From Genotype to Functional Phenotype: Unraveling the Metabolomic Features of Colorectal Cancer


Abstract
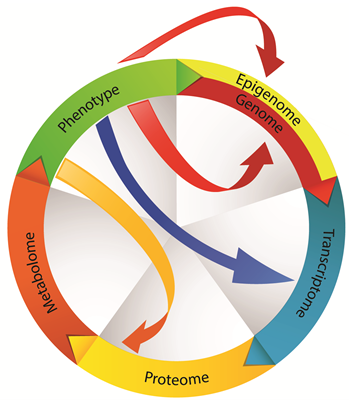
:
1. Introduction
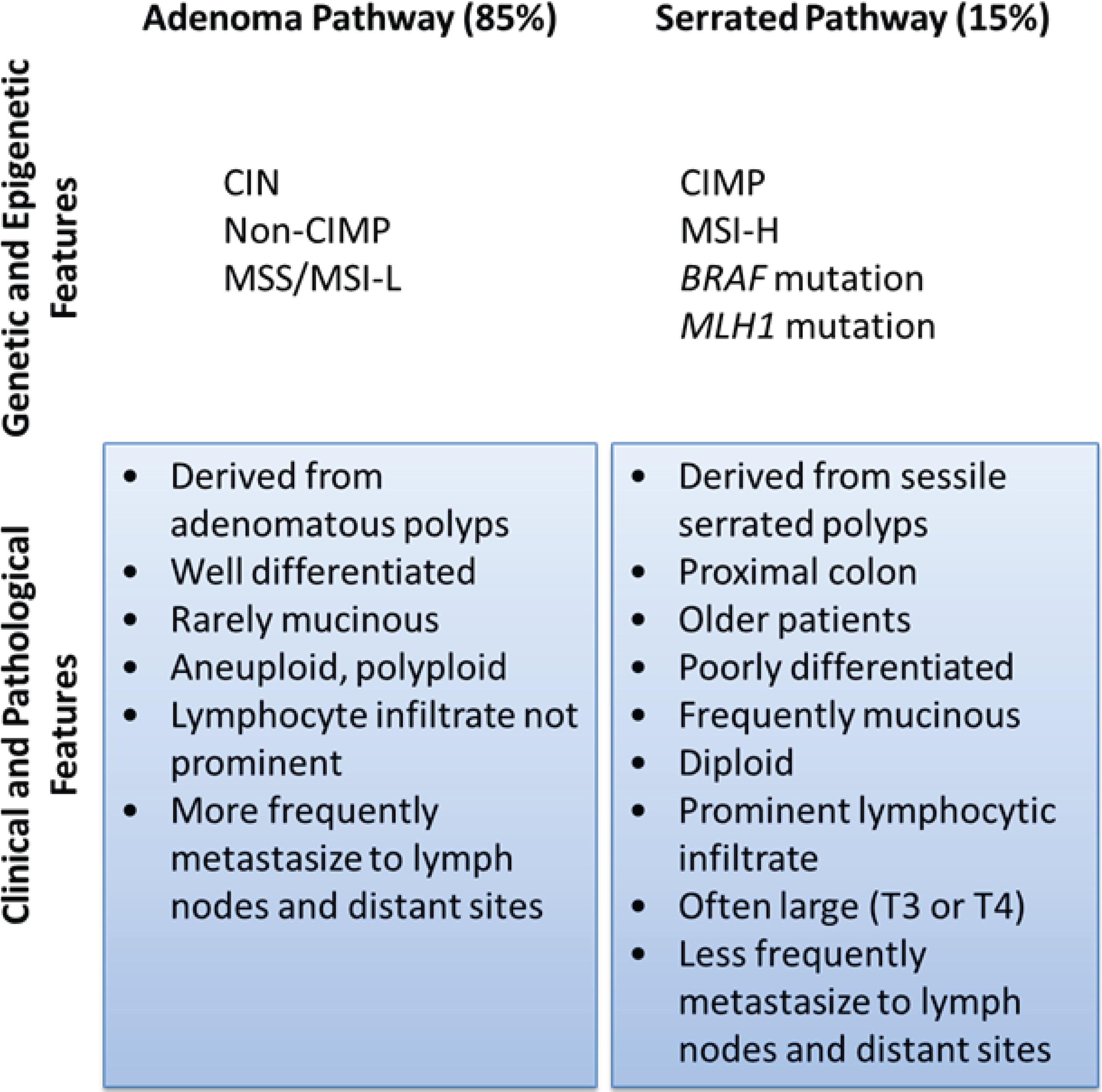
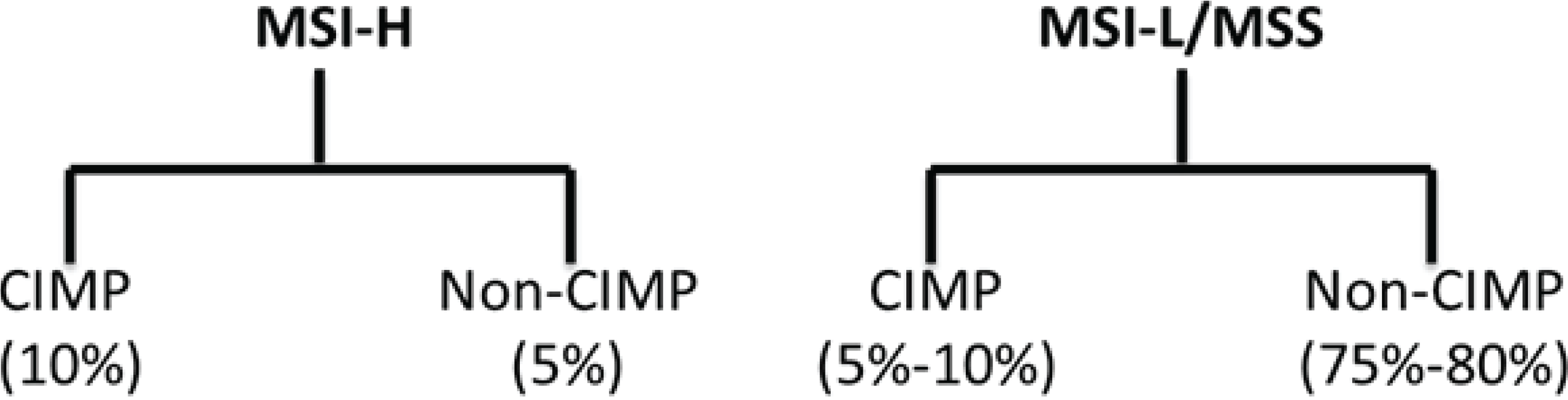
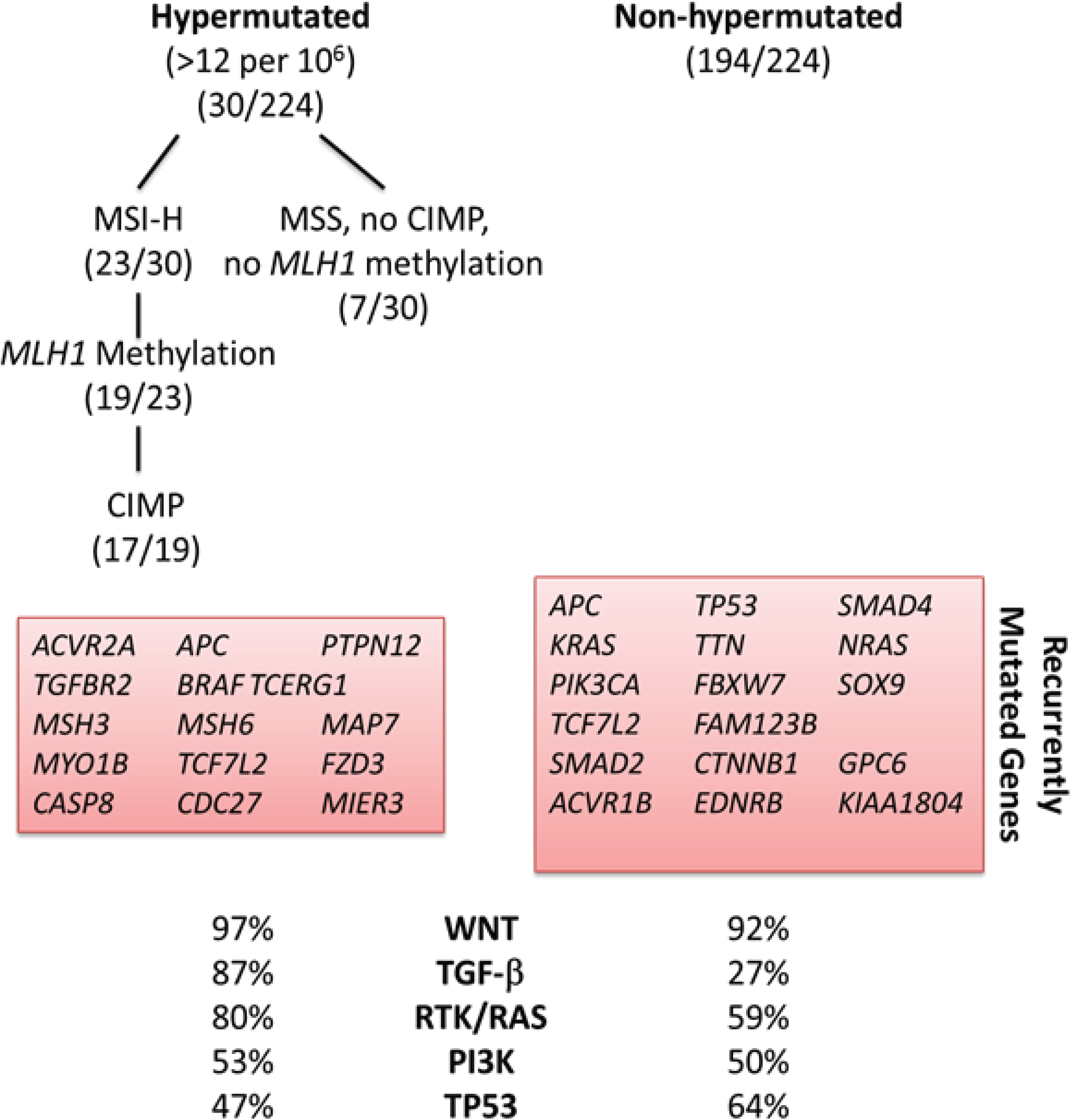
2. Molecular Subclassification of CRC: From Genomics to Phenotype



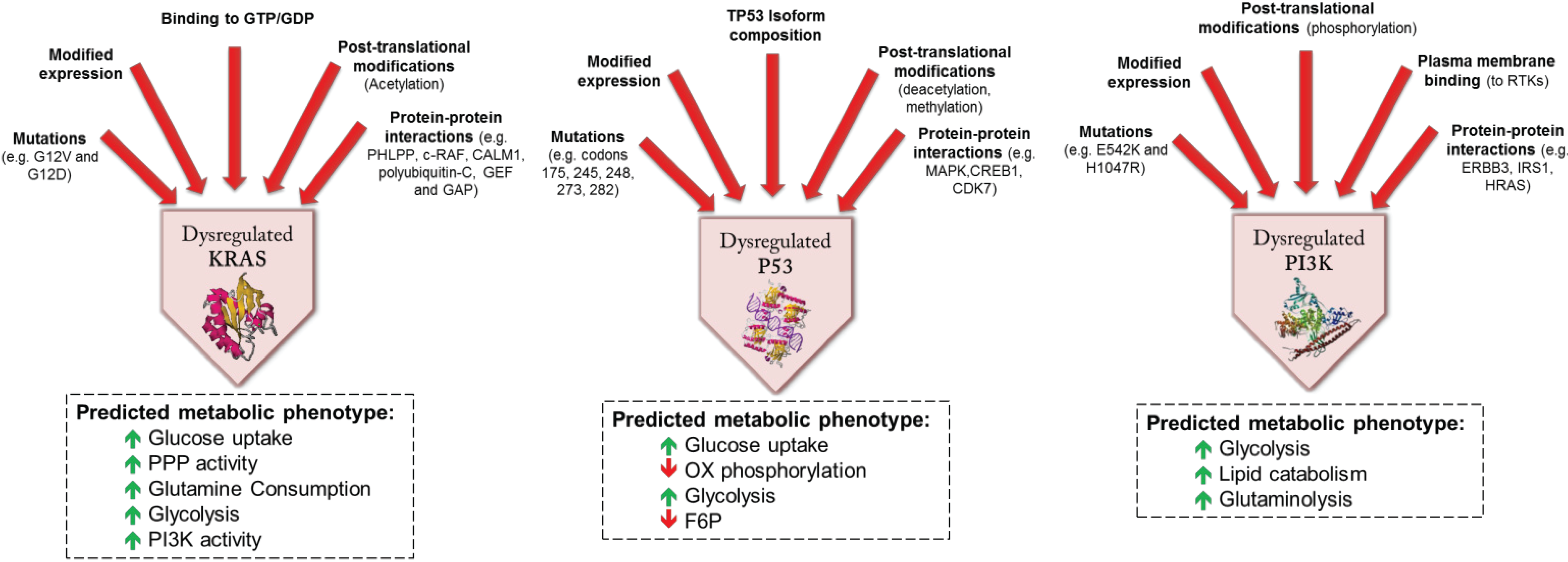
3. Functional Genomics: Defining the Biological Impact of the CRC Genome

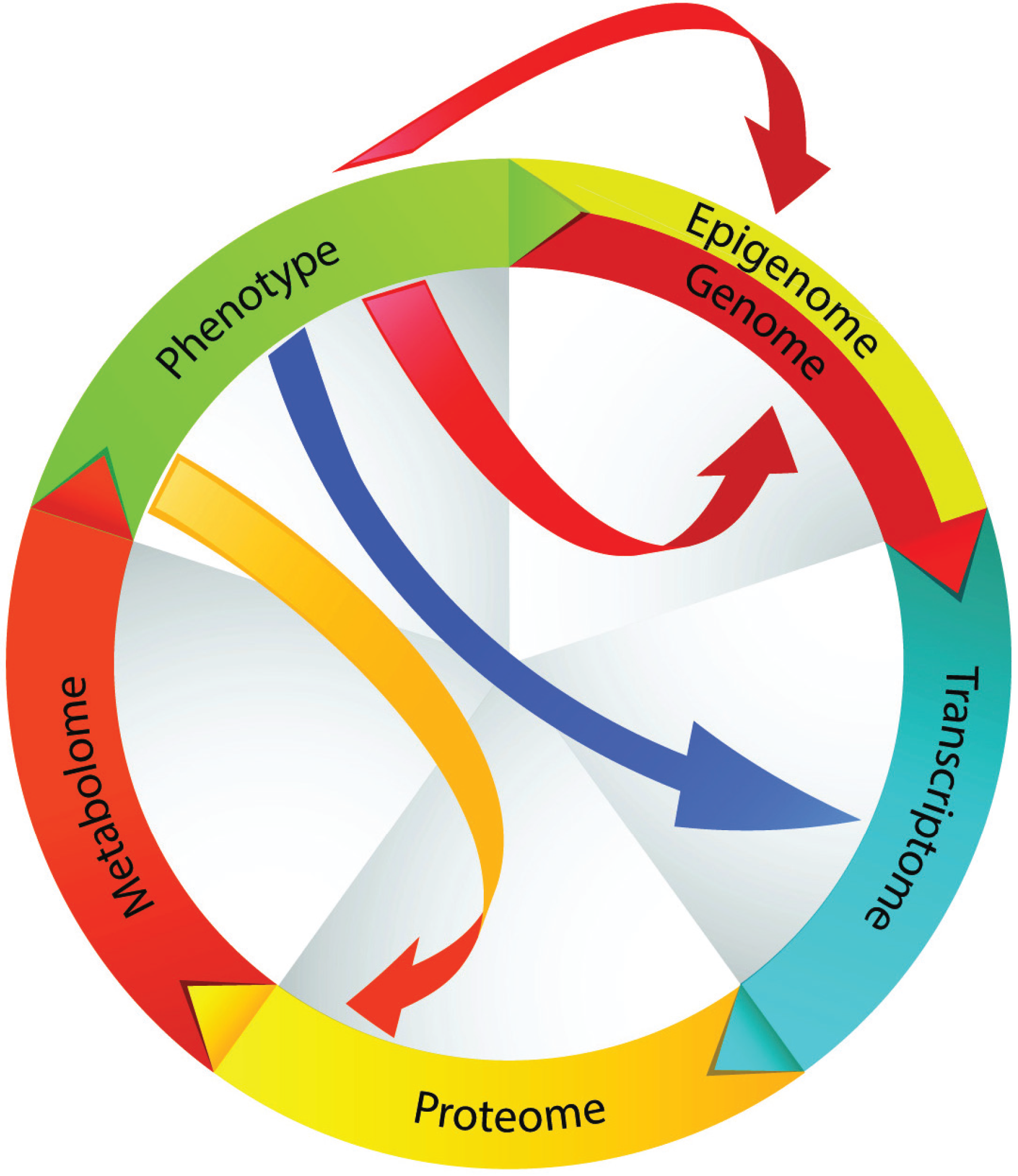
4. Metabolism: A Terminal Function Reflecting Phenotype
4.1. Disordered Metabolism Is a Hallmark of Cancer
4.2. Genomic and Molecular Events Influencing Metabolism in CRC


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Product | Mechanism of Change in Function | Metabolic Effect |
|---|---|---|---|
| TGFBR2 | TGF-beta receptor type-2 | Inactivating mutation, overexpression | Activation of MAPK/ERK and TGF-β-SMAD pathway; inactivation leads to increased proliferation and decreased apoptosis |
| TP53 | Tumor protein p53 | Inactivating mutation or SNP in tumor suppressor | Inhibition of glucose transporters, inhibition of insulin receptor, activation of TCA cycle and oxidative phosphorylation |
| KRAS | GTPase kras | Activating mutation | Increased glucose uptake. Increased glycolysis, activation of PI3K pathway |
| PI3KCA | Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha | Activating mutation | Increased lipid metabolism, growth-factor independence, increased glycolysis and glutaminolysis |
| SMAD4 | Mothers against decapentaplegic homolog 4 | Inactivating mutation | TGF-β signaling |
| TCF7L2 | Transcription factor 7-like 2 | Activating mutation | Increased Wnt signaling, increased glycolysis and lactate production |
| SMAD2 | Mothers against decapentaplegic homolog 2 | Inactivating mutation in tumor suppressor | TGF-β signaling |
| CTNNB1 | Catenin beta-1 | Activating mutation | Wnt signaling pathway |
| SOX9 | SRY (sex determining region Y)-box 9 | Mutation or Overexpression of transcription factor | Wnt signaling pathway, inactivation of insulin signaling, anti-proliferation |
| SOX9 | SRY (sex determining region Y)-box 9 | Mutation or Overexpression of transcription factor | Wnt signaling pathway, inactivation of insulin signaling, anti-proliferation |
| ACVR1B | Activin receptor type-1B | Mutation, Overexpression | Activation of TGF-β signaling |
| EDNRB | Endothelin B receptor | Mutation, hypermethylation, Overexpression | Response to peptide hormonal stimuli |
| FASN | Fatty acid synthase | Overexpression | Production of fatty acids from Acetyl-CoA |
| PTGS2 (COX2) | Prostaglandin G/H synthase 2 | Overexpression | Modulated by HIF-2α, inducing TGF-β pathway |
| E-Cadherin (CDH1) | Cadherin 1, type 1, E-cadherin | Mutation, Overexpression | Activates Wnt signaling and lipid metabolism pathway |
| CDKN2A (p16-INK4a) | Cyclin dependent kinase inhibitor 2A | Mutation, deletion, Methylation | Leads to mitochondrial dysfunction and impaired phosphorylative oxidation, increased glycolysis |
| THBS1/TSP1 | Thrombospondin 1 | Methylation | Regulator of TGF-β signaling, increased inflammation in adipose tissue |
