
Genetics of Type 2 Diabetes—Pitfalls and Possibilities


1
Department of Clinical Sciences, Diabetes and Endocrinology, Lund University Diabetes Centre, Lund University, CRC, Skåne University Hospital SUS, SE-205 02 Malmö, Sweden
2
Finnish Institute of Molecular Medicine (FIMM), Helsinki University, Helsinki 00014, Finland
*
Authors to whom correspondence should be addressed.
Genes 2015, 6(1), 87-123; https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010087
Submission received: 9 December 2014
/
Revised: 28 January 2015
/
Accepted: 27 February 2015
/
Published: 12 March 2015
(This article belongs to the Special Issue Genetics of Diabetes)
Abstract
:Type 2 diabetes (T2D) is a complex disease that is caused by a complex interplay between genetic, epigenetic and environmental factors. While the major environmental factors, diet and activity level, are well known, identification of the genetic factors has been a challenge. However, recent years have seen an explosion of genetic variants in risk and protection of T2D due to the technical development that has allowed genome-wide association studies and next-generation sequencing. Today, more than 120 variants have been convincingly replicated for association with T2D and many more with diabetes-related traits. Still, these variants only explain a small proportion of the total heritability of T2D. In this review, we address the possibilities to elucidate the genetic landscape of T2D as well as discuss pitfalls with current strategies to identify the elusive unknown heritability including the possibility that our definition of diabetes and its subgroups is imprecise and thereby makes the identification of genetic causes difficult.
1. The Diabetes Epidemic
Diabetes refers to a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both [1]. The chronic hyperglycemia of diabetes is associated with long-term damage, dysfunction, and failure of different organs, especially the eyes, kidneys, nerves, heart, and blood vessels. Diabetes is currently the fastest-growing epidemic and has been ascribed to a collision between genes and the environment. Worldwide prevalence figures estimate that there were 382 million people living with diabetes in 2013 and that by 2035 this number will have risen to 592 million [2]. India and China have the highest reported prevalences of diabetes with 65 and 98 million in 2013, respectively, More than 90% of these cases are considered as T2D. In Europe, ~8% of the population is affected by diabetes, 90% of which is accounted for by T2D, making T2D the fastest-increasing disease in Europe and worldwide [2,3].
The T2D epidemic can largely be ascribed to the worldwide increase in obesity during the last 30 years, for instance, more than 60% of individuals older than 15 in the UK and US are overweight (BMI > 25) [4]. This has been ascribed to a collision between genes and the environment. The social determinants of environmental factors tend to vary across populations and have changed rapidly over the last decades. A traditional high energy-burning lifestyle has been replaced by a Western sedentary lifestyle with little or no exercise and consumption of an energy-dense diet. Meanwhile, genetic factors evolve at a slower rate across generations, and tend to favor selection of “energy-saving thrifty genotypes,” which might have been beneficial for individuals living in times of unstable food supply by storing energy in times of surplus [5]. While this hypothesis provides an appealing explanation to the obesity and T2D epidemic, formal proof for this hypothesis is still lacking.
Advances in genomic technology have initiated a myriad of novel genetic discoveries including more than 2,000 common variants contributing to risk of complex disease. While we have learned a great deal, a new biology about pathogenesis of these variants can only explain only a small portion of the total heritability of diabetes, leaving much of the underlying mechanisms unknown. The current review will try to explore the pitfalls with current strategies and elucidate possibilities to use genetics to enhance our understanding of the disease.
2. The Diabetes Spectrum
Diabetes encompasses a range of heterogeneous metabolic disorders characterized by the inability of the body to assimilate glucose and maintain glucose homeostasis. Diabetes has been traditionally subdivided into type 2 diabetes (T2D) and type 1 diabetes (T1D). However, this is a gross oversimplification of a rather complex situation. The concept of diabetes has grown over the past decades to the understanding that several different overlapping contributions from genetics and environment can lead to manifestations of varying forms disease. Contrary to being dichotomously distinct disorders, T1D and T2D can be considered rather as the two ends of a diabetes spectrum, with the intermediates comprising of maturity-onset diabetes of the young (MODY), latent autoimmune diabetes in adults (LADA) and other subtypes.
Type 1 diabetes (T1D), also known as juvenile diabetes or insulin-dependent diabetes, is a chronic condition which is due to autoimmune destruction of pancreatic beta cells and is characterized by (nearly) complete absence of insulin secretion, and presence of autoantibodies including glutamic acid decarboxylase (GAD) antibodies, leading to dependence on insulin injections. It is most often diagnosed in children, adolescents or young adults less than 35 years old. The incidence of T1D varies based upon geography, age, gender, and family history [2]. Only 10% to 15% of newly diagnosed patients have a positive family history of T1D [6]. However, increased susceptibility to T1D can be inherited, because the average prevalence risk is 0.4% for children with no family history whereas ~6% when either parent has T1D, >30% when both parents are affected. There is a great difference in recurrence risk between dizygotic (8%) and monozygotic (50%) twins (30% risk within 10 years of diagnosis of the first twin) [7,8]. Interestingly, the risk in offspring of an affected mother is 2%–4%, whereas the risk of an affected father is as high as 5%–8% [9,10]. The sibling relative risk of T1D is estimated at 15 [7,11,12]. T1D results from interplay between genetic, epigenetic and environmental factors [13]. Genetic studies have been able to explain 80% of the heritability of T1D [14]. The main susceptibility genes currently accepted for T1D are the HLA class II alleles, which account for up to 50% of genetic T1D risk and non-HLA loci including the insulin gene, CTLA4, PTPN22, interleukin 2 receptor a (IL2RA), and others [15]. Environmental factors suggested so far include enterovirus infections including viruses of the picoRNA family, as they are seen more often among newly diagnosed T1D individuals than in the general population, and they precede the appearance of autoantibodies, environmental pollutants, gut flora variations and vitamin D exposure [16,17,18].
LADA (Latent Autoimmune Diabetes in Adults) is a common subgroup of diabetes accounting for about 7% of all diabetic patients in Europe. LADA is usually defined as GAD antibody-positive diabetes with onset greater than 35 years of age and no insulin requirement during the first 6 months after diagnosis [19,20,21]. The cutoff for defining GAD ab positivity is the same as for T1D. Therefore, LADA with high ab titers are found to the left of the spectrum close to T1D, whereas LADA with lower titers are to the right of the spectrum close to T2D [22]. A family history of any form of diabetes is a strong risk factor for the development of LADA [23].
MODY (Maturity-Onset-Diabetes of the Young) refers to monogenic forms of diabetes with well-defined mutations in more than 10 different genes, and this number is still increasing. The disease is characterized by autosomal dominant transmission of early-onset (<25 years) diabetes and varying degree of beta-cell dysfunction [24]. It was long debated whether the MODY genes would harbor common less-penetrant variants increasing risk of T2D; now, this seems to be the case for most of them including HNF1A, HNF4A, HNF1B, GCK, and PDX1 [25,26,27]. MODY shows extreme allelic heterogeneity meaning that most MODY mutations are unique; to date, there are more than 200 mutations described in the GCK (MODY2) and HNF1A (MODY3) genes [28,29]. The appropriate diagnosis of MODY requires sequencing. With the advent of next-generation sequencing technologies, accurate MODY diagnoses are much more feasible today.
Maternally inherited diabetes and deafness (MIDD) is due to the A3242G mutation in mitochondrial DNA (mtDNA) [28,30]. As mtDNA is only transmitted from the mother, MIDD shows maternal transmission. In addition to hearing loss, many patients also display neurological problems similar to patients with the MELAS syndrome (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke), which is also caused by the same mutation in mtDNA.
Neonatal diabetes is defined as diabetes with onset at birth or during the first 6 months of life with both transient and permanent forms [28]. Mutations in several genes have been shown to cause neonatal diabetes (KCJN11, SUR1, GCK, INS, etc.), and an appropriate genetic diagnosis is a prerequisite for optimal treatment. Patients with mutations in the KCJN11 gene do not only have severe diabetes but also developmental defects (DEND). These conditions are improved after switching from insulin treatment to treatment with large doses of sulfonylureas [31].
Gestational Diabetes mellitus (GDM) is a transitory form of diabetes which manifests as hyperglycemia during pregnancy which is clearly not overt diabetes and resolves itself post-partum. An estimated sibling risk ratio of 1.75 was reported for GDM [32,33]; however, changes in the diagnostic criteria have complicated retrospective identification of GDM cases and correct estimation of heritability. It has been observed that many of the GDM-associated variants overlap with T2D risk variants [34,35,36] which may partly explain the increased risk for T2D in women with previous GDM [37,38]. Despite being a transient condition, women with GDM are at increased risk for adverse pregnancy outcomes and fetal hyperinsulinism, and infants with macrosomia [39,40].
Type 2 diabetes is the most prevailing form, constituting 80%–90% of all reported diabetes cases. T2D is the result of a complex interplay between genetic, epigenetic and environmental factors. T2D develops when pancreatic beta cells can no longer produce enough insulin to compensate for the insulin resistance imposed by increasing obesity. There is no formal definition of T2D; patients who do not fulfill criteria of T1D, LADA, secondary diabetes (see below), or monogenic forms of diabetes are considered to have T2D. T2D is more often associated with increased age, wherein age of onset is usually over 35 years [14]. However, it is increasingly reported in adolescents in high risk countries such as India AND China [3]. Heritability of T2D is discussed below.
Finally, diabetes can develop secondary to pancreatic disease or other endocrine disorders and referred to as secondary diabetes.
All these forms of diabetes represent a range of genetic etiologies from the monogenic MODY variants to T2D, which is a complex heterogeneous polygenic disease with a strong environmental component. The ANDIS (All New Diabetics in Scania) project in southern Sweden represents a new attempt to reclassify diabetes into subgroups based upon genetic markers and biomarkers (Figure 1). The aim of ANDIS is to register all new cases of diabetes in Scania and improve diagnosis and treatment strategies. At the time of registration, blood samples are drawn to determine the presence of GAD antibodies, measure C-peptide, biobanking and for genetic analysis. The genetic data is used to classify the disease into subtypes and to study genetic causes of diabetes, diabetic complications and other disorders related to diabetes (http://andis.ludc.med.lu.se/all-new-diabetics-in-scania-andis/). A similar project has been initiated in Uppsala with the same goal (ANDIU—All new diabetics in Uppsala). The ANDIU study design is similar to the ANDIS study (http://www.andiu.se/english/).
3. Heritability of T2D
T2D clusters in families and it is well established that the risk of developing T2D depends on both genetic and environmental factors. However, heritability estimates have varied between 25%–80% in different studies; the highest estimates are seen in those studies with the longest follow-up periods. The lifetime risk of developing T2D is 40% for individuals who have one parent with T2D and almost 70% if both parents are affected [41]. Furthermore, the concordance rate of T2D in monozygotic twins is about 70%, while the concordance in dizygotic twins is only 20%–30%. The proband-wise concordance rates (number of affected twins having a diabetic co-twin) for monozygotic twins vary between 34% and 100% [42,43,44,45].
The relative risk for first degree relatives, i.e., the risk of developing T2D if you have an affected parent or sibling compared to the general population, is approximately 3, and ~6 if both parents are affected [46]. However, these figures vary depending on the cohort and population studied.
The prevalence of T2D varies widely among populations, from a few percent among Caucasians in Europe to as high as 50% among Pima Indians in Arizona [47]. While part of the observed ethnic variability could be attributed to environmental and cultural factors, some of the variation seems to depend on genetic differences.
In spite of these reservations, there is no doubt the risk of T2D is partly determined by genetic factors, many of which have already been identified, and while each identified variant explains only a very small proportion of the risk of T2D in the human population, they have contributed to our understanding of disease pathogenesis. One should also keep in mind that the variance explained by a risk allele in a population is not necessarily an indicator of its importance in specific patients, nor is it proportional to the affected pathway’s importance or potential as a therapeutic target.
Figure 1.
The spectrum of diabetes subgroups. The data is from the ANDIS project (All New Diabetics in Scania) (http://andis.ludc.med.lu.se) in April 2012 which at that time included 5,800 newly diagnosed diabetic patients aged 0–100 years. The criteria used for diagnosis are as follows: T1D: age at onset <35 years, C-peptide < 0.2nmol/L and GAD antibodies >20; T1D with relative insulin deficiency if C-peptide 0.2–0.6 nmol/L. T2D: age at onset >35 years, C-peptide > 0.6 nmol/L, GAD antibodies <10; T2D with relative insulin deficiency C-peptide 0.2–0.6 nmol/L. LADA (latent autoimmune diabetes in adults): age at onset > 5 years, GAD antibodies >20; LADA light if GAD antibodies 10–20. The data clearly illustrates the difficulty classifying diabetic patients at diagnosis with 19% unclassifiable.
Figure 1.
The spectrum of diabetes subgroups. The data is from the ANDIS project (All New Diabetics in Scania) (http://andis.ludc.med.lu.se) in April 2012 which at that time included 5,800 newly diagnosed diabetic patients aged 0–100 years. The criteria used for diagnosis are as follows: T1D: age at onset <35 years, C-peptide < 0.2nmol/L and GAD antibodies >20; T1D with relative insulin deficiency if C-peptide 0.2–0.6 nmol/L. T2D: age at onset >35 years, C-peptide > 0.6 nmol/L, GAD antibodies <10; T2D with relative insulin deficiency C-peptide 0.2–0.6 nmol/L. LADA (latent autoimmune diabetes in adults): age at onset > 5 years, GAD antibodies >20; LADA light if GAD antibodies 10–20. The data clearly illustrates the difficulty classifying diabetic patients at diagnosis with 19% unclassifiable.

4. Possibilities
4.1. Search for the Genetic Basis of T2D
4.1.1. Linkage Studies
The CAPN10 gene on chromosome 10 encoding calpain 10, a cysteine protease with largely unknown functions in glucose metabolism, was the first T2D susceptibility gene to be identified through linkage studies [48]. Unfortunately, this locus has been difficult to replicate in subsequent studies. The greatest success in linkage studies relates to the discovery of variants in the TCF7L2 as being associated with T2D. The DeCode team observed a rather modest linkage at a 10.5 Mb region on chromosome 10q, but decided to pursue and fine-map it thereby identifying the variant until the date showing the strongest association with T2D, an intronic variant (rs7903146) in the TCF7L2 gene contributing to, but not fully explaining, the original linkage [49,50,51]. This association has since been confirmed in African, Asian and European populations rendering it the most consistently replicated genetic association with T2D to date, conferring a relative risk of ~1.4 [52].
4.1.2. Candidate Genes for T2D
The first candidate gene reproducibly associated with T2D was PPARG, encoding the nuclear receptor PPAR-γ [53]. The PPAR-γ receptor is a molecular target for thiazolidinediones, a class of insulin-sensitizing drugs used to treat T2D, making it a very compelling candidate gene. The transcript expressed in adipose tissue has an extra exon B and a substitution of a proline for alanine at position 12 of this protein, which is seen in about 15% of the European population. This variant has been shown to be associated with increased transcriptional activity, increased insulin sensitivity and protection against T2D [53].
T2D risk variants in KCJN11 were also discovered through candidate association studies [54,55]. KCNJ11 codes for four subunits of the ATP-sensitive potassium (K-ATP) channel, the other four coded by another gene (ABCC8). The E23K polymorphisms in KCNJ11 and P12A in PPARG putatively acted in an additive manner to increase T2D risk [56]. In pancreatic beta cells, ATP−potassium channels are crucial for the regulation of glucose-stimulated insulin secretion and are the target for the sulfonylureas which are oral hypoglycemic agents widely used in the treatment of T2D and for diazoxide, a potassium channel opener. Activating mutations in this gene also caused neonatal diabetes. Additionally, loss-of-function mutations in KCNJ11 and ABCC8 caused hyperinsulinemia in infancy [57].
4.1.3. Genome Wide Association Studies (GWAS)
The development of new genotyping technologies and the realization that we inherit stretches of the genome together as haplotypes facilitated the cataloging of common variants (HAPMAP, 1000 genomes) and allowed for new possibilities to apply unbiased global approaches to screen millions of common variants for association with complex diseases. Several GWAS for diabetes were published in 2007, coined “Breakthrough of the Year” by Science magazine. The first was a GWAS on early-onset T2D reporting two new diabetes loci: HHEX and SLC30A8 [58]. The ultimate proof of the value of GWAS for T2D came from three GWAS published back to back in Science in 2007; for the first time in the genetics of T2D, three different studies reported the same top findings [59,60,61]!
The first wave of GWAS was followed by a second wave combining existing or new GWAS into meta-analyses of >50,000 individuals [25]. A prerequisite for this was that many research groups could work together in consortia like DIAGRAM (DIAbetes Genetics Replication and Meta-analysis Consortium) and MAGIC (Meta-Analyses of Glucose-and Insulin-related traits Consortium). GWAS do not inevitably lead to identification of a gene or genes in a given locus associated with disease. Since the most strongly associated single nucleotide polymorphisms (SNPs) are often only markers for the functional variant responsible for the observed genetic effect and most associated regions harbor several genes, additional fine mapping of the loci in even larger sample sets is often necessary. To do this cost efficiently, a custom-designed chip, the so-called CardioMetabochip (Illumina) was been developed for metabolic/cardiovascular gene mapping. This chip contains ~200,000 polymorphisms selected to cover association signals from a wide range of metabolic disorders (T2D, lipid disorders, obesity and cardiovascular disease), and was designed to perform both deep replication of major disease signals and fine mapping of established loci. Meta-analysis of previous GWAS by the DIAGRAM consortium with an additional 22,669 T2D cases and 58,119 controls genotyped using the CardioMetabochