; Tsalikian, E.; Tan, K. Risk Variants Disrupting Enhancers of TH1 and TREG Cells in Type 1 Diabetes. Proc. Natl. Acad. Sci. USA 2019, 116, 7581–7590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerna, M. Epigenetic Regulation in Etiology of Type 1 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 36. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.K.; Vanderlinden, L.A.; Dong, F.; Carry, P.M.; Seifert, J.; Waugh, K.; Shorrosh, H.; Fingerlin, T.; Frohnert, B.I.; Yang, I.V.; et al. Longitudinal DNA Methylation Differences Precede Type 1 Diabetes. Sci. Rep. 2020, 10, 3721. [Google Scholar] [CrossRef] [Green Version]
- Fasolino, M.; Goldman, N.; Wang, W.; Cattau, B.; Zhou, Y.; Petrovic, J.; Link, V.M.; Cote, A.; Chandra, A.; Silverman, M.; et al. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 2020, 52, 257–274.e11. [Google Scholar] [CrossRef]
- Dieter, C.; Lemos, N.E.; de Faria Corrêa, N.R.; Assmann, T.S.; Crispim, D. The Impact of LncRNAs in Diabetes Mellitus: A Systematic Review and In Silico Analyses. Front. Endocrinol. 2021, 12, 602597. [Google Scholar] [CrossRef]
- Grieco, G.E.; Fignani, D.; Formichi, C.; Nigi, L.; Licata, G.; Maccora, C.; Brusco, N.; Sebastiani, G.; Dotta, F. Extracellular Vesicles in Immune System Regulation and Type 1 Diabetes: Cell-to-Cell Communication Mediators, Disease Biomarkers, and Promising Therapeutic Tools. Front. Immunol. 2021, 12, 682948. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Yao, H. Signature RNAS and Related Regulatory Roles in Type 1 Diabetes Mellitus Based on Competing Endogenous RNA Regulatory Network Analysis. BMC Med. Genom. 2021, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Tesovnik, T.; Kovač, J.; Pohar, K.; Hudoklin, S.; Dovč, K.; Bratina, N.; Trebušak Podkrajšek, K.; Debeljak, M.; Veranič, P.; Bosi, E.; et al. Extracellular Vesicles Derived Human-MiRNAs Modulate the Immune System in Type 1 Diabetes. Front. Cell Dev. Biol. 2020, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Giri, K.R.; de Beaurepaire, L.; Jegou, D.; Lavy, M.; Mosser, M.; Dupont, A.; Fleurisson, R.; Dubreil, L.; Collot, M.; Van Endert, P.; et al. Molecular and Functional Diversity of Distinct Subpopulations of the Stressed Insulin-Secreting Cell’s Vesiculome. Front. Immunol. 2020, 11, 1814. [Google Scholar] [CrossRef]
- Garavelli, S.; Bruzzaniti, S.; Tagliabue, E.; Prattichizzo, F.; Di Silvestre, D.; Perna, F.; La Sala, L.; Ceriello, A.; Mozzillo, E.; Fattorusso, V.; et al. Blood Co-Circulating Extracellular Micrornas and Immune Cell Subsets Associate with Type 1 Diabetes Severity. Int. J. Mol. Sci. 2020, 21, 477. [Google Scholar] [CrossRef] [Green Version]
- Jankauskas, S.S.; Gambardella, J.; Sardu, C.; Lombardi, A.; Santulli, G. Functional Role of MiR-155 in the Pathogenesis of Diabetes Mellitus and Its Complications. Noncoding RNA 2021, 7, 39. [Google Scholar] [CrossRef]
- Mattke, J.; Vasu, S.; Darden, C.M.; Kumano, K.; Lawrence, M.C.; Naziruddin, B. Role of Exosomes in Islet Transplantation. Front. Endocrinol. 2021, 12, 681600. [Google Scholar] [CrossRef]
- Mishto, M.; Mansurkhodzhaev, A.; Rodriguez-Calvo, T.; Liepe, J. Potential Mimicry of Viral and Pancreatic β Cell Antigens Through Non-Spliced and Cis-Spliced Zwitter Epitope Candidates in Type 1 Diabetes. Front. Immunol. 2021, 12, 656451. [Google Scholar] [CrossRef]
- Liu, C.; Li, N.; Liu, G. The Role of MicroRNAs in Regulatory T Cells. J. Immunol. Res. 2020, 2020, 3232061. [Google Scholar] [CrossRef]
- Januszewski, A.S.; Cho, Y.H.; Joglekar, M.V.; Farr, R.J.; Scott, E.S.; Wong, W.K.M.; Carroll, L.M.; Loh, Y.W.; Benitez-Aguirre, P.Z.; Keech, A.C.; et al. Insulin Micro-Secretion in Type 1 Diabetes and Related MicroRNA Profiles. Sci. Rep. 2021, 11, 11727. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, C.; Zheng, Y. Involvement of CircRNAs in Proinflammatory Cytokines-Mediated β-Cell Dysfunction. Mediat. Inflamm. 2021, 2021, 5566453. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.L.; Filipp, S.L.; Grajo, J.R.; Nambam, B.; Beegle, R.; Middlebrooks, E.H.; Gurka, M.J.; Atkinson, M.A.; Schatz, D.A.; Haller, M.J. Relative Pancreas Volume Is Reduced in First-Degree Relatives of Patients with Type 1 Diabetes. Diabetes Care 2019, 42, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-jørgensen, K.; Flodström-Tullberg, M.; Hyöty, H.; Insel, R.A.; Lernmark, Å.; Lloyd, R.E.; et al. Rationale for Enteroviral Vaccination and Antiviral Therapies in Human Type 1 Diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyöty, H.; Leon, F.; Knip, M. Developing a Vaccine for Type 1 Diabetes by Targeting Coxsackievirus B. Expert Rev. Vaccines 2018, 17, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.A.S.; Yassin, E.; Al-Maraghi, A.; Aliyev, E.; Al-Malki, K.; Fakhro, K.A. Diagnosis and Treatment of Type 1 Diabetes at the Dawn of the Personalized Medicine Era. J. Transl. Med. 2021, 19, 137. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene Name | Gene Function(s) | References |
