
Clinical Phenotypes of CDHR1-Associated Retinal Dystrophies

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Identification
2.2. Molecular Diagnosis
2.3. Clinical Assessment
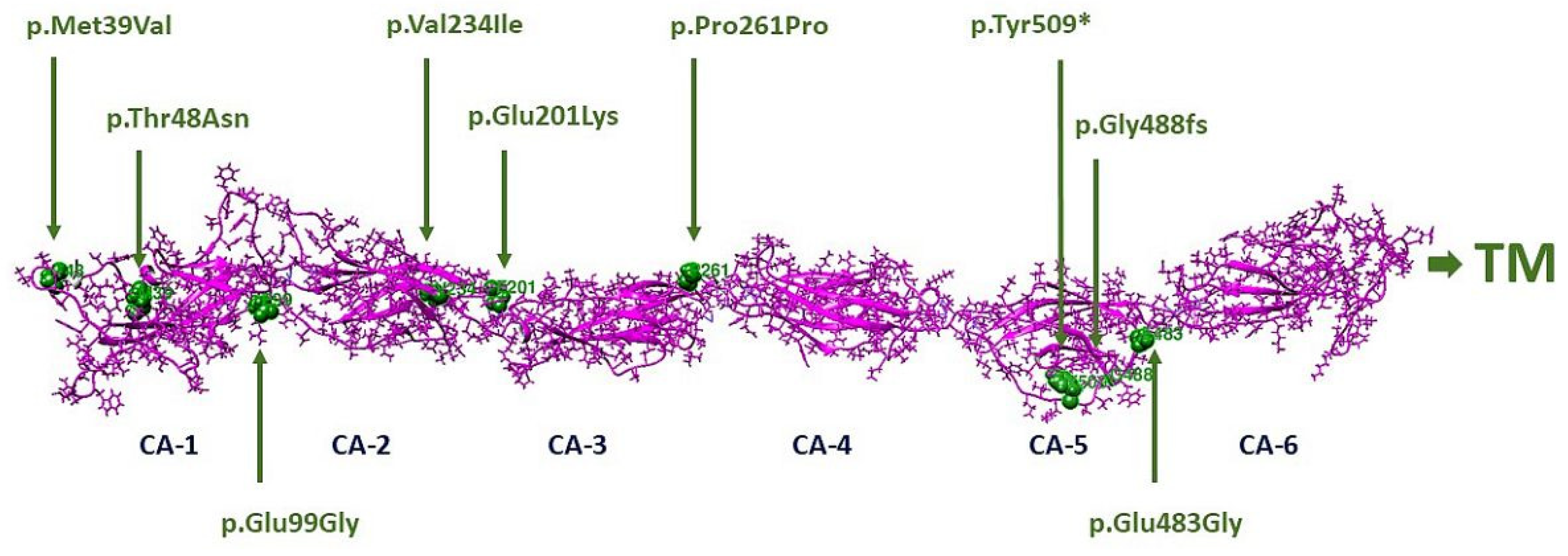
2.4. Molecular Modeling
3. Results
3.1. Patient Characteristics
3.2. Genetic Analysis/Molecular Findings
3.3. Clinical Subgroups and Age at First Manifestation
3.4. Clinical Findings
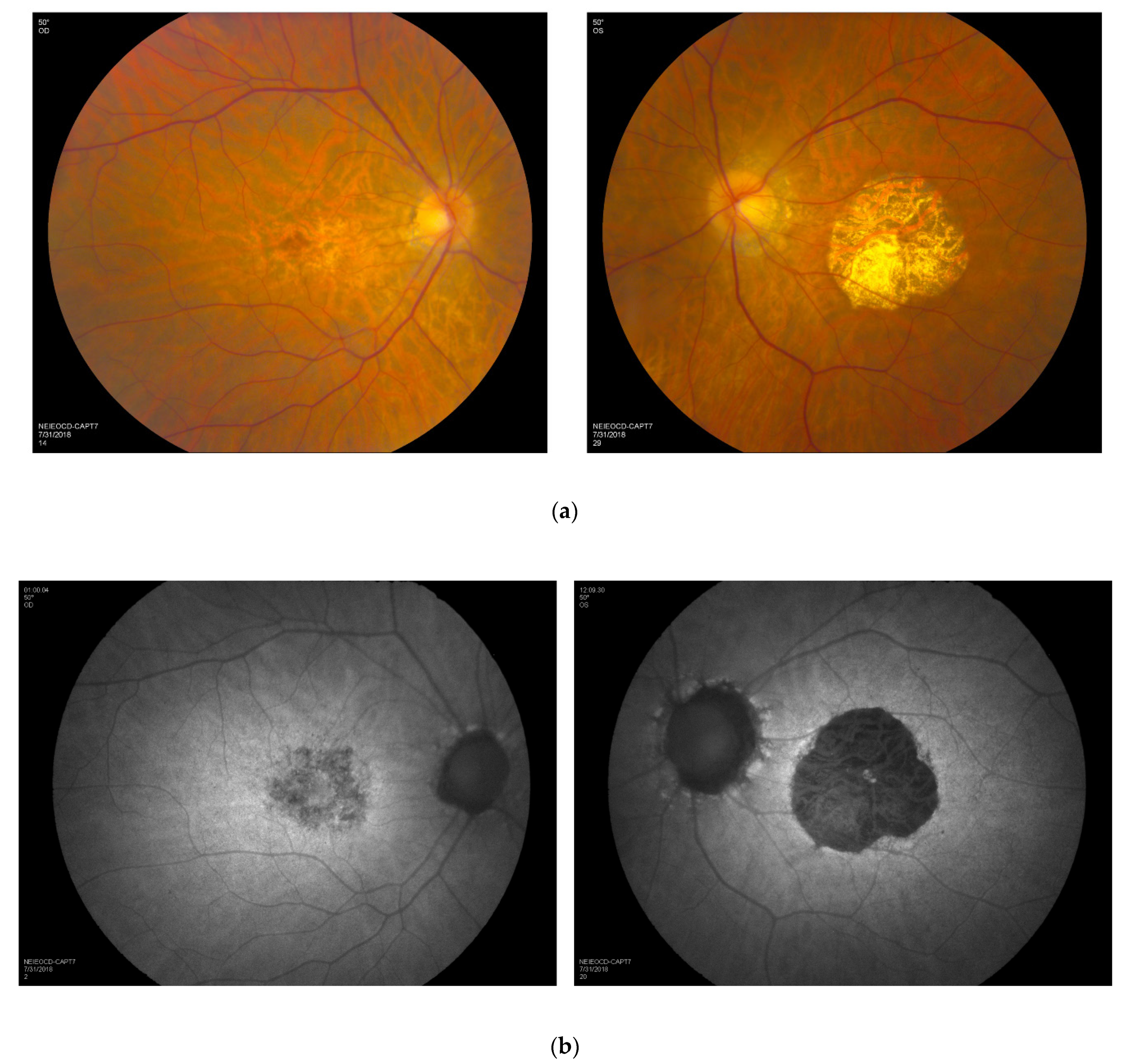
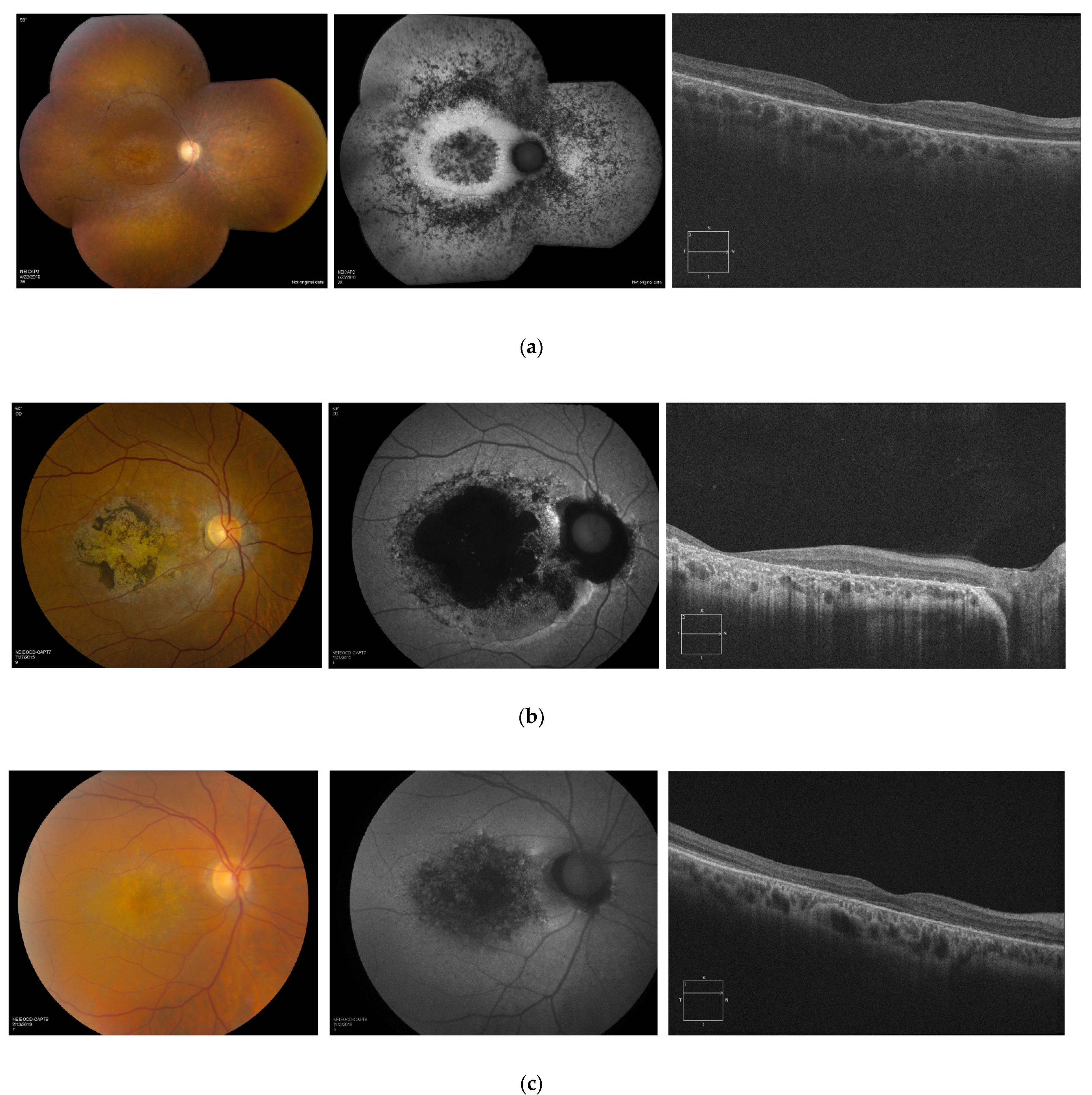
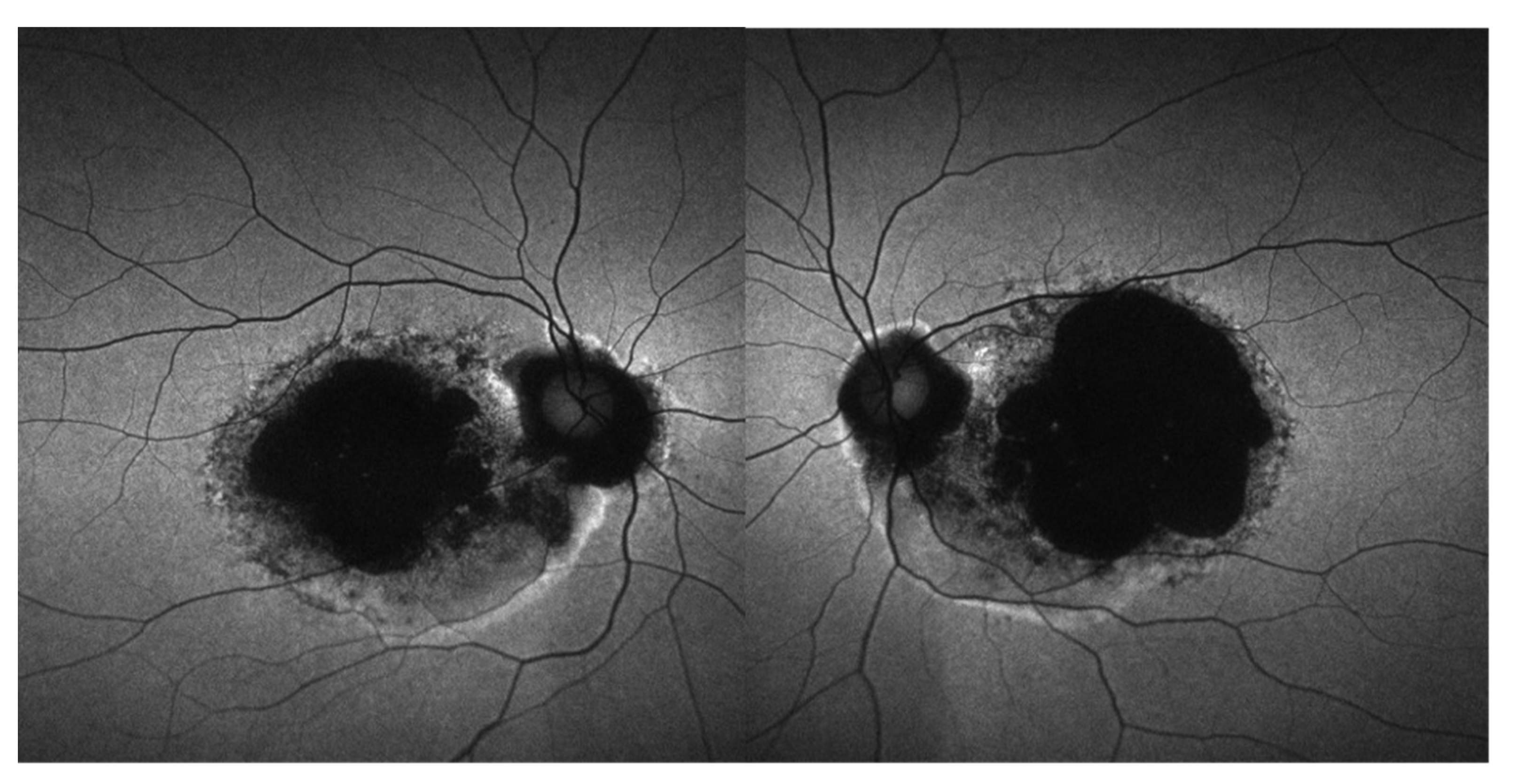
3.5. Retinal Imaging
3.6. Longitudinal Data
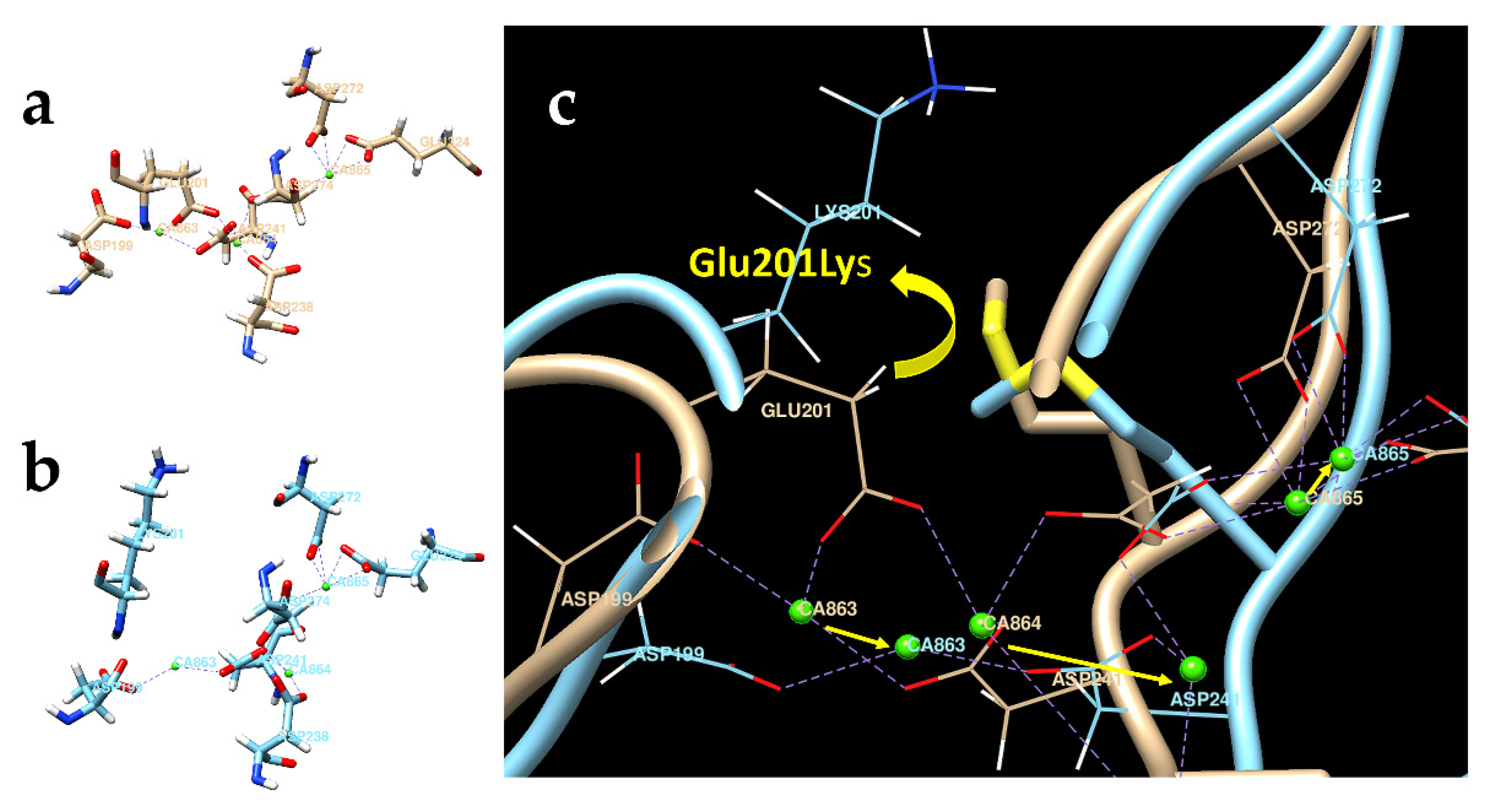
3.7. Molecular Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crewe, J.M.; Morlet, N.; Morgan, W.H.; Spilsbury, K.; Mukhtar, A.S.; Clark, A.; Semmens, J.B. Mortality and hospital morbidity of working-age blind. Br. J. Ophthalmol. 2013, 97, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Heath Jeffery, R.C.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Smallwood, P.M.; Williams, J.; Cooke, C.; Savchenko, A.; Lyubarsky, A.; Pugh, E.N.; Nathans, J. A Photoreceptor-Specific Cadherin Is Essential for the Structural Integrity of the Outer Segment and for Photoreceptor Survival. Neuron 2001, 32, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Yagi, T.; Takeichi, M. Cadherin superfamily genes: Functions, genomic organization, and neurologic diversity. Genes Dev. 2000, 14, 1169–1180. [Google Scholar] [CrossRef]
