
Complex Presentation of Hao-Fountain Syndrome Solved by Exome Sequencing Highlighting Co-Occurring Genomic Variants

 , ,
, ,
Abstract
:1. Introduction
2. Methods
3. Results
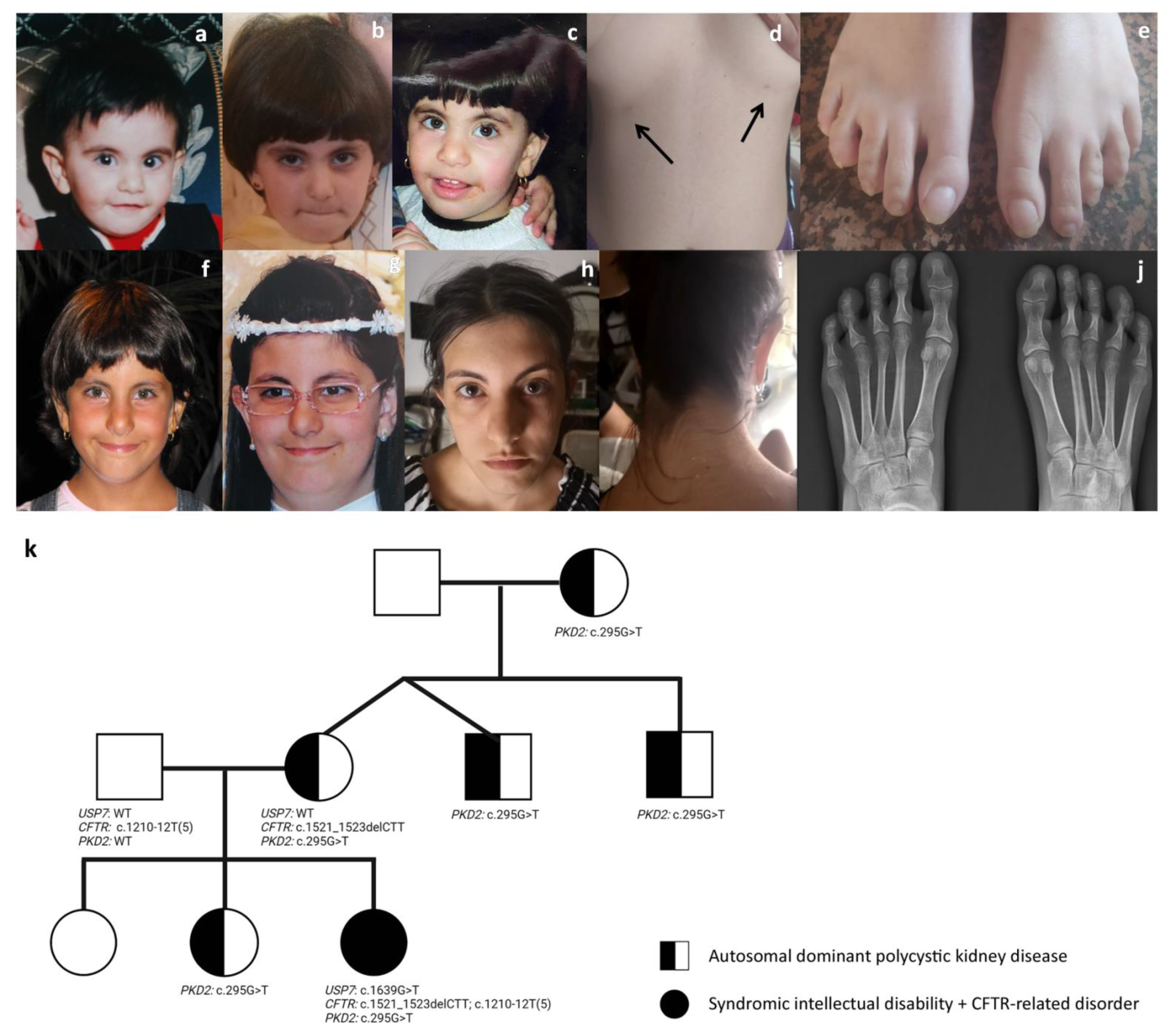
3.1. Case Presentation
3.2. Genetic Analysis Co-Occurring Pathogenic Variants in USP7, CFTR and PKD2 Genes
3.3. Facial Features Assessment in Hao-Fountain Syndrome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Posey, J.E.; Harel, T.; Liu, P.; Pengfel, L.; Rosenfeld, J.; James, R.A.; Akdemir, Z.H.C.; Walklewitz, M.; Bi, W.; Xiao, R.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Karaca, E.; Posey, J.E.; Coban Akdemir, Z.; Pehlivan, D.; Harel, T.; Jhangiani, S.N.; Bayram, Y.; Song, X.; Bahrambeigi, V.; Yuregir, O.O.; et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 2018, 20, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T.; et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.; Blanco, K.; Sayan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A retrospective review of multiple findings in diagnostic exome sequencing: Half are distinct and half are overlapping diagnoses. Genet. Med. 2019, 21, 2199–2207. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.H.; Fountain, M.D., Jr.; FonTacer, K.; Xia, F.; Bi, W.; Kang, S.H.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell. 2015, 59, 956–969. [Google Scholar] [CrossRef] [Green Version]
- Fountain, M.D.; Oleson, D.S.; Rech, M.E.; Segebrecht, L.; Hunter, J.V.; McCarthy, J.M.; Lupo, P.J.; Holtgrewe, M.; Moran, R.; Rosenfeld, J.A.; et al. Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies. Genet. Med. 2019, 21, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- Capra, A.P.; Agolini, E.; La Rosa, M.A.; Novelli, A.; Briuglia, S. Correspondence on “Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies” by Fountain et al. Genet. Med. 2021, 23, 421–422. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, A.; Cimbalo, C.; Castaldo, R.J.; D’Antonio, M.; Scorza, M.; Salvadori, L.; Sepe, A.; Raia, V.; Tosco, A. Cystic Fibrosis-Screening Positive Inconclusive Diagnosis: Newborn Screening and Long-Term Follow-Up Permits to Early Identify Patients with CFTR-Related Disorders. Diagnostics 2020, 10, 570. [Google Scholar] [CrossRef]
- Welsh, M.J.; Ramsey, B.W.; Accurso, F.; Cutting, G.R. Cystic fibrosis. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill Inc.: New York, NY, USA, 2001; pp. 5121–5188. [Google Scholar]
- WHO. Classification of Cystic Fibrosis and Related Disorders; Report of a Joint WHO/ICF(M)AECFTN Meeting; World Health Organization: Geneva, Switzerland, 2001; Reprinted in J. Cyst. Fibros. 2002, 1, 5–8. [Google Scholar]
