
Role of Mecp2 in Experience-Dependent Epigenetic Programming

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Make-Up of Epigenetic Marks
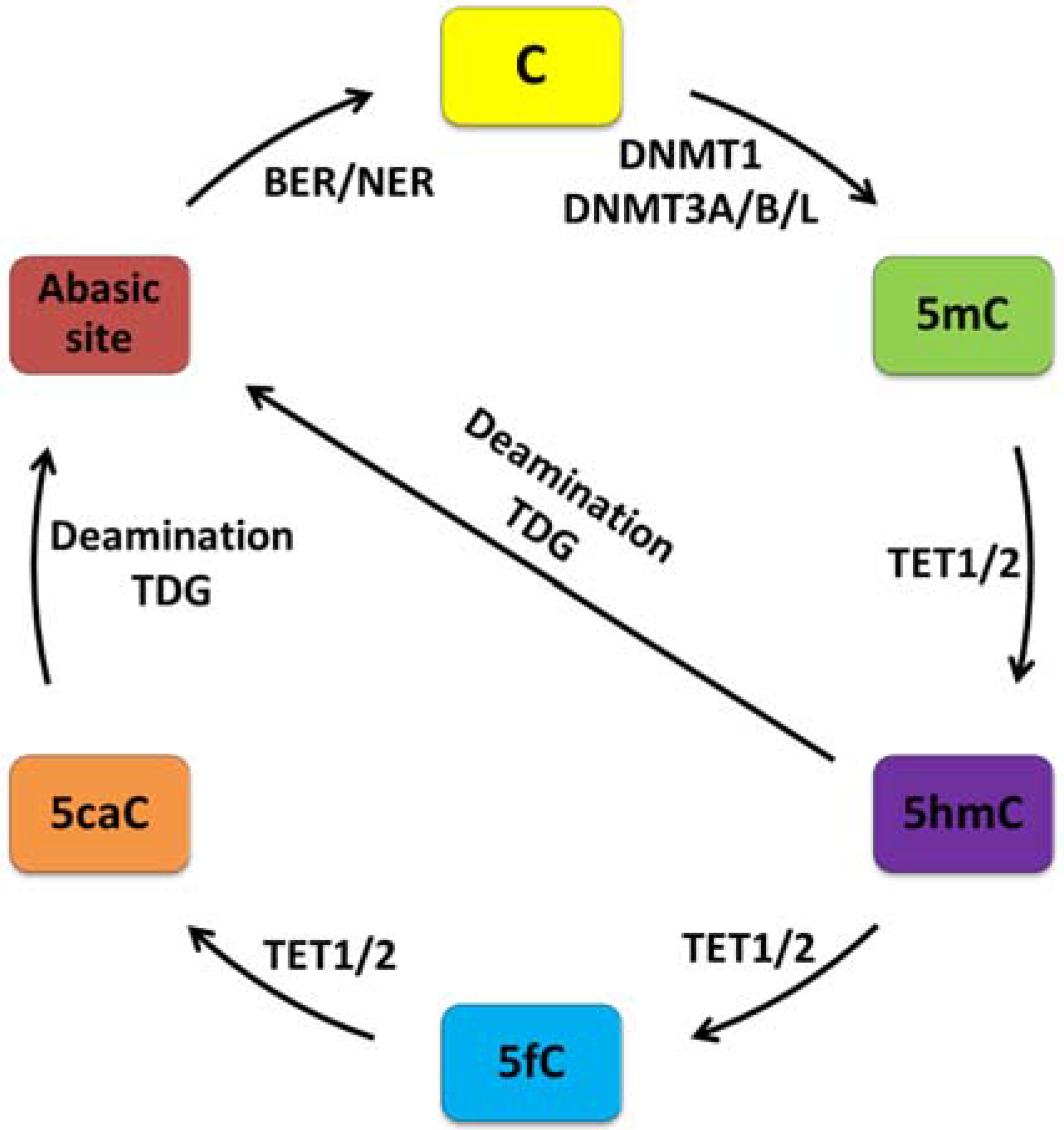
2.1. DNA Modifications

2.2. Histone Modifications
3. MECP2 Mutations Encode a Heritable Neurodevelopmental Disorder
3.1. MECP2 Mutations and Rett Syndrome
3.2. MECP2 Expression and Neuropathological Changes
3.3. MECP2 DNA Binding and Transcriptional Regulation
3.4. MECP2 Target Genes
4. Neuronal Activity Controls Mecp2 via Posttranslational Modifications
4.1. Activity-Dependent (De-)Phosphorylation of Mecp2
4.2. Mecp2 Phosphorylation in Learning and Memory
4.3. Mecp2 Phosphorylation in Drug Addiction
4.4. Mecp2 Phosphorylation in Mood Disorders
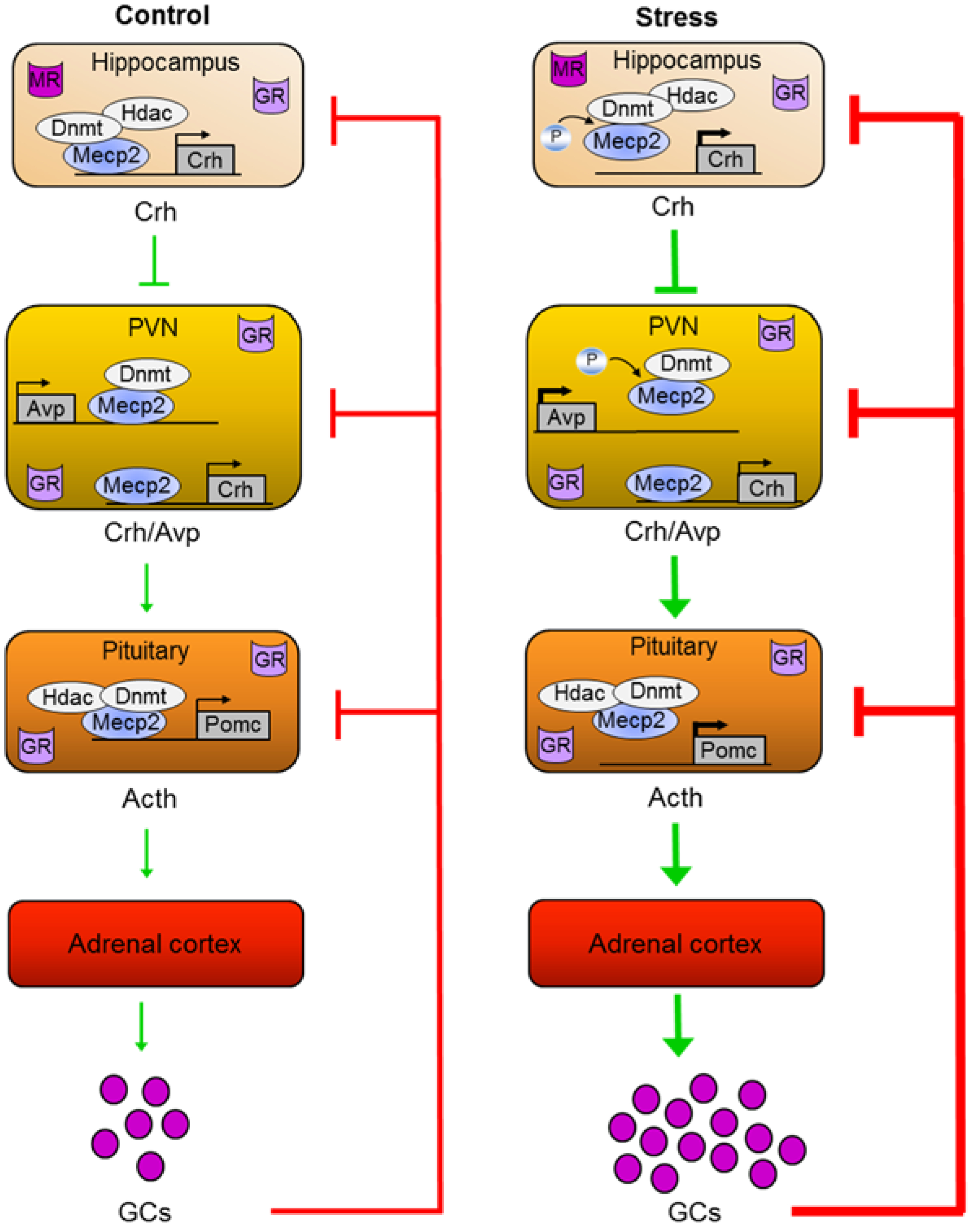
5. Mecp2 Mediates Early-Life Stress
5.1. Early-Life Stress
5.2. The HPA Axis Mediates Early-Life Stress

5.3. A Role for Mecp2 in ELS-Dependent Epigenetic Programming of Avp
5.4. A Role for Mecp2 in ELS-Dependent Epigenetic Programming of Crh
5.5. A Role of Mecp2 in ELS-Dependent Epigenetic Programming of Pomc
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Waddington, C.H. The Strategy of the Genes. A Discussion of Some Aspects of Theoretical Biology; Routledge: London, UK, 2014. [Google Scholar]
- Russo, V.E.A.; Martienssen, R.A.; Riggs, A.D. Epigenetic Mechanisms of Gene Regulation; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1996; Volume 32. [Google Scholar]
- Allis, C.D.; Jenuwein, T.; Reinberg, D. Epigenetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2007. [Google Scholar]
- Ausió, J.; de Paz, A.M.; Esteller, M. MeCP2: The long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Spengler, D. Genetic variation in the epigenetic machinery and mental health. Curr. Psychiatry Rep. 2012, 14, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-Y.; Meaney, M.J. Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 2010, 61, 439–466. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Wu, Y.; Bockmühl, Y.; Spengler, D. Genes learn from stress: How infantile trauma programs us for depression. Epigenetics 2010, 5, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. DNA memories of early social life. Neuroscience 2014, 264, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Eccleston, A.; DeWitt, N.; Gunter, C.; Marte, B.; Nath, D. Insight: Introduction Epigenetics. Nature 2007, 447, 395. [Google Scholar] [CrossRef]
- Tollefsbol, T.O. Handbook of Epigenetics: The New Molecular and Medical Genetics, 1 edition; Academic Press: London, UK, 2010. [Google Scholar]
- Armstrong, L. Epigenetics; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; O’Donnell, A.H.; Bestor, T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009, 122, 2787–2791. [Google Scholar] [CrossRef] [PubMed]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chédin, F. Reconstitution and mechanism of the stimulation of de Novo methylation by human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar]
- Gibbs, J.R.; van der Brug, M.P.; Hernandez, D.G.; Traynor, B.J.; Nalls, M.A.; Lai, S.-L.; Arepalli, S.; Dillman, A.; Rafferty, I.P.; Troncoso, J.; et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6, e1000952. [Google Scholar]