- Bolz, H.; Ebermann, I.; Gal, A. Protocadherin-21 (PCDH21), a candidate gene for human retinal dystrophies. Mol. Vis. 2005, 11, 929–933. [Google Scholar]
- Henderson, R.H.; Li, Z.; El Aziz, M.M.A.; Mackay, D.S.; Eljinini, M.A.; Zeidan, M.; Moore, A.T.; Bhattacharya, S.S.; Webster, A.R. Biallelic mutation of Protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol. Vis. 2010, 16, 46–52. [Google Scholar]
- Yusuf, I.H.; McClements, M.E.; MacLaren, R.E.; Issa, P.C. Deep phenotyping of the Cdhr1(−/−) mouse validates its use in pre-clinical studies for human CDHR1-associated retinal degeneration. Exp. Eye Res. 2021, 208, 108603. [Google Scholar] [CrossRef]
- Ostergaard, E.; Batbayli, M.; Duno, M.; Vilhelmsen, K.; Rosenberg, T. Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. J. Med. Genet. 2010, 47, 665–669. [Google Scholar] [CrossRef]
- Bessette, A.P.; DeBenedictis, M.; Traboulsi, E.I. Clinical characteristics of recessive retinal degeneration due to mutations in the CDHR1 gene and a review of the literature. Ophthalmic Genet. 2017, 39, 51–55. [Google Scholar] [CrossRef]