| SDH | Succinate dehydrogenase complex, subunit B, iron sulfur | Underexpression (mechanism unclear) | Enzyme for TCA cycle, phosphorylative oxidation activity, decreased glucose uptake |
| PTEN | Phosphatase and tensin homolog | Inactivating mutation in tumor suppressor | Suppressor of PI3K/Akt pathway. Inactivation leads to increased glycolysis, lipogenesis and glycogenesis. |
| HIF-1α | Hypoxia-inducible factor 1-alpha | Overexpression and molecular stabilization | Activates glycolysis, deactivates TCA cycle and phosphorylative oxidation |
4.3. Metabolomic Studies Related to CRC
5. Linking Genotypic Subsets with Functional Subsets of CRC
6. Metabolomics as a Means to Discover Novel Therapeutic Targets
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Miranda, E.; Destro, A.; Malesci, A.; Balladore, E.; Bianchi, P.; Baryshnikova, E.; Franchi, G.; Morenghi, E.; Laghi, L.; Gennari, L.; et al. Genetic and epigenetic changes in primary metastatic and nonmetastatic colorectal cancer. Br. J. Cancer 2006, 95, 1101–1107. [Google Scholar] [CrossRef]
- Oliveira, C.; Velho, S.; Moutinho, C.; Ferreira, A.; Preto, A.; Domingo, E.; Capelinha, A.F.; Duval, A.; Hamelin, R.; Machado, J.C.; et al. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 2007, 26, 158–163. [Google Scholar] [CrossRef]
- Gunther, K.; Leier, J.; Henning, G.; Dimmler, A.; Weissbach, R.; Hohenberger, W.; Forster, R. Prediction of lymph node metastasis in colorectal carcinoma by expression of chemokine receptor CCR7. Int. J. Cancer 2005, 116, 726–733. [Google Scholar] [CrossRef]
- Artinyan, A.; Essani, R.; Lake, J.; Kaiser, A.M.; Vukasin, P.; Danenberg, P.; Danenberg, K.; Haile, R.; Beart, R.W., Jr. Molecular predictors of lymph node metastasis in colon cancer: Increased risk with decreased thymidylate synthase expression. J. Gastrointest. Surg. 2005, 9, 1216–1221. [Google Scholar] [CrossRef]
- Lin, Y.M.; Furukawa, Y.; Tsunoda, T.; Yue, C.T.; Yang, K.C.; Nakamura, Y. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene 2002, 21, 4120–4128. [Google Scholar] [CrossRef]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Aranes, M.J.; Nicholas, C.; Lesser, M.; Mariadason, J.M.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Arango, D.; Shi, Q.; Wilson, A.J.; Corner, G.A.; Nicholas, C.; Aranes, M.J.; Lesser, M.; Schwartz, E.L.; Augenlicht, L.H. Gene expression profiling-based prediction of response of colon carcinoma cells to 5-fluorouracil and camptothecin. Cancer Res. 2003, 63, 8791–8812. [Google Scholar]
- Li, M.; Lin, Y.M.; Hasegawa, S.; Shimokawa, T.; Murata, K.; Kameyama, M.; Ishikawa, O.; Katagiri, T.; Tsunoda, T.; Nakamura, Y.; et al. Genes associated with liver metastasis of colon cancer, identified by genome-wide cDNA microarray. Int. J. Oncol. 2004, 24, 305–312. [Google Scholar]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Ogino, S.; Goel, A. Molecular classification and correlates in colorectal cancer. J. Mol. Diagn. 2008, 10, 13–27. [Google Scholar] [CrossRef]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef]
- Ogino, S.; Cantor, M.; Kawasaki, T.; Brahmandam, M.; Kirkner, G.J.; Weisenberger, D.J.; Campan, M.; Laird, P.W.; Loda, M.; Fuchs, C.S. Cpg island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006, 55, 1000–1006. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Albertsen, H.; Herrick, J.; Levin, T.R.; Sweeney, C.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Evaluation of a large, population-based sample supports a cpg island methylator phenotype in colon cancer. Gastroenterology 2005, 129, 837–845. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. Cpg island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with braf mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Ogino, S.; Odze, R.D.; Kawasaki, T.; Brahmandam, M.; Kirkner, G.J.; Laird, P.W.; Loda, M.; Fuchs, C.S. Correlation of pathologic features with CpG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am. J. Surg. Pathol. 2006, 30, 1175–1183. [Google Scholar]
- Soreide, K.; Janssen, E.A.M.; Soiland, H.; Korner, H.; Baak, J.P.A. Microsatellite instability in colorectal cancer. Br. J. Surg. 2006, 93, 395–406. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Guastadisegni, C.; Colafranceschi, M.; Ottini, L.; Dogliotti, E. Microsatellite instability as a marker of prognosis and response to therapy: A meta-analysis of colorectal cancer survival data. Eur. J. Cancer 2010, 46, 2788–2798. [Google Scholar] [CrossRef]
- Laghi, L.; Malesci, A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig. Dis. 2012, 30, 304–309. [Google Scholar] [CrossRef]
- Ward, R.L.; Cheong, K.; Ku, S.L.; Meagher, A.; O’Connor, T.; Hawkins, N.J. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J. Clin. Oncol. 2003, 21, 3729–3736. [Google Scholar] [CrossRef]
- Ogino, S.; Meyerhardt, J.A.; Kawasaki, T.; Clark, J.W.; Ryan, D.P.; Kulke, M.H.; Enzinger, P.C.; Wolpin, B.M.; Loda, M.; Fuchs, C.S. CpG island methylation, response to combination chemotherapy, and patient survival in advanced microsatellite stable colorectal carcinoma. Virchows Arch. 2007, 450, 529–537. [Google Scholar] [CrossRef]
- Watanabe, T.; Wu, T.T.; Catalano, P.J.; Ueki, T.; Satriano, R.; Haller, D.G.; Benson, A.B., 3rd; Hamilton, S.R. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2001, 344, 1196–1206. [Google Scholar] [CrossRef]
- Jung, B.; Smith, E.J.; Doctolero, R.T.; Gervaz, P.; Alonso, J.C.; Miyai, K.; Keku, T.; Sandler, R.S.; Carethers, J.M. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. Int. J. Cancer 2006, 118, 2509–2513. [Google Scholar] [CrossRef]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef]
- Matsui, M.; Chu, Y.; Zhang, H.; Gagnon, K.T.; Shaikh, S.; Kuchimanchi, S.; Manoharan, M.; Corey, D.R.; Janowski, B.A. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res. 2013, 41, 10086–10109. [Google Scholar] [CrossRef]
- Svoboda, M.; Slyskova, J.; Schneiderova, M.; Makovicky, P.; Bielik, L.; Levy, M.; Lipska, L.; Hemmelova, B.; Kala, Z.; Protivankova, M.; et al. HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis 2014, 35, 1510–1515. [Google Scholar] [CrossRef]
- Qi, P.; Xu, M.D.; Ni, S.J.; Shen, X.H.; Wei, P.; Huang, D.; Tan, C.; Sheng, W.Q.; Zhou, X.Y.; Du, X. Down-regulation of ncRAN, a long non-coding RNA, contributes to colorectal cancer cell migration and invasion and predicts poor overall survival for colorectal cancer patients. Mol. Carcinog. 2014. [Google Scholar] [CrossRef]