|---|---|---|
| BACH2 | Regulating proinflammatory cytokine-induced apoptotic pathways in pancreatic β-cells (crosstalk with PTPN2) | [46] |
| C1QTNF6 | Participating in the BCR signalling pathway/cytotoxicity | [50] |
| CCR5 | Th cell development/chemokine-induced signalling | [50] |
| CD226 | Modulating thymic T cell selection Impact on peripheral memory/effector CD8+ T cell activation and function Reducing regulatory functions of Foxp3+ Tregs | [51,52] |
| CD69 | Participating in early lymphocyte activation Limiting the inflammatory response Influencing the signalling of NK cells | [53] |
| CLEC16A | Regulating the mitophagy for mitochondrial quality control Possible involvement in β-cell fragility | [54] |
| COL6A6 | / | [55] |
| CTLA4 | Controlling the proliferation of Tregs in the periphery Regulating pancreas autoimmunity | [56] |
| CTSH | Regulating cytokines inside β-cells for proapoptotic signal transduction | [49] |
| ERBB3 | Modulating antigen presentation Modulating cytokine-induced β-cell apoptosis | [53] |
| GLIS3 | Implication in the generation of β-cells, insulin expression Maintaining β-cell functions and mass Exerting antiapoptotic effects | [46,50,57] |
| HIP14 | Regulating β-cell apoptosis and insulin secretion | [50] |
| IFIH1 | Mediating the innate immune system’s interferon response to certain viruses Participating in β-cell response to viral dsRNA | [46,49] |
| IKZF1 | Regulating immune cell development | [50] |
| IL2/IL21 | Influencing T(h) cell differentiation and inflammatory response | [50] |
| IL27 | Modulating T cell subsets and regulating inflammatory response | [53] |
| IL2RA | Variants causing abnormalities in sensitivity to IL2, which is critical to T-regulatory cell function Potential altering of the balance between Tregs and Teffs | [46] |
| IL7R | Involvement in antigen binding, Ig production, and cytotoxicity | [50] |
| MRPS21-PRPF3 | / | [55] |
| NRIR | Negative regulator of interferon response | [55] |
| PRKCQ | Influencing T cell function/apoptosis/innate immune response | [50] |
| PTPN2 | Inducing β-cell apoptosis after interaction with increased local levels of interferon Influencing β-cell response to viral dsRNA | [46,49] |
| PTPN22 | Participating in T cell receptor signalling pathway | [53] |
| SH2B3 | Participating in growth factor and cytokine signalling | [50] |
| STX4 | Associated with insulin secretion Downregulating the expression of chemokine genes associated with inflammation and the apoptosis of pancreatic islets Decreasing the translocation and activation of NF-kB, thus decreasing the apoptosis | [50] |
| TASP1 | Cleaving the MLL protein, which is required for proper HOX gene expression | [55] |
| TNFAIP3 | Downregulating the intrinsic apoptotic pathway Regulating the expression levels of ZnT8 Essential for insulin production and secretion | [50] |
| TYK2 | Regulating the effects of cytokines inside β-cells for proapoptotic signal transduction Mediating interferon response in connection to resistance to various infections Mediating Th1- and Th17-type immune reactions | [50,58,59] |
| UBASH3A | Downregulating the NF-kB signalling pathway upon T cell receptor stimulation, thus reducing the IL2 expression | [46,60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zajec, A.; Trebušak Podkrajšek, K.; Tesovnik, T.; Šket, R.; Čugalj Kern, B.; Jenko Bizjan, B.; Šmigoc Schweiger, D.; Battelino, T.; Kovač, J. Pathogenesis of Type 1 Diabetes: Established Facts and New Insights. Genes 2022, 13, 706. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040706
Zajec A, Trebušak Podkrajšek K, Tesovnik T, Šket R, Čugalj Kern B, Jenko Bizjan B, Šmigoc Schweiger D, Battelino T, Kovač J. Pathogenesis of Type 1 Diabetes: Established Facts and New Insights. Genes. 2022; 13(4):706. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040706
Chicago/Turabian StyleZajec, Ana, Katarina Trebušak Podkrajšek, Tine Tesovnik, Robert Šket, Barbara Čugalj Kern, Barbara Jenko Bizjan, Darja Šmigoc Schweiger, Tadej Battelino, and Jernej Kovač. 2022. "Pathogenesis of Type 1 Diabetes: Established Facts and New Insights" Genes 13, no. 4: 706. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040706