- Stingl, K.; Mayer, A.K.; Llavona, P.; Mulahasanovic, L.; Rudolph, G.; Jacobson, S.G.; Zrenner, E.; Kohl, S.; Wissinger, B.; Weisschuh, N. CDHR1 mutations in retinal dystrophies. Sci. Rep. 2017, 7, 6992. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, I.H.; MacLaren, R.E.; Issa, P.C. CDHR1-related late-onset macular dystrophy: Further insights. Eye 2020, 35, 2901–2902. [Google Scholar] [CrossRef] [PubMed]
- Ba-Abbad, R.; Robson, A.G.; Mahroo, O.A.; Wright, G.; Schiff, E.; Duignan, E.S.; Michaelides, M.; Arno, G.; Webster, A.R. A clinical study of patients with novel CDHR1 genotypes associated with late-onset macular dystrophy. Eye 2020, 35, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Holladay, J.T. Visual acuity measurements. J. Cataract. Refract. Surg. 2004, 30, 287–290. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2014, 130, 1–12. [Google Scholar] [CrossRef]
- Issa, P.C.; Gliem, M.; Yusuf, I.H.; Birtel, J.; Müller, P.; Mangold, E.; Downes, S.M.; MacLaren, R.E.; Betz, C.; Bolz, H.J. A Specific Macula-Predominant Retinal Phenotype Is Associated with the CDHR1 Variant c.783G>A, a Silent Mutation Leading to In-Frame Exon Skipping. Investig. Opthalmol. Vis. Sci. 2019, 60, 3388–3397. [Google Scholar] [CrossRef] [Green Version]
- Ba-Abbad, R.; Sergouniotis, P.I.; Plagnol, V.; Robson, A.G.; Michaelides, M.; Holder, G.E.; Webster, A.R. Clinical characteristics of early retinal disease due to CDHR1 mutation. Mol. Vis. 2013, 19, 2250–2259. [Google Scholar]
- Cohen, B.; Chervinsky, E.; Jabaly-Habib, H.; Shalev, S.A.; Briscoe, D.; Ben-Yosef, T. A novel splice site mutation of CDHR1 in a consanguineous Israeli Christian Arab family segregating autosomal recessive cone-rod dystrophy. Mol. Vis. 2012, 18, 2915–2921. [Google Scholar]
- Nikopoulos, K.; Avila-Fernandez, A.; Corton, M.; Lopez-Molina, M.I.; Perez-Carro, R.; Bontadelli, L.; Di Gioia, S.A.; Zurita, O.; Garcia-Sandoval, B.; Rivolta, C.; et al. Identification of two novel mutations in CDHR1 in consanguineous Spanish families with autosomal recessive retinal dystrophy. Sci. Rep. 2015, 5, 13902. [Google Scholar] [CrossRef] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Roorda, A.; Navani, M.; Vishweswaraiah, S.; Syed, R.; Soudry, S.Z.; Ratnam, K.; Gudiseva, H.V.; Lee, P.; Gaasterland, T.; et al. Identification of a Novel Mutation in the CDHR1 Gene in a Family With Recessive Retinal Degeneration. Arch. Ophthalmol. 2012, 130, 1301–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collison, F.T.; Fishman, G.A.; Nagasaki, T.; Zernant, J.; McAnany, J.J.; Park, J.C.; Allikmets, R. Characteristic Ocular Features in Cases of Autosomal Recessive PROM1 Cone-Rod Dystrophy. Investig. Opthalmol. Vis. Sci. 2019, 60, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Arno, G.; Hull, S.; Carss, K.; Dev-Borman, A.; Chakarova, C.; Bujakowska, K.; Born, L.I.V.D.; Robson, A.G.; Holder, G.E.; Michaelides, M.; et al. Reevaluation of the Retinal Dystrophy Due to Recessive Alleles of RGR With the Discovery of a Cis-Acting Mutation in CDHR1. Investig. Opthalmol. Vis. Sci. 2016, 57, 4806–4813. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.; Scheetz, T.; Sheffield, V.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Fu, J.; Ma, L.; Cheng, J.; Yang, L.; Wei, C.; Fu, S.; Lv, H.; Chen, R.; Fu, J. A novel, homozygous nonsense variant of the CDHR1 gene in a Chinese family causes autosomal recessive retinal dystrophy by NGS-based genetic diagnosis. J. Cell. Mol. Med. 2018, 22, 5662–5669. [Google Scholar] [CrossRef]
- Gan, L.; Yang, C.; Shu, Y.; Liu, F.; Sun, R.; Deng, B.; Xu, J.; Huang, G.; Qu, C.; Gong, B.; et al. Identification of a novel homozygous nonsense mutation in the CDHR1 gene in a Chinese family with autosomal recessive retinitis pigmentosa. Clin. Chim. Acta 2020, 507, 17–22. [Google Scholar] [CrossRef]
- Haque, M.N.; Kurata, K.; Hosono, K.; Ohtsubo, M.; Ohishi, K.; Sato, M.; Minoshima, S.; Hotta, Y. A Japanese family with cone-rod dystrophy of delayed onset caused by a compound heterozygous combination of novel CDHR1 frameshift and known missense variants. Hum. Genome Var. 2019, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, A.; Lemke, J.; Altmueller, J.; Thiele, H.; Glaus, E.; Fleischhauer, J.; Nürnberg, P.; Neidhardt, J.; Berger, W. Identification of Novel and Recurrent Disease-Causing Mutations in Retinal Dystrophies Using Whole Exome Sequencing (WES): Benefits and Limitations. PLoS ONE 2016, 11, e0158692. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDHR1 Variant | Protein Change | ACMG Classification |
|---|---|---|
| c.115A > G | p.Met39Val | VUS (PM2) |
| c.143C > A | p.Thr48Asn | VUS (PM2) |
| c.525 + 1G > A | Abnormal splicing | Pathogenic (PVS1, PM2, PP5) |
| c.601G > A | p.Glu201Lys | VUS (PS4-moderate, PM2, PP3) |
| c.700G > A | p.Val234Ile | VUS (PM2, BP4) |
| c.783G > A | p.Pro261= | Pathogenic (PS4, PS3, PM3, PP3) |
| c.1463delG | p.Gly488fs | Pathogenic (PVS1, PS4-moderate, PM2) |
| c.1485 + 2T > C | Abnormal splicing | Pathogenic (PVS1, PS4-moderate, PS3, PM3) |
| c.1527T > G | p.Tyr509* | Pathogenic (PVS1, PS4-moderate, PM2) |
| c.2522_2528delTCTCTGA | p.Ile841Serfs*119 | Pathogenic (PVS1, PS4-moderate, PM2) |
| 7.38 Mb del including CDHR1 | - | Pathogenic (PVS1, PM2, PM3) |
| Phenotype | Ethnicity/Race | Gender | Age at First Symptoms (years) | Age at First NEI Visit (years) | BCVA OD | BCVA OS | GVF OD | GVF OS | D-15 OD (Number of Errors) | D-15 OS (Number of Errors) | Scotopic 0 dB b-Wave (µV) | Photopic 30 Hz Flicker (µV) | CDHR1 Variant 1 | CDHR1 Variant 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCD | Caucasian | F | 28 | 33 | 20/50 | 20/63 | severely constricted | severely constricted | multiple | multiple | 0 | 0 | c.783G > A | 7.38 Mb del including CDHR1 |
| RCD | Greek | F | ~26 | 31 | 20/50 | 20/50 | constricted | constricted | unable to distinguish colors | unable to distinguish colors | 0 | 12 | c.783G > A | c.2522_2528delTCTCTGA |
| RCD | Bangladeshi | M | ~5 | 26 | 20/32 | 20/25 | central scotoma | central scotoma | multiple | multiple | 0 | 5 | c.1463delG | c.1463delG |
| CRD | Chinese | M | 35 | 49 | CF < 8″ | CF < 8″ | constricted | constricted | multiple | multiple | 162 | 31 | c.601G > A | c.700G > A |
| CRD | Romanian | M | ~15 | 49 | 20/20 | 20/500 | central scotoma | central scotoma | two | multiple | 256 | 37 | c.783G > A | c.1527T > G |
| CRD | Puerto Rican | F | ~30 | 49 | 20/63 | 20/63 | central scotoma | central scotoma | unable to distinguish colors | unable to distinguish colors | 278 | 11.5 | c.1485+2T > C | c.525+1G > A |
| CRD | African American | M | ~10 | 45 | 20/200 | 20/200 | central scotoma | central scotoma | three | no errors | 236 | 44.5 | c.143C > A | c.143C > A |
| CRD | African American | M | 45 | 54 | 20/400+2 | 20/400+3 | central scotoma (HVF) | central scotoma (HVF) | two | multiple | 247 | 22 | c.143C > A | c.143C > A |
| CRD | Greek | M | ~45 | 63 | 20/16 | 20/63 | central scotoma | central scotoma | multiple | multiple | 251 | 70.5 | c.115A > G | c.2522_2528delTCTCTGA |
| LOMD | Greek | F | ~40 | 53 | 20/100 | 20/80 | central scotoma | central scotoma | multiple | multiple | 363 | 70 | c.783G > A | c.2522_2528delTCTCTGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malechka, V.V.; Cukras, C.A.; Chew, E.Y.; Sergeev, Y.V.; Blain, D.; Jeffrey, B.G.; Ullah, E.; Hufnagel, R.B.; Brooks, B.P.; Huryn, L.A.; et al. Clinical Phenotypes of CDHR1-Associated Retinal Dystrophies. Genes 2022, 13, 925. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050925
Malechka VV, Cukras CA, Chew EY, Sergeev YV, Blain D, Jeffrey BG, Ullah E, Hufnagel RB, Brooks BP, Huryn LA, et al. Clinical Phenotypes of CDHR1-Associated Retinal Dystrophies. Genes. 2022; 13(5):925. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050925
Chicago/Turabian StyleMalechka, Volha V., Catherine A. Cukras, Emily Y. Chew, Yuri V. Sergeev, Delphine Blain, Brett G. Jeffrey, Ehsan Ullah, Robert B. Hufnagel, Brian P. Brooks, Laryssa A. Huryn, and et al. 2022. "Clinical Phenotypes of CDHR1-Associated Retinal Dystrophies" Genes 13, no. 5: 925. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050925