- Chen, T.; Yao, L.Q.; Shi, Q.; Ren, Z.; Ye, L.C.; Xu, J.M.; Zhou, P.H.; Zhong, Y.S. MicroRNA-31 contributes to colorectal cancer development by targeting factor inhibiting HIF-1alpha (FIH-1). Cancer Biol. Ther. 2014, 15, 516–523. [Google Scholar] [CrossRef]
- Pichler, M.; Ress, A.L.; Winter, E.; Stiegelbauer, V.; Karbiener, M.; Schwarzenbacher, D.; Scheideler, M.; Ivan, C.; Jahn, S.W.; Kiesslich, T.; et al. MIR-200a regulates epithelial to mesenchymal transition-related gene expression and determines prognosis in colorectal cancer patients. Br. J. Cancer 2014, 110, 1614–1621. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Sacconi, A.; Landi, L.; Ludovini, V.; Biagioni, F.; D’Incecco, A.; Capodanno, A.; Salvini, J.; Corgna, E.; Cupini, S.; et al. Microrna signature in metastatic colorectal cancer patients treated with anti-EGFR monoclonal antibodies. Clin. Colorectal Cancer 2014, 13, 37–45. [Google Scholar] [CrossRef]
- Nosho, K.; Igarashi, H.; Nojima, M.; Ito, M.; Maruyama, R.; Yoshii, S.; Naito, T.; Sukawa, Y.; Mikami, M.; Sumioka, W.; et al. Association of microRNA-31 with BRAF mutation, colorectal cancer survival and serrated pathway. Carcinogenesis 2014, 35, 776–783. [Google Scholar] [CrossRef]
- Pizzini, S.; Bisognin, A.; Mandruzzato, S.; Biasiolo, M.; Facciolli, A.; Perilli, L.; Rossi, E.; Esposito, G.; Rugge, M.; Pilati, P.; et al. Impact of micrornas on regulatory networks and pathways in human colorectal carcinogenesis and development of metastasis. BMC Genomics 2013, 14. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mrnas regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Tay, Y.; Kats, L.; Salmena, L.; Weiss, D.; Tan, S.M.; Ala, U.; Karreth, F.; Poliseno, L.; Provero, P.; di Cunto, F.; et al. Coding-independent regulation of the tumor suppressor pten by competing endogenous mrnas. Cell 2011, 147, 344–357. [Google Scholar]
- Matassa, D.S.; Amoroso, M.R.; Agliarulo, I.; Maddalena, F.; Sisinni, L.; Paladino, S.; Romano, S.; Romano, M.F.; Sagar, V.; Loreni, F.; et al. Translational control in the stress adaptive response of cancer cells: A novel role for the heat shock protein trap1. Cell Death Dis. 2013, 4, e851. [Google Scholar]
- Dixon, D.A. Dysregulated post-transcriptional control of COX-2 gene expression in cancer. Curr. Pharm. Des. 2004, 10, 635–646. [Google Scholar] [CrossRef]
- Pedersen, J.W.; Blixt, O.; Bennett, E.P.; Tarp, M.A.; Dar, I.; Mandel, U.; Poulsen, S.S.; Pedersen, A.E.; Rasmussen, S.; Jess, P.; et al. Seromic profiling of colorectal cancer patients with novel glycopeptide microarray. Int. J. Cancer 2011, 128, 1860–1871. [Google Scholar] [CrossRef]
- Nakagawa, H.; Chadwick, R.B.; Peltomaki, P.; Plass, C.; Nakamura, Y.; de la Chapelle, A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 591–596. [Google Scholar]
- Levine, A.J.; Ihenacho, U.; Lee, W.; Figueiredo, J.C.; Vandenberg, D.J.; Edlund, C.K.; Davis, B.D.; Stern, M.C.; Haile, R.W. Genetic variation in insulin pathway genes and distal colorectal adenoma risk. Int. J. Colorectal Dis. 2012, 27, 1587–1595. [Google Scholar] [CrossRef]
- Guo, S.T.; Jiang, C.C.; Wang, G.P.; Li, Y.P.; Wang, C.Y.; Guo, X.Y.; Yang, R.H.; Feng, Y.; Wang, F.H.; Tseng, H.Y.; et al. Microrna-497 targets insulin-like growth factor 1 receptor and has a tumour suppressive role in human colorectal cancer. Oncogene 2013, 32, 1910–1920. [Google Scholar] [CrossRef]
- Janku, F. Tumor heterogeneity in the clinic: Is it a real problem? Ther. Adv. Med. Oncol. 2014, 6, 43–51. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27C, 16–25. [Google Scholar]
- Bathe, O.; Dalyot-Herman, N.; Malek, T. Therapeutic limitations in tumor-specific CD8+ memory T cell engraftment. BMC Cancer 2003, 3. [Google Scholar] [CrossRef] [Green Version]
- Schwitalla, S. Tumor cell plasticity: The challenge to catch a moving target. J. Gastroenterol. 2014, 49, 618–627. [Google Scholar] [CrossRef]
- Navin, N.E. Tumor evolution in response to chemotherapy: Phenotype versus genotype. Cell Rep. 2014, 6, 417–419. [Google Scholar] [CrossRef]
- Wu, D.; Wu, P.; Huang, Q.; Liu, Y.; Ye, J.; Huang, J. Interleukin-17: A promoter in colorectal cancer progression. Clin. Dev. Immunol. 2013. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar]
- Pozza, A.; Scarpa, M.; Ruffolo, C.; Polese, L.; Erroi, F.; Bridda, A.; Norberto, L.; Frego, M. Colonic carcinogenesis in ibd: Molecular events. Ann. Ital. Chir. 2011, 82, 19–28. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Lee, H.S.; Kim, H.O.; Hong, Y.S.; Kim, T.W.; Kim, J.C.; Yu, C.S.; Kim, J.S. Prognostic value of metabolic parameters in patients with synchronous colorectal cancer liver metastasis following curative-intent colorectal and hepatic surgery. J. Nucl. Med. 2014, 55, 582–589. [Google Scholar] [CrossRef]
- Miles, K.A.; Ganeshan, B.; Rodriguez-Justo, M.; Goh, V.J.; Ziauddin, Z.; Engledow, A.; Meagher, M.; Endozo, R.; Taylor, S.A.; Halligan, S.; et al. Multifunctional imaging signature for V-Ki-RAS2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations in colorectal cancer. J. Nucl. Med. 2014, 55, 386–391. [Google Scholar] [CrossRef]
- Babbar, M.; Sheikh, M.S. Metabolic stress and disorders related to alterations in mitochondrial fission or fusion. Mol. Cell. Pharmacol. 2013, 5, 109–133. [Google Scholar]
- Grills, C.; Jithesh, P.V.; Blayney, J.; Zhang, S.D.; Fennell, D.A. Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS One 2011, 6, e14635. [Google Scholar]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Ino, K.; Yamamoto, E.; Shibata, K.; Kajiyama, H.; Yoshida, N.; Terauchi, M.; Nawa, A.; Nagasaka, T.; Takikawa, O.; Kikkawa, F. Inverse correlation between tumoral indoleamine 2,3-dioxygenase expression and tumor-infiltrating lymphocytes in endometrial cancer: Its association with disease progression and survival. Clin. Cancer Res. 2008, 14, 2310–2317. [Google Scholar] [CrossRef]
- Sucher, R.; Kurz, K.; Weiss, G.; Margreiter, R.; Fuchs, D.; Brandacher, G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int. J. Tryptophan Res. 2010, 3, 113–120. [Google Scholar]
- Cai, F.; Dupertuis, Y.M.; Pichard, C. Role of polyunsaturated fatty acids and lipid peroxidation on colorectal cancer risk and treatments. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 99–106. [Google Scholar] [CrossRef]