- Gutierrez-Arcelus, M.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Ongen, H.; Yurovsky, A.; Bryois, J.; Giger, T.; Romano, L.; Planchon, A.; et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2013, 2, e00523. [Google Scholar]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and Biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.; Song, H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011, 10, 2662–2668. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Drohat, A.C. Thymine DNA Glycosylase Can Rapidly Excise 5-formylcytosine and 5-carboxylcytosine potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011, 286, 35334–35338. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.T.C.; Bauer, C.; Münzel, M.; Wagner, M.; Müller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Beyond the sequence: Cellular organization of genome function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Cairns, B.R. Chromatin remodeling: Insights and intrigue from single-molecule studies. Nat. Struct. Mol. Biol. 2007, 14, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian Methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.-T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Fu, D.D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Adkins, N.L.; Georgel, P.T. MeCP2: structure and function. Biochem. Cell. Biol. 2010, 89, 1–11. [Google Scholar] [CrossRef]
- Pelka, G.J.; Watson, C.M.; Christodoulou, J.; Tam, P.P.L. Distinct expression profiles of Mecp2 transcripts with different lengths of 3'UTR in the brain and visceral organs during mouse development. Genomics 2005, 85, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Na, E.S.; Monteggia, L.M. The role of MeCP2 in CNS development and function. Horm. Behav. 2011, 59, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; McNae, I.W.; Schmiedeberg, L.; Klose, R.J.; Bird, A.P.; Walkinshaw, M.D. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell 2008, 29, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Sarraf, S.A.; Schmiedeberg, L.; McDermott, S.M.; Stancheva, I.; Bird, A.P. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 2005, 19, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Jan Veenstra, G.C.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Shi, X.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Woodcock, C.L. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 2007, 27, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The Methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, L.; Nakielny, S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 2004, 279, 49479–49487. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.W.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Georgel, P.T.; Horowitz-Scherer, R.A.; Adkins, N.; Woodcock, C.L.; Wade, P.A.; Hansen, J.C. Chromatin compaction by human MeCP2 assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 2003, 278, 32181–32188. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; LaSalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Harikrishnan, K.N.; Bayles, R.; Ciccotosto, G.D.; Maxwell, S.; Cappai, R.; Pelka, G.J.; Tam, P.P.L.; Christodoulou, J.; El-Osta, A. Alleviating transcriptional inhibition of the norepinephrine Slc6a2 transporter gene in depolarized neurons. J. Neurosci. 2010, 30, 1494–1501. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Jiang, Y.; Tremblay, D.C.; Bonvissuto, A.C.; Eubanks, J.H.; Mann, M.R.W.; Bérubé, N.G. ATRX Partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 2010, 18, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Tudor, M.; Akbarian, S.; Chen, R.Z.; Jaenisch, R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15536–15541. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Cheval, H.; Selfridge, J.; Bird, A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 2011, 27, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 2012, e415825. [Google Scholar]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.; Francke, U. Ube3a expression is not altered in Mecp2 mutant mice. Hum. Mol. Genet. 2006, 15, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Paterson, A.; Curtis, J.; Guy, J.; MacLeod, N.; Bird, A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of rett syndrome. Mol. Cell. Biol. 2006, 26, 5033–5042. [Google Scholar] [CrossRef] [PubMed]
- Nuber, U.A.; Kriaucionis, S.; Roloff, T.C.; Guy, J.; Selfridge, J.; Steinhoff, C.; Schulz, R.; Lipkowitz, B.; Ropers, H.H.; Holmes, M.C.; et al. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005, 14, 2247–2256. [Google Scholar]
- Lang, F.; Strutz-Seebohm, N.; Seebohm, G.; Lang, U.E. Significance of SGK1 in the regulation of neuronal function. J. Physiol. 2010, 588, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Storer, C.L.; Dickey, C.A.; Galigniana, M.D.; Rein, T.; Cox, M.B. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol. Metab. 2011, 22, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Spengler, D. Epigenetic programming of the HPA axis: Early life decides. Stress 2011, 14, 581–589. [Google Scholar] [PubMed]
- Wu, Y.; Patchev, A.V.; Daniel, G.; Almeida, O.F.X.; Spengler, D. Early-life stress reduces DNA methylation of the Pomc gene in male mice. Endocrinology 2014, 155, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.P.; Thaller, C.; Zoghbi, H.Y. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.B.; Uhr, M.; Holsboer, F.; Keck, M.E. Hypothalamic-pituitary-adrenocortical system and mood disorders: Highlights from mutant mice. Neuroendocrinology 2004, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.; Ho, H.H.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar]
- Samaco, R.C.; Mandel-Brehm, C.; McGraw, C.M.; Shaw, C.A.; McGill, B.E.; Zoghbi, H.Y. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat. Genet. 2012, 44, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.B.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Bellini, E.; Pavesi, G.; Barbiero, I.; Bergo, A.; Chandola, C.; Nawaz, M.S.; Rusconi, L.; Stefanelli, G.; Strollo, M.; Valente, M.M.; et al. MeCP2 post-translational modifications: A mechanism to control its involvement in synaptic plasticity and homeostasis? Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Li, H.; Chang, Q. Regulation and function of stimulus-induced phosphorylation of MeCP2. Front. Biol. 2014, 9, 367–375. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Hu, K.; Chang, Q.; Wu, H.; Sherman, N.E.; Martinowich, K.; Klose, R.J.; Schanen, C.; Jaenisch, R.; Wang, W.; et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. USA 2009, 106, 4882–4887. [Google Scholar]