- Bombieri, C.; Claustres, M.; De BoecK, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Wilschanski, M.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10, S86–S102. [Google Scholar] [CrossRef] [Green Version]
- Casals, T.; De-Garcia, J.; Gallego, M.; Dorca, J.; Rodríguez-Sanchón, B.; Ramos, M.D.; Giménez, J.; Cisteró-Bahima, A.; Olveira, C.; Estivill, X. Bronchiectasis in adult patients: An expression of heterozyosity for CFTR gene mutations? Clin. Genet. 2004, 65, 490–495. [Google Scholar] [CrossRef]
- Pignatti, P.F.; Bombieri, C.; Benetazzo, M.; Casartelli, A.; Trabetti, E.; Gilè, L.S.; Martinati, L.C.; Boner, A.L.; Luisetti, M. CFTR gene variant IVS8-5T in disseminatedbronchiectasis. Am. J. Hum. Genet. 1996, 58, 889–892. [Google Scholar]
- Girodon, E.; Cazeneuve, C.; Lebargy, F.; Chinet, T.; Costes, B.; Ghanem, N.; Martin, J.; Lemay, S.; Scheid, P.; Housset, B.; et al. CFTR gene mutations in adults with disseminated bronchiectasis. Eur. J. Hum. Genet. 1997, 5, 149–155. [Google Scholar] [CrossRef]
- Divac, A.; Nikolic, A.; Mitic-Milikic, M.; Nagorni-Obradovic, L.; Petrovic-Stanojevic, N.; Dopudja-Pantic, V.; Nadaskic, R.; Savic, A.; Radojkovic, D. CFTR mutations and polymorphisms in adult with disseminated bronchiectasis: A controversial issue. Thorax 2005, 60, 85. [Google Scholar]
- Zielenski, J.; Patrizio, P.; Corey, M.; Handelin, B.; Markiewicz, D.; Asch, R.; Tsui, L.C. CFTR gene variant for patients with congenital absence of vas deferens. Am. J. Hum. Genet. 1995, 57, 958–960. [Google Scholar]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.R.; Grantham, J.J. Polycystic kidney disease: Etiology, pathogenesis, and treatment. Dis. Mon. 1995, 41, 693–765. [Google Scholar] [CrossRef]
- Rundle, D.R.; Gorbsky, G.; Tsiokas, L. PKD2 interacts and co-localizes with mDia1 to mitotic spindles of dividing cells: Role of mDia1 in PKD2 localization to mitotic spindles. J. Biol. Chem. 2004, 279, 29728–29739. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef]
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Flex, E.; Martinelli, S.; Van Dijck, A.; Ciolfi, A.; Cecchetti, S.; Coluzzi, E.; Pannone, L.; Andreoli, C.; Radio, F.C.; Pizzi, S.; et al. Aberrant Function of the C-terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes Premature Aging. Am. J. Hum. Genet. 2019, 105, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human nonsynonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Jagadeesh, K.; Wenger, A.; Berger, M.; Guturu, H.; Stenson, P.; Cooper, D.; Bernstein, J.; Bejerano, G. M-CAP eliminates a majority of variants with VoUS in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 2, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Havrilla, J.M.; Fang, L.; Chen, Y.; Peng, J.; Liu, C.; Wu, C.; Sarmady, M.; Botas, P.; Isla, J.; et al. Phen2Gene: Rapid phenotype-driven gene prioritization for rare diseases. NAR Genom. Bioinform. 2020, 2, lqaa032. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Nicholson, B.; Suresh Kumar, K.G. The multifaceted roles of USP7: New therapeutic opportunities. Cell. Biochem. Biophys. 2011, 60, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, J.B.; Morgan, D.O. Protein-linked ubiquitin chain structure restricts activity of deubiquitinating enzymes. J. Biol. Chem. 2011, 286, 45186–45196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavelle, L.P.; McEvoy, S.H.; Ni Mhurchu, E.; Gibney, R.G.; McMahon, C.J.; Heffernan, E.J.; Malone, D.E. Cystic Fibrosis below the Diaphragm: Abdominal Findings in Adult Patients. Radiographics 2015, 35, 680–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecder, T.; Schrier, R.W. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat. Rev. Nephrol. 2009, 5, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.E.; Cai, Y.; Chen, X.I.; Wu, G.Q.; Geng, L.; Cleghorn, K.A.; Johnson, C.M.; Somlo, S. Vascular expression of polycystin-2. J. Am. Soc. Nephrol. 2001, 12, 1–9. [Google Scholar] [CrossRef]


{kind=link}
| Reported Cases | N = 8 | N = 5 | N = 8 | N = 3 | Present Report | Total, N = 25 |
|---|---|---|---|---|---|---|
| USP7 variant | gene deletions a,b | truncating variants b,c | missense variants b | splice site variants b | truncating variant | |
| Sex | 5M, 3F | 1M, 4F | 2M, 6F | 3M | F | 11M, 14F |
| Dysmorphic facial features | 4/6 | 5/5 | 7/7 | 3/3 | + | 20/22 |
| DEVELOPMENT | ||||||
| Developmental delay/intellectual disability | 8/8 | 5/5 | 7/8 | 3/3 | + | 24/25 |
| Decreased fetal movement | 0/6 | 1/3 | 1/3 | 0/3 | + | 3/16 |
| Neonatal hypotonia | 1/6 | 5/5 | 4/7 | 0/3 | + | 11/22 |
| Hypotonia | 4/7 | 5/5 | 5/7 | 1/3 | + | 16/22 |
| Speech delay | 8/8 | 5/5 | 8/8 | 3/3 | + | 25/25 |
| Nonverbal | 1/8 | 0/3 | 3/8 | 0/3 | - | 4/22 |
| Walking milestone age (months) | 28 (mean) | 25 (mean) | 57 (mean) | 21 (mean) | 20 | |
| Sitting without support (months) | 14 (mean) | 11.6 (mean) | 17 (mean) | 11 (mean) | 12 | |
| NEUROLOGICAL | ||||||
| Abnormal MRI | 3/4 | 2/4 | 7/7 | 0/1 | + | 13/17 |
| Seizures | 4/8 | 3/5 | 3/7 | 0/3 | - | 10/24 |
| Abnormal gait | 2/5 | 0/2 | 4/4 | 0/3 | + | 7/15 |
| BEHAVIOR | ||||||
| Behavioral anomalies | 7/8 | 1/3 | 2/7 | 2/3 | + | 15/22 |
| Autism spectrum disorder | 7/7 | 0/3 | 2/4 | 0/3 | - | 9/17 |
| attention deficit-hyperactivity disorder | 3/7 | 0/2 | 1/4 | 2/3 | + | 7/17 |
| Skin picking | 2/8 | 0/4 | 1/5 | 0/3 | - | 3/21 |
| GASTROINTESTINAL | ||||||
| Feeding problems, need for special feeding tools | 4/7 | 2/4 | 4/8 | 2/3 | + | 13/23 |
| Gastroesophageal reflux | 3/5 | 2/3 | 4/6 | 1/3 | + | 11/18 |
| Difficulty in gaining weight | 1/6 | 1/3 | 5/7 | 2/3 | + | 10/20 |
| Chronic constipation | 3/5 | 1/2 | 1/6 | 0/3 | + | 6/17 |
| Neonatal poor suck | 2/6 | 2/5 | 1/4 | 0/3 | + | 6/19 |
| Excessive weight gain | 0/6 | 0/4 | 0/6 | 3/3 | - | 3/20 |
| RESPIRATORY | ||||||
| Asthma | 1/4 | 2/3 | 1/4 | 2/3 | - | 6/15 |
| Sleep apnea/sleep disturbance | 3/7 | 1/3 | 1/7 | 0/3 | + | 6/21 |
| SKELETAL | ||||||
| Short stature | 2/7 | 1/4 | 3/6 | 0/3 | - | 6/21 |
| Scoliosis/kyphosis | 2/6 | 0/4 | 1/7 | 3/3 | + | 7/21 |
| Contractures | 2/6 | 2/4 | 0/4 | 0/3 | + | 5/18 |
| Small hands | 2/6 | 2/4 | 0/5 | 0/3 | + | 5/19 |
| Small feet | 1/6 | 2/4 | 1/6 | 0/3 | + | 5/20 |
| Hip dysplasia | 0/6 | 0/4 | 2/8 | 0/3 | + | 3/22 |
| SENSORY SYSTEM | ||||||
| Eye abnormalities | 6/8 | 4/5 | 4/8 | 1/3 | + | 16/25 |
| Hearing difficulties | 1/7 | 1/4 | 0/8 | 0/3 | + | 3/23 |
| Dysmorphic Features (Human Phenotype Ontology Term) | Fountain et al., 2019 [6] | Capra et al., 2020 [7] | Present Case | Total |
|---|---|---|---|---|
| Low-set eyebrows with respect to the upper eyelid (NA) | (11/14) | NA | + | 12/15(80%) |
| Deeply set eyes (HP:0000490) | 14/14 | NA | + | 15/15 (100%) |
| Prominent nasal septum (HP:0005322) | 12/14 | NA | + | 13/15 (87%) |
| Low hanging septum of nose (HP:0005322) | 14/14 | + | + | 16/16 (100%) |
| Long palpebral fissures (HP:0000637) | 12/14 | + | + | 14/16 (87%) |
| Long eyelashes (HP:0000527) | NA | NA | + | |
| Protruding ears (HP:0000411) | NA | NA | + | |
| Short philtrum (HP:0000322) | 12/14 | - | + | 13/16 81% |
| Prominent philtrum (HP:0002002) | 10/14 | - | + | 11/16 68% |
| Thin upper lip (HP:0000219) | 9/14 | + | + | 11/16 68% |
| Low posterior hairline (HP:0002162) | NA | NA | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priolo, M.; Mancini, C.; Pizzi, S.; Chiriatti, L.; Radio, F.C.; Cordeddu, V.; Pintomalli, L.; Mammì, C.; Dallapiccola, B.; Tartaglia, M. Complex Presentation of Hao-Fountain Syndrome Solved by Exome Sequencing Highlighting Co-Occurring Genomic Variants. Genes 2022, 13, 889. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050889
Priolo M, Mancini C, Pizzi S, Chiriatti L, Radio FC, Cordeddu V, Pintomalli L, Mammì C, Dallapiccola B, Tartaglia M. Complex Presentation of Hao-Fountain Syndrome Solved by Exome Sequencing Highlighting Co-Occurring Genomic Variants. Genes. 2022; 13(5):889. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050889
Chicago/Turabian StylePriolo, Manuela, Cecilia Mancini, Simone Pizzi, Luigi Chiriatti, Francesca Clementina Radio, Viviana Cordeddu, Letizia Pintomalli, Corrado Mammì, Bruno Dallapiccola, and Marco Tartaglia. 2022. "Complex Presentation of Hao-Fountain Syndrome Solved by Exome Sequencing Highlighting Co-Occurring Genomic Variants" Genes 13, no. 5: 889. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050889