- McMillan, D.C.; Crozier, J.E.; Canna, K.; Angerson, W.J.; McArdle, C.S. Evaluation of an inflammation-based prognostic score (GPS) in patients undergoing resection for colon and rectal cancer. Int. J. Colorectal Dis. 2007, 22, 881–886. [Google Scholar] [CrossRef]
- Crozier, J.E.; McKee, R.F.; McArdle, C.S.; Angerson, W.J.; Anderson, J.H.; Horgan, P.G.; McMillan, D.C. Preoperative but not postoperative systemic inflammatory response correlates with survival in colorectal cancer. Br. J. Surg. 2007, 94, 1028–1032. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Chatr-aryamontri, A.; Breitkreutz, B.-J.; Heinicke, S.; Boucher, L.; Winter, A.; Stark, C.; Nixon, J.; Ramage, L.; Kolas, N.; O’Donnell, L.; et al. The biogrid interaction database: 2013 Update. Nucl. Acids Res. 2013, 41, D816–D823. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Magrane, M.; Consortium, U. Uniprot knowledgebase: A hub of integrated protein data. Database 2011, 2011. [Google Scholar] [CrossRef]
- Furlan, D.; Sahnane, N.; Carnevali, I.; Cerutti, R.; Uccella, S.; Bertolini, V.; Chiaravalli, A.M.; Capella, C. Up-regulation and stabilization of HIF-1alpha in colorectal carcinomas. Surg. Oncol. 2007, 16, S25–S27. [Google Scholar]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Bluemlein, K.; Gruning, N.M.; Feichtinger, R.G.; Lehrach, H.; Kofler, B.; Ralser, M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget 2011, 2, 393–400. [Google Scholar]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Vousden, K.H. Metabolic regulation by p53. J. Mol. Med. (Berl.) 2011, 89, 237–245. [Google Scholar] [CrossRef]
- Sinha, S.; Ghildiyal, R.; Mehta, V.S.; Sen, E. ATM-NFkappab axis-driven tigar regulates sensitivity of glioma cells to radiomimetics in the presence of TNFalpha. Cell Death Dis. 2013, 4, e615. [Google Scholar] [CrossRef]
- Molinari, F.; Frattini, M. Functions and regulation of the PTEN gene in colorectal cancer. Front. Oncol. 2013, 3, e326. [Google Scholar]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. AKT stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of AKT-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef]
- Ogino, S.; Kawasaki, T.; Ogawa, A.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. TGFBR2 mutation is correlated with CpG island methylator phenotype in microsatellite instability-high colorectal cancer. Hum. Pathol. 2007, 38, 614–620. [Google Scholar] [CrossRef]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef]
- Bellam, N.; Pasche, B. TGF-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010, 155, 85–103. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yi, Y.; Choi, S.G.; Kim, S.J. Development of TGF-beta resistance during malignant progression. Arch. Pharm. Res. 1999, 22, 1–8. [Google Scholar] [CrossRef]
- Chan, E.C.; Koh, P.K.; Mal, M.; Cheah, P.Y.; Eu, K.W.; Backshall, A.; Cavill, R.; Nicholson, J.K.; Keun, H.C. Metabolic profiling of human colorectal cancer using high-resolution magic angle spinning nuclear magnetic resonance (HR-MAS NMR) spectroscopy and gas chromatography mass spectrometry (GC/MS). J. Proteome Res. 2009, 8, 352–361. [Google Scholar] [CrossRef]
- Denkert, C.; Budczies, J.; Weichert, W.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Niesporek, S.; Noske, A.; Buckendahl, A.; Dietel, M.; et al. Metabolite profiling of human colon carcinoma—Deregulation of TCA cycle and amino acid turnover. Mol. Cancer 2008, 7. [Google Scholar] [CrossRef]
- Tessem, M.B.; Selnaes, K.M.; Sjursen, W.; Trano, G.; Giskeodegard, G.F.; Bathen, T.F.; Gribbestad, I.S.; Hofsli, E. Discrimination of patients with microsatellite instability colon cancer using 1H HR MAS MR spectroscopy and chemometric analysis. J. Proteome Res. 2010, 9, 3664–3670. [Google Scholar] [CrossRef]
- Monleon, D.; Morales, J.M.; Barrasa, A.; Lopez, J.A.; Vazquez, C.; Celda, B. Metabolite profiling of fecal water extracts from human colorectal cancer. NMR Biomed. 2009, 22, 342–348. [Google Scholar] [CrossRef]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 2013, 8, e70803. [Google Scholar]
- Qiu, Y.; Cai, G.; Su, M.; Chen, T.; Zheng, X.; Xu, Y.; Ni, Y.; Zhao, A.; Xu, L.X.; Cai, S.; et al. Serum metabolite profiling of human colorectal cancer using GC-TOFMS and UPLC-QTOFMS. J. Proteome Res. 2009, 8, 4844–4850. [Google Scholar] [CrossRef]
- Kondo, Y.; Nishiumi, S.; Shinohara, M.; Hatano, N.; Ikeda, A.; Yoshie, T.; Kobayashi, T.; Shiomi, Y.; Irino, Y.; Takenawa, T.; et al. Serum fatty acid profiling of colorectal cancer by gas chromatography/mass spectrometry. Biomark. Med. 2011, 5, 451–460. [Google Scholar] [CrossRef]
- Ludwig, C.; Ward, D.G.; Martin, A.; Viant, M.R.; Ismail, T.; Johnson, P.J.; Wakelam, M.J.; Gunther, U.L. Fast targeted multidimensional NMR metabolomics of colorectal cancer. Magn. Reson. Chem. 2009, 47, S68–S73. [Google Scholar] [CrossRef]
- Leichtle, A.B.; Nuoffer, J.M.; Ceglarek, U.; Kase, J.; Conrad, T.; Witzigmann, H.; Thiery, J.; Fiedler, G.M. Serum amino acid profiles and their alterations in colorectal cancer. Metabolomics 2012, 8, 643–653. [Google Scholar] [CrossRef]
- Nishiumi, S.; Kobayashi, T.; Ikeda, A.; Yoshie, T.; Kibi, M.; Izumi, Y.; Okuno, T.; Hayashi, N.; Kawano, S.; Takenawa, T.; et al. A novel serum metabolomics-based diagnostic approach for colorectal cancer. PLoS One 2012, 7, e40459. [Google Scholar] [CrossRef]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.; Buie, W.D.; Maclean, A.; Dixon, E.; Sutherland, F.R.; Molckovsky, A.; Vogel, H.J.; Bathe, O.F. Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome Med. 2012, 4. [Google Scholar] [CrossRef]
- Tan, B.; Qiu, Y.; Zou, X.; Chen, T.; Xie, G.; Cheng, Y.; Dong, T.; Zhao, L.; Feng, B.; Hu, X.; et al. Metabonomics identifies serum metabolite markers of colorectal cancer. J. Proteome Res. 2013, 12, 3000–3009. [Google Scholar] [CrossRef]
- Bertini, I.; Cacciatore, S.; Jensen, B.V.; Schou, J.V.; Johansen, J.S.; Kruhoffer, M.; Luchinat, C.; Nielsen, D.L.; Turano, P. Metabolomic NMR fingerprinting to identify and predict survival of patients with metastatic colorectal cancer. Cancer Res. 2012, 72, 356–364. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. Metaboanalyst: A web server for metabolomic data analysis and interpretation. Nucl. Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Ingenuity Systems Pathway Analysis. Available online: http://www.ingenuity.com/ (aceessed on 23 December 2014).
- Li, C.; Han, J.; Yao, Q.; Zou, C.; Xu, Y.; Zhang, C.; Shang, D.; Zhou, L.; Zou, C.; Sun, Z.; et al. Subpathway-GM: Identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucl. Acids Res. 2013, 41, e101. [Google Scholar] [CrossRef]
- Nibbe, R.K.; Koyutürk, M.; Chance, M.R. An integrative-omics approach to identify functional sub-networks in human colorectal cancer. PLoS Comput. Biol. 2010, 6, e1000639. [Google Scholar] [CrossRef]
- Kanani, H.; Dutta, B.; Klapa, M.I. Individual vs. Combinatorial effect of elevated CO2 conditions and salinity stress on arabidopsis thaliana liquid cultures: Comparing the early molecular response using time-series transcriptomic and metabolomic analyses. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef]
- Grimplet, J.; Cramer, G.R.; Dickerson, J.A.; Mathiason, K.; van Hemert, J.; Fennell, A.Y. Vitisnet: “Omics” integration through grapevine molecular networks. PLoS One 2009, 4, e8365. [Google Scholar]
- Oberbach, A.; Bluher, M.; Wirth, H.; Till, H.; Kovacs, P.; Kullnick, Y.; Schlichting, N.; Tomm, J.M.; Rolle-Kampczyk, U.; Murugaiyan, J.; et al. Combined proteomic and metabolomic profiling of serum reveals association of the complement system with obesity and identifies novel markers of body fat mass changes. J. Proteome Res. 2011, 10, 4769–4788. [Google Scholar] [CrossRef]
- Wong, C.K.; Vaske, C.J.; Ng, S.; Sanborn, J.Z.; Benz, S.C.; Haussler, D.; Stuart, J.M. The UCSC interaction browser: Multidimensional data views in pathway context. Nucl. Acids Res. 2013, 41, W218–W224. [Google Scholar] [CrossRef]
- Cbioportal for cancer genomics. Available online: http://www.cbioportal.org/public-portal/ (accessed on 14 January 2014).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Interaction browser. Available online: http://sysbio.soe.ucsc.edu/nets/ (accessed on 14 January 2014).
- Qiu, Y.; Cai, G.; Zhou, B.D.O.M.; Li, D.; Zhao, A.; Xie, G.; Li, H.; Cai, S.; Xie, D.; Huang, C.; et al. A distinct metabolic signature of human colorectal cancer with prognostic potential. Clin. Cancer Res. 2014, 20, 2136–2146. [Google Scholar] [CrossRef]
- Manna, S.K.; Tanaka, N.; Krausz, K.W.; Haznadar, M.; Xue, X.; Matsubara, T.; Bowman, E.D.; Fearon, E.R.; Harris, C.C.; Shah, Y.M.; et al. Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology 2014, 146, 1313–1324. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Mitochondrial ion channels as oncological targets. Oncogene 2014. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297–3302. [Google Scholar] [CrossRef]
- Higurashi, T.; Takahashi, H.; Endo, H.; Hosono, K.; Yamada, E.; Ohkubo, H.; Sakai, E.; Uchiyama, T.; Hata, Y.; Fujisawa, N.; et al. Metformin efficacy and safety for colorectal polyps: A double-blind randomized controlled trial. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Jardim, D.L.; Wheler, J.J.; Hess, K.; Tsimberidou, A.M.; Zinner, R.; Janku, F.; Subbiah, V.; Naing, A.; Piha-Paul, S.A.; Westin, S.N.; et al. FBXW7 mutations in patients with advanced cancers: Clinical and molecular characteristics and outcomes with mtor inhibitors. PLoS One 2014, 9, e89388. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. mTOR Pathway in colorectal cancer: An update. Oncotarget 2014, 5, 49–66. [Google Scholar]
- Francipane, M.G.; Lagasse, E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 2013, 4, 1948–1962. [Google Scholar]
- Ewing, G.P.; Goff, L.W. The insulin-like growth factor signaling pathway as a target for treatment of colorectal carcinoma. Clin. Colorectal Cancer 2010, 9, 219–223. [Google Scholar] [CrossRef]
- Golan, T.; Javle, M. Targeting the insulin growth factor pathway in gastrointestinal cancers. Oncology (Williston Park) 2011, 25, 518–526, 529. [Google Scholar]
- Gaglio, D.; Soldati, C.; Vanoni, M.; Alberghina, L.; Chiaradonna, F. Glutamine deprivation induces abortive S-phase rescued by deoxyribonucleotides in K-ras transformed fibroblasts. PLoS One 2009, 4, e4715. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bathe, O.F.; Farshidfar, F. From Genotype to Functional Phenotype: Unraveling the Metabolomic Features of Colorectal Cancer. Genes 2014, 5, 536-560. https://0-doi-org.brum.beds.ac.uk/10.3390/genes5030536
Bathe OF, Farshidfar F. From Genotype to Functional Phenotype: Unraveling the Metabolomic Features of Colorectal Cancer. Genes. 2014; 5(3):536-560. https://0-doi-org.brum.beds.ac.uk/10.3390/genes5030536
Chicago/Turabian StyleBathe, Oliver F., and Farshad Farshidfar. 2014. "From Genotype to Functional Phenotype: Unraveling the Metabolomic Features of Colorectal Cancer" Genes 5, no. 3: 536-560. https://0-doi-org.brum.beds.ac.uk/10.3390/genes5030536