- Bracaglia, G.; Conca, B.; Bergo, A.; Rusconi, L.; Zhou, Z.; Greenberg, M.E.; Landsberger, N.; Soddu, S.; Kilstrup‐Nielsen, C. Methyl‐CpG‐binding protein 2 is phosphorylated by homeodomain‐interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009, 10, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.L.; Adams, S.; Dunaway, K.W.; LaSalle, J.M. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 2012, 32, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011, 72, 72–85. [Google Scholar]
- Li, H.; Zhong, X.; Chau, K.F.; Williams, E.C.; Chang, Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat. Neurosci. 2011, 14, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J. Molecular basis of long-term plasticity underlying addiction. Nat. Rev. Neurosci. 2001, 2, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.E.; Malenka, R.C.; Nestler, E.J. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu. Rev. Neurosci. 2006, 29, 565–598. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.; Carouge, D.; Gensburger, C.; Anglard, P.; Burgun, C.; Dietrich, J.-B.; Aunis, D.; Zwiller, J. Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol. Pharmacol. 2006, 70, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.V.; Rodriguiz, R.M.; Hutchinson, A.N.; Kim, I.-H.; Wetsel, W.C.; West, A.E. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat. Neurosci. 2010, 13, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.-M.; Horton, E.; Guo, M.-L.; Xue, B.; Jin, D.-Z.; Fibuch, E.E.; Wang, J.Q. Cocaine increases phosphorylation of MeCP2 in the rat striatum in vivo: A differential role of NMDA receptors. Neurochem. Int. 2011, 59, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.V.; Wan, Y.; Wang, X.; Cohen, S.; Wetsel, W.C.; Greenberg, M.E.; Kenny, P.J.; Calakos, N.; West, A.E. MeCP2 phosphorylation limits psychostimulant-induced behavioral and neuronal plasticity. J. Neurosci. 2014, 34, 4519–4527. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, A.N.; Deng, J.V.; Aryal, D.K.; Wetsel, W.C.; West, A.E. Differential regulation of MeCP2 phosphorylation in the CNS by dopamine and serotonin. Neuropsychopharmacology 2012, 37, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Schloesser, R.J.; Martinowich, K.; Manji, H.K. Mood-stabilizing drugs: Mechanisms of action. Trends Neurosci. 2012, 35, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Hikosaka, O. The habenula: from stress evasion to value-based decision-making. Nat. Rev. Neurosci. 2010, 11, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; McLaughlin, K.A.; Berglund, P.A.; et al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication i: Associations with first onset of DSM-IV disorders. Arch. Gen. Psychiatry 2010, 67, 113–123. [Google Scholar]
