
A High-Quality Genome Assembly of Striped Catfish (Pangasianodon hypophthalmus) Based on Highly Accurate Long-Read HiFi Sequencing Data

 , , ,
, , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Library Construction and DNA Sequence
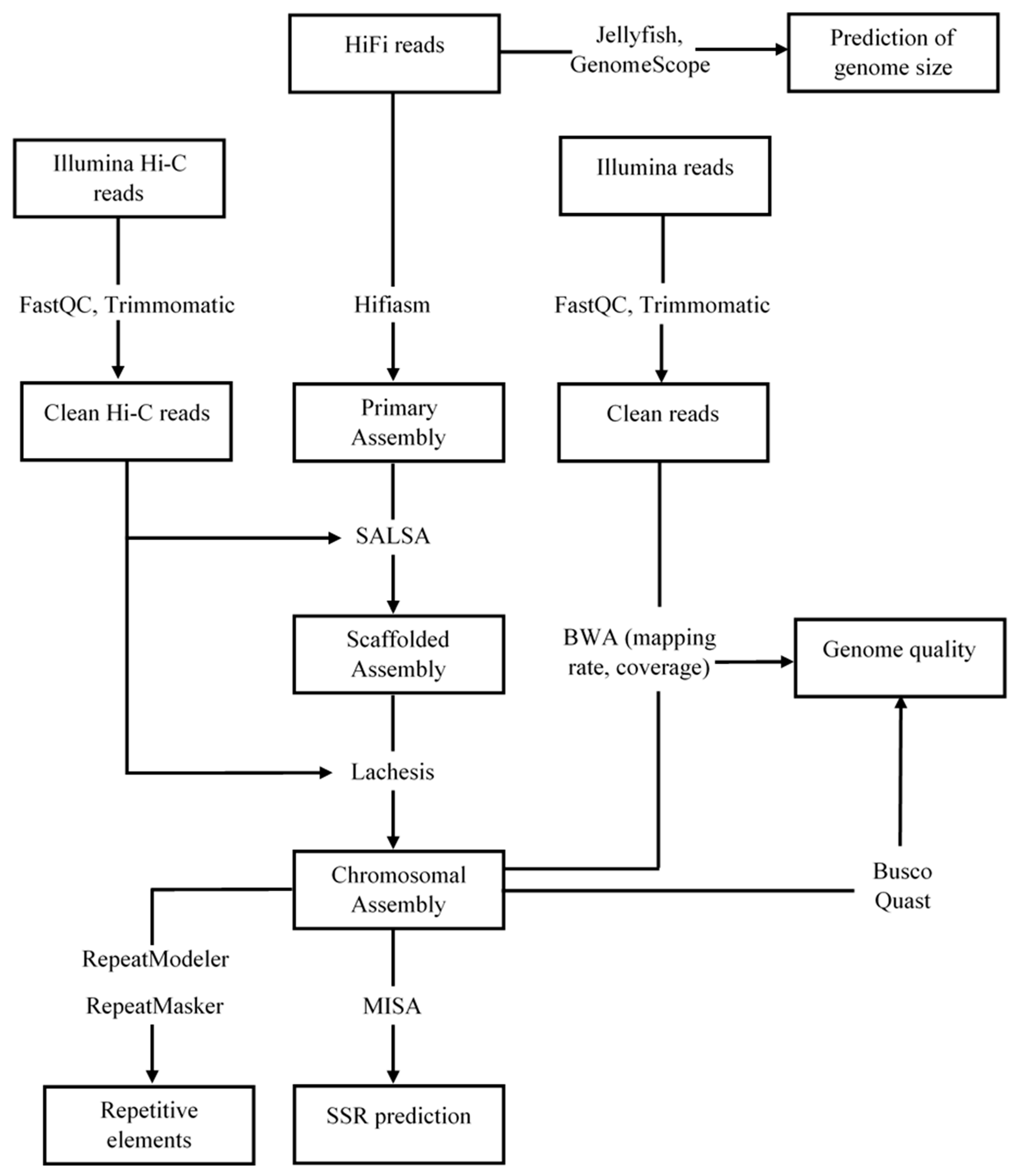
2.3. Sequence Data Processing and Genome Assembly
2.4. Genome Quality Assessment
- BUSCO Actinopterygii Genes—Quality assessment was conducted using BUSCO software version 5.2.2 [56] with default parameters by searching the genome against 3640 single-copy orthologues from actinopterygii_odb10 database (https://busco-data.ezlab.org/v5/data/lineages/) (accessed on 10 November 2021);
- Illumina short DNA reads—To assess the accuracy of our genome assembly, we aligned the Illumina short reads to the assembly using BWA version 0.7.17 [53] to evaluate the mapping and covering rate;
- Comparison to other assemblies—Contig N50 values were calculated for comparison with those of previous studies on striped catfish [18,19] and other Siluriformes species by QUAST version 5.0.2 [57]. In addition, we also mapped our newly assembled genome against the previously published one: Genbank accession no. GCA_003671635.1 [18] and GCA_016801045.1 [19] to identify the gaps using MUMMER version 3.23 [58].
2.5. Identification of Repetitive Elements and Simple Sequence Repeat Markers
3. Results
3.1. DNA Sequencing
3.2. Genome Assembly
3.3. Genome Quality Evaluation
3.4. Repetitive Genome Elements and SSR Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phuong, N.T.; Oanh, D.T.H. Striped Catfish Aquaculture in Vietnam: A Decade of Unprecedented Development. In Success Stories in Asian Aquaculture; Silva, S.S., Davy, F.B., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 131–147. [Google Scholar] [CrossRef]
- Phan, L.T.; Bui, T.M.; Nguyen, T.T.; Gooley, G.J.; Ingram, B.A.; Nguyen, H.V.; Nguyen, P.T.; De Silva, S.S. Current Status of Farming Practices of Striped Catfish, Pangasianodon hypophthalmus in the Mekong Delta, Vietnam. Aquaculture 2009, 296, 227–236. [Google Scholar] [CrossRef]
- Vietnam Association of Seafood Exporters and Producers (VASEP). Available online: http://vasep.com.vn (accessed on 25 February 2022).
- De Silva, S.S.; Phuong, N.T. Striped Catfish Farming in the Mekong Delta, Vietnam: A Tumultuous Path to a Global Success. Rev. Aquac. 2011, 3, 45–73. [Google Scholar] [CrossRef]
- Hai, T.N.; Phuong, N.T.; Van, H.N.; Viet, L.Q. Promoting Coastal Aquaculture for Adaptation to Climate Change and Saltwater Intrusion in the Mekong Delta, Vietnam. World Aquac. 2020, 51, 19–26. [Google Scholar]
- Hoa, T.T.T.; Boerlage, A.S.; Duyen, T.T.M.; Thy, D.T.M.; Hang, N.T.T.; Humphry, R.W.; Phuong, N.T. Nursing Stages of Striped Catfish (Pangasianodon hypophthalmus) in Vietnam: Pathogens, Diseases and Husbandry Practices. Aquaculture 2021, 533, 736114. [Google Scholar] [CrossRef]
- Yue, G.H.; Wang, L. Current Status of Genome Sequencing and Its Applications in Aquaculture. Aquaculture 2017, 468, 337–347. [Google Scholar] [CrossRef]
- Abdelrahman, H.; ElHady, M.; Alcivar-Warren, A.; Allen, S.; Al-Tobasei, R.; Bao, L.; Beck, B.; Blackburn, H.; Bosworth, B.; Buchanan, J.; et al. Aquaculture Genomics, Genetics and Breeding in the United States: Current Status, Challenges, and Priorities for Future Research. BMC Genom. 2017, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liu, S.; Yao, J.; Bao, L.; Zhang, J.; Li, Y.; Jiang, C.; Sun, L.; Wang, R.; Zhang, Y.; et al. The Channel Catfish Genome Sequence Provides Insights into the Evolution of Scale Formation in Teleosts. Nat. Commun. 2016, 7, 757. [Google Scholar] [CrossRef]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic Salmon Genome Provides Insights into Rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Brawand, D.; Wagner, C.E.; Li, Y.I.; Malinsky, M.; Keller, I.; Fan, S.; Simakov, O.; Ng, A.Y.; Lim, Z.W.; Bezault, E.; et al. The Genomic Substrate for Adaptive Radiation in African Cichlid Fish. Nature 2015, 513, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noël, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The Rainbow Trout Genome Provides Novel Insights into Evolution after Whole-Genome Duplication in Vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef] [Green Version]
- Star, B.; Nederbragt, A.J.; Jentoft, S.; Grimholt, U.; Malmstrøm, M.; Gregers, T.F.; Rounge, T.B.; Paulsen, J.; Solbakken, M.H.; Sharma, A.; et al. The Genome Sequence of Atlantic Cod Reveals a Unique Immune System. Nature 2011, 477, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Griot, R.; Allal, F.; Brard-Fudulea, S.; Morvezen, R.; Haffray, P.; Phocas, F.; Vandeputte, M. APIS: An Auto-Adaptive Parentage Inference Software That Tolerates Missing Parents. Mol. Ecol. Resour. 2020, 20, 579–590. [Google Scholar] [CrossRef]
- Pang, M.; Fu, B.; Yu, X.; Liu, H.; Wang, X.; Yin, Z.; Xie, S.; Tong, J. Quantitative Trait Loci Mapping for Feed Conversion Efficiency in Crucian Carp (Carassius auratus). Sci. Rep. 2017, 7, 16971. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.P.; Yáñez, J.M.; Fukui, S.; Swift, B.; Davidson, W.S. Genome-Wide Association Study (GWAS) for Growth Rate and Age at Sexual Maturation in Atlantic Salmon (Salmo Salar). PLoS ONE 2015, 10, e0119730. [Google Scholar] [CrossRef] [Green Version]
- Moen, T.; Torgersen, J.; Santi, N.; Davidson, W.S.; Baranski, M.; Ødegård, J.; Kjøglum, S.; Velle, B.; Kent, M.; Lubieniecki, K.P.; et al. Epithelial Cadherin Determines Resistance to Infectious Pancreatic Necrosis Virus in Atlantic Salmon. Genetics 2015, 200, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.T.P.; Nguyen, P.T.; Shoguchi, E.; Hisata, K.; Vo, T.T.B.; Inoue, J.; Shinzato, C.; Le, B.T.N.; Nishitsuji, K.; Kanda, M.; et al. A Draft Genome of the Striped Catfish, Pangasianodon hypophthalmus, for Comparative Analysis of Genes Relevant to Development and a Resource for Aquaculture Improvement. BMC Genom. 2018, 19, 733. [Google Scholar] [CrossRef]
- Gao, Z.; You, X.; Zhang, X.; Chen, J.; Xu, T.; Huang, Y.; Lin, X.; Xu, J.; Bian, C.; Shi, Q.A. Chromosome-Level Genome Assembly of the Striped Catfish (Pangasianodon hypophthalmus). Genomics 2021, 113, 3349–3356. [Google Scholar] [CrossRef]
- Derakhshani, H.; Bernier, S.P.; Marko, V.A.; Surette, M.G. Completion of Draft Bacterial. Genomes by Long-Read Sequencing of Synthetic Genomic Pools. BMC Genom. 2020, 21, 519. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Shaw, D.E.; White, M.A. Improved Contiguity of the Threespine Stickleback Genome Using Long-Read Sequencing. G3 Genes Genomes Genet. 2021, 11, jkab007. [Google Scholar] [CrossRef]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long Reads: Their Purpose and Place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, J.; Kolmogorov, M.; Shen, M.W.; Chaisson, M.; Pevzner, P.A. Assembly of Long Error-Prone Reads Using de Bruijn Graphs. Proc. Natl. Acad. Sci. USA 2016, 113, E8396–E8405. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA Sequencing at 40: Past, Present and Future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.P.; Wilson, R.K.; Eichler, E.E. Genetic Variation and the de Novo Assembly of Human Genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef]
- Nowak, R.M.; Jastrzębski, J.P.; Kuśmirek, W.; Sałamatin, R.; Rydzanicz, M.; Sobczyk-Kopcioł, A.; Sulima-Celińska, A.; Paukszto, Ł.; Makowczenko, K.G.; Płoski, R.; et al. Hybrid de Novo Whole-Genome Assembly and Annotation of the Model Tapeworm Hymenolepis diminuta. Sci. Data 2019, 6, 302. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Austin, C.M.; Hammer, M.P.; Lee, Y.P.; Croft, L.J.; Gan, H.M. Finding Nemo: Hybrid Assembly with Oxford Nanopore and Illumina Reads Greatly Improves the Clownfish (Amphiprion ocellaris) Genome Assembly. GigaScience 2018, 7, gix137. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A Fast and Efficient Genome Polishing Tool for Long- Read Assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and Its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Vollger, M.R.; Logsdon, G.A.; Audano, P.A.; Sulovari, A.; Porubsky, D.; Peluso, P.; Wenger, A.M.; Concepcion, G.T.; Kronenberg, Z.N.; Munson, K.M.; et al. Improved Assembly and Variant Detection of a Haploid Human Genome Using Single-molecule, High-fidelity Long Reads. Ann. Hum. Genet. 2020, 84, 125–140. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, X.; Qu, S.; Jia, P.; Wang, B.; Gao, S.; Xu, T.; Zhang, W.; Huang, J.; Ye, K. Haplotype-Resolved Chinese Male Genome Assembly Based on High-Fidelity Sequencing. Fundam. Res. 2022. [Google Scholar] [CrossRef]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate Assembly of Segmental Duplications, Satellites, and Allelic Variants from High-Fidelity Long Reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef]
- Di Genova, A.; Buena-Atienza, E.; Ossowski, S.; Sagot, M.-F. Efficient Hybrid de Novo Assembly of Human Genomes with WENGAN. Nat. Biotechnol. 2021, 39, 422–430. [Google Scholar] [CrossRef]
- Gavrielatos, M.; Kyriakidis, K.; Spandidos, D.A.; Michalopoulos, I. Benchmarking of next and Third Generation Sequencing Technologies and Their Associated Algorithms for de Novo Genome Assembly. Mol. Med. Rep. 2021, 23, 251. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Highly-Accurate Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Huddleston, J.; Chaisson, M.J.P.; Steinberg, K.M.; Warren, W.; Hoekzema, K.; Gordon, D.; Graves-Lindsay, T.A.; Munson, K.M.; Kronenberg, Z.N.; Vives, L.; et al. Discovery and Genotyping of Structural Variation from Long-Read Haploid Genome Sequence Data. Genome Res. 2017, 27, 677–685. [Google Scholar] [CrossRef]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing Genomics to Fast-Track Genetic Improvement in Aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef]
- Mehrotra, S.; Goyal, V. Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genom. Proteom. Bioinform. 2014, 12, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Malhis, N.; Jones, S.J.M. High Quality SNP Calling Using Illumina Data at Shallow Coverage. Bioinformatics 2010, 26, 1029–1035. [Google Scholar] [CrossRef] [Green Version]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing Genomic Information for Livestock Improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Zenger, K.R.; Khatkar, M.S.; Jones, D.B.; Khalilisamani, N.; Jerry, D.R.; Raadsma, H.W. Genomic Selection in Aquaculture: Application, Limitations and Opportunities with Special Reference to Marine Shrimp and Pearl Oysters. Front. Genet. 2019, 10, 693. [Google Scholar] [CrossRef]
- Benevenuto, J.; Ferrão, L.F.V.; Amadeu, R.R.; Munoz, P. How Can a High-Quality Genome Assembly Help Plant Breeders? GigaScience 2019, 8, giz068. [Google Scholar] [CrossRef]
- Wen, M.; Pan, Q.; Jouanno, E.; Montfort, J.; Zahm, M.; Cabau, C.; Klopp, C.; Iampietro, C.; Roques, C.; Bouchez, O.; et al. An Ancient Truncated Duplication of the Anti-Mullerian Hormone Receptor Type 2 Gene Is a Potential Conserved Master Sex Determinant in the Pangasiidae Catfish Family. Mol. Ecol. Resour. 2022, 1–18. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics-FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 August 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Ghurye, J.; Rhie, A.; Walenz, B.P.; Schmitt, A.; Selvaraj, S.; Pop, M.; Phillippy, A.M.; Koren, S. Integrating Hi-C Links with Assembly Graphs for Chromosome-Scale Assembly. PLoS Comput. Biol. 2019, 15, e1007273. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-Scale Scaffolding of de Novo Genome Assemblies Based on Chromatin Interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4–10. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum Vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Tang, B.; Wang, Z.; Liu, Q.; Wang, Z.; Ren, Y.; Guo, H.; Qi, T.; Li, Y.; Zhang, H.; Jiang, S.; et al. Chromosome-Level Genome Assembly of Paralithodes Platypus Provides Insights into Evolution and Adaptation of King Crabs. Mol. Ecol. Resour. 2021, 21, 511–525. [Google Scholar] [CrossRef]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling Large Genomes with Single-Molecule Sequencing and Locality-Sensitive Hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef]
- Molina-Mora, J.A.; Campos-Sánchez, R.; Rodríguez, C.; Shi, L.; García, F. High Quality 3C de Novo Assembly and Annotation of a Multidrug Resistant ST-111 Pseudomonas Aeruginosa Genome: Benchmark of Hybrid and Non-Hybrid Assemblers. Sci. Rep. 2020, 10, 1392. [Google Scholar] [CrossRef]
- Ellis, E.A.; Storer, C.G.; Kawahara, A.Y. De Novo Genome Assemblies of Butterflies. GigaScience 2021, 10, giab041. [Google Scholar] [CrossRef]
- Sreeputhorn, K.; Mangumphan, K.; Muanphet, B.; Tanomtong, A.; Supiwong, W.; Kaewmad, P. The First Report on Chromosome Analysis of F1 Hybrid Catfish: Mekong Giant Catfish (Pangasianodon gigas) × Striped Catfish (Pangasianodon hypophthalmus) and Spot Pangasius (Pangasius larnaudii) × Pangasianodon Hypophthalmus (Siluriformes, pangasiidae). Cytologia 2017, 82, 457–463. [Google Scholar] [CrossRef]
- Sharma, P.; Al-Dossary, O.; Alsubaie, B.; Al-Mssallem, I.; Nath, O.; Mitter, N.; Margarido, G.R.A.; Topp, B.; Murigneux, V.; Kharabian Masouleh, A.; et al. Improvements in the Sequencing and Assembly of Plant Genomes. Gigabyte 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Chin, C.-S.; Khalak, A. Human Genome Assembly in 100 Minutes. bioRxiv 2019, 705616. [Google Scholar] [CrossRef]
- Driguez, P.; Bougouffa, S.; Carty, K.; Putra, A.; Jabbari, K.; Reddy, M.; Soppe, R.; Cheung, M.S.; Fukasawa, Y.; Ermini, L. LeafGo: Leaf to Genome, a Quick Workflow to Produce High-Quality de Novo Plant Genomes Using Long-Read Sequencing Technology. Genome Biol. 2021, 22, 256. [Google Scholar] [CrossRef]
- Zimin, A.V.; Salzberg, S.L. The SAMBA Tool Uses Long Reads to Improve the Contiguity of Genome Assemblies. PLoS Comput. Biol. 2022, 18, e1009860. [Google Scholar] [CrossRef]
- Austin, C.M.; Tan, M.H.; Harrisson, K.A.; Lee, Y.P.; Croft, L.J.; Sunnucks, P.; Pavlova, A.; Gan, H.M. De Novo Genome Assembly and Annotation of Australia’s Largest Freshwater Fish, the Murray Cod (Maccullochella peelii), from Illumina and Nanopore Sequencing Read. GigaScience 2017, 6, gix063. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Magadan, S.; Waldbieser, G.C.; Youngblood, R.C.; Wheeler, P.A.; Scheffler, B.E.; Thorgaard, G.H.; Palti, Y. A Long Reads-Based de-Novo Assembly of the Genome of the Arlee Homozygous Line Reveals Chromosomal Rearrangements in Rainbow Trout. G3 Genes Genomes Genet. 2021, 11, jkab052. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Libraries | Insert Size (bp) | Total Data (Gb) | Read Length (bp) | Sequence Coverage (×) * |
|---|---|---|---|---|
| Illumina reads | 350 | 51.2 | 150 | 71.8 |
| Illumina reads | 550 | 49.5 | 150 | 69.3 |
| PacBio (HiFi) reads | 16,400 | 17.28 | 14,791 | 24.2 |
| Total | 118.0 | 165.3 |
| Category | Contig | Scaffold | ||
|---|---|---|---|---|
| Length (bp) | Number | Length (bp) | Number | |
| Total | 788,121,403 | 845 | 788,355,903 | 381 |
| Largest | 30,145,618 | NA | 35,439,358 | NA |
| N50 | 14,675,983 | 20 | 21,837,136 | 15 |
| N60 | 11,176,154 | 26 | 19,579,817 | 19 |
| N70 | 6,525,798 | 35 | 17,728,427 | 24 |
| N80 | 2,330,994 | 55 | 14,533,765 | 28 |
| N90 | 459,384 | 139 | 1,263,789 | 54 |
| Chr ID | Length (bp) | Chr ID | Length (bp) | Chr ID | Length (bp) |
|---|---|---|---|---|---|
| Chr1 | 57,614,776 | Chr11 | 30,308,490 | Chr21 | 20,241,537 |
| Chr2 | 57,569,486 | Chr12 | 30,160,703 | Chr22 | 20,107,636 |
| Chr3 | 47,895,633 | Chr13 | 27,700,161 | Chr23 | 19,020,310 |
| Chr4 | 40,963,295 | Chr14 | 26,554,853 | Chr24 | 18,006,869 |
| Chr5 | 37,055,990 | Chr15 | 26,362,405 | Chr25 | 16,472,023 |
| Chr6 | 33,881,059 | Chr16 | 25,536,520 | Chr26 | 12,481,112 |
| Chr7 | 33,777,686 | Chr17 | 25,407,408 | Chr27 | 6,262,317 |
| Chr8 | 33,526,538 | Chr18 | 22,657,484 | Chr28 | 2,381,093 |
| Chr9 | 32,744,715 | Chr19 | 22,587,380 | Chr29 | 1,580,235 |
| Chr10 | 32,245,061 | Chr20 | 20,661,286 | Chr30 | 1,373,485 |
| Total chromosome-level length | 783,137,546 | ||||
| Total length | 788,355,903 | ||||
| Chromosome length/total length | 99.3% | ||||
| Index | Number |
|---|---|
| Complete BUSCOs (C) | 3497 |
| Complete and single-copy BUSCOs (S) | 3405 |
| Complete and duplicated BUSCOs (D) | 92 |
| Fragmented BUSCOs (F) | 35 |
| Missing BUSCOs (M) | 108 |
| Total BUSCO groups searched (n) | 3640 |
| C: 96.0% [S: 93.5%, D: 2.5%], F: 1.0%, M: 3.0%, n: 3640 | |
| Species | Genome Size (Mb) | Number of Contig | Number |
|---|---|---|---|
| Pangasianodon hypophthalmus (from the present study) | 788.4 | 845 | 14.7 |
| Clarias macrocephalus (GCA_011419295.1) | 883.3 | 44,869 | 0.05 |
| Clarias batrachus (GCA_003987875.1) | 821.7 | 78,047 | 0.02 |
| Ictalurus punctatus (GCA_004006655.2) | 1002.3 | 5816 | 2.7 |
| Ageneiosus marmoratus (GCA_003347165.1) | 1030 | 169,048 | 0.007 |
| Ompok bimaculatus (GCA_009108245.1) | 718.1 | 27,068 | 0.08 |
| Bagarius yarrelli (GCA_005784505.1) | 570.8 | 928 | 1.9 |
| Tachysurus fulvidraco (GCA_003724035.1) | 713.8 | 2402 | 1.0 |
| Genomic Feature | This Study | Gao et al. [19] | Kim et al. [18] |
|---|---|---|---|
| The size of genome (Mb) | 788.4 | 742.6 | 715.7 |
| Number of contigs | 845 | 821 | 23,340 |
| Contig N50 (Mb) | 14.7 | 3.5 | 0.06 |
| Longest contig (Mb) | 30.1 | 16.1 | 0.5 |
| GC content (%) | 38.9 | 38.9 | 38.7 |
| Repetitive regions (%) | 39.1 | 36.9 | 33.8 |
| Complete BUSCOs (C) (%) | 96.0 | 93.3 | 92.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, D.M.; Yen, D.T.; Liem, P.T.; Tam, B.M.; Huong, D.T.T.; Hang, B.T.B.; Hieu, D.Q.; Garigliany, M.-M.; Coppieters, W.; Kestemont, P.; et al. A High-Quality Genome Assembly of Striped Catfish (Pangasianodon hypophthalmus) Based on Highly Accurate Long-Read HiFi Sequencing Data. Genes 2022, 13, 923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050923
Hai DM, Yen DT, Liem PT, Tam BM, Huong DTT, Hang BTB, Hieu DQ, Garigliany M-M, Coppieters W, Kestemont P, et al. A High-Quality Genome Assembly of Striped Catfish (Pangasianodon hypophthalmus) Based on Highly Accurate Long-Read HiFi Sequencing Data. Genes. 2022; 13(5):923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050923
Chicago/Turabian StyleHai, Dao Minh, Duong Thuy Yen, Pham Thanh Liem, Bui Minh Tam, Do Thi Thanh Huong, Bui Thi Bich Hang, Dang Quang Hieu, Mutien-Marie Garigliany, Wouter Coppieters, Patrick Kestemont, and et al. 2022. "A High-Quality Genome Assembly of Striped Catfish (Pangasianodon hypophthalmus) Based on Highly Accurate Long-Read HiFi Sequencing Data" Genes 13, no. 5: 923. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050923