- U.S. Department of Health and Human Services; Administration for Children and Families; Administration on Children, Youth and Families. Children’s Bureau. Child. Maltreatment 2009. Available online: http://www.acf.hhs.gov/programs/cb/stats_research/index.htm#can (accessed on 2 December 2014).
- U.S. Department of Health and Human Services; Administration for Children and Families; Administration on Children, Youth and Families; Children’s Bureau. Child. Maltreatment 2010. Available online: http://www.acf.hhs.gov/programs/cb/stats_research/index.htm#can (accessed on 2 December 2014).
- Briere, J.; Berliner, L.; Bulkley, J.A.; Jenny, C.; Reid, T. The APSAC Handbook on Child Maltreatment; American Professional Society on the Abuse of Children, Ed.; SAGE Publications: Thousand Oaks, CA, USA, 1996. [Google Scholar]
- Olson, S. From Neurons to Neighborhoods: An Update: Workshop Summary; Institute of Medicine (U.S.), National Research Council (U.S.), Eds.; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- England, M.J.; Sim, L.J. Depression in Parents, Parenting, and Children: Opportunities to Improve Identification, Treatment, and Prevention; National Research Council (U.S.), Institute of Medicine (U.S.), Ed.; National Academies Press: Washington, DC, USA, 2009. [Google Scholar]
- Edwards, V.J.; Holden, G.W.; Felitti, V.J.; Anda, R.F. Relationship between multiple forms of childhood maltreatment and adult mental health in community respondents: results from the adverse childhood experiences study. Am. J. Psychiatry 2003, 160, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Felitti, V.J.; Anda, R.F.; Nordenberg, D.; Williamson, D.F.; Spitz, A.M.; EdwardS, V.; Koss, M.P.; Marks, J.S. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults: The adverse childhood experiences (ACE) study. Am. J. Prev. Med. 1998, 14, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.P.; Whitfield, C.L.; Felitti, V.J.; Dube, S.R.; Edwards, V.J.; Anda, R.F. Adverse childhood experiences and the risk of depressive disorders in adulthood. J. Affect. Disord. 2004, 82, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Dube, S.R.; Anda, R.F.; Felitti, V.J.; Chapman, D.P.; Williamson, D.F.; Giles, W.H. Childhood abuse, household dysfunction, and the risk of attempted suicide throughout the life span: Findings from the adverse childhood experiences study. JAMA 2001, 286, 3089–3096. [Google Scholar] [CrossRef] [PubMed]
- Raabe, F.J.; Spengler, D. Epigenetic risk factors in PTSD and depression. Front. Mol. Psychiatry 2013, 4, 80. [Google Scholar]
- Lupien, S.J.; McEwen, B.S.; Gunnar, M.R.; Heim, C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 2009, 10, 434–445. [Google Scholar] [CrossRef] [PubMed]
- McCrory, E.; De Brito, S.A.; Viding, E. Research Review: The neurobiology and genetics of maltreatment and adversity. J. Child. Psychol. Psychiatry 2010, 51, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Lupien, S.J.; Fiocco, A.; Wan, N.; Maheu, F.; Lord, C.; Schramek, T.; Tu, M.T. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology 2005, 30, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Spengler, D. The lasting legacy of social stress on the epigenome of the hypothalamic–pituitary–adrenal axis. Epigenomics 2012, 4, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Patchev, A.V.; Rodrigues, A.J.; Sousa, N.; Spengler, D.; Almeida, O.F.X. The future is now: early life events preset adult behaviour. Acta. Physiol. 2014, 210, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Fink, G. Encyclopedia of Stress, 2nd ed.; Academic Press: Waltham, MA, USA, 2007. [Google Scholar]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, N.D. Stress responsiveness of the hypothalamic-pituitary-adrenal axis: Age-related features of the vasopressinergic regulation. Front. Endocrinol. 2013, 4. [Google Scholar] [CrossRef]
- Makara, G.B.; Varga, J.; Barna, I.; Pintér, O.; Klausz, B.; Zelena, D. The vasopressin-deficient brattleboro rat: Lessons for the hypothalamo-pituitary-adrenal axis regulation. Cell. Mol. Neurobiol. 2012, 32, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.V.; Sterlemann, V.; Wagner, K.; Niederleitner, B.; Ganea, K.; Liebl, C.; Deussing, J.M.; Berger, S.; Schütz, G.; Holsboer, F.; et al. Postnatal glucocorticoid excess due to pituitary glucocorticoid receptor deficiency: Differential short- and long-term consequences. Endocrinology 2009, 150, 2709–2716. [Google Scholar]
- Murgatroyd, C.; Spengler, D. Polycomb binding precedes early-life stress responsive DNA methylation at the avp enhancer. PLOS ONE 2014, 9, e90277. [Google Scholar] [CrossRef] [PubMed]
- Menger, Y.; Bettscheider, M.; Murgatroyd, C.; Spengler, D. Sex differences in brain epigenetics. Epigenomics 2010, 2, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bender, R.A.; Frotscher, M.; Baram, T.Z. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: A quantitative spatiotemporal analysis. J. Neurosci. 2001, 21, 7171–7181. [Google Scholar] [PubMed]
- Chen, Y.; Andres, A.; Frotscher, M.; Baram, T.Z. Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front. Cell. Neurosci. 2012, 6, 13. [Google Scholar]
- Maras, P.M.; Baram, T.Z. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012, 35, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Nie, W.; Li, H.; Hou, Y.; Yu, Z.; Fan, Q.; Sun, R. Epigenetic upregulation of corticotrophin-releasing hormone mediates postnatal maternal separation-induced memory deficiency. PLOS ONE 2014, 9, e94394. [Google Scholar] [PubMed]
- Murat, B.; Devost, D.; Andrés, M.; Mion, J.; Boulay, V.; Corbani, M.; Zingg, H.H.; Guillon, G. V1b and CRHR1 receptor heterodimerization mediates synergistic biological actions of vasopressin and CRH. Mol. Endocrinol. 2012, 26, 502–520. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, C.A.; Hoffmann, A.; Raabe, F.; Spengler, D. Role of Mecp2 in Experience-Dependent Epigenetic Programming. Genes 2015, 6, 60-86. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010060
Zimmermann CA, Hoffmann A, Raabe F, Spengler D. Role of Mecp2 in Experience-Dependent Epigenetic Programming. Genes. 2015; 6(1):60-86. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010060
Chicago/Turabian StyleZimmermann, Christoph A., Anke Hoffmann, Florian Raabe, and Dietmar Spengler. 2015. "Role of Mecp2 in Experience-Dependent Epigenetic Programming" Genes 6, no. 1: 60-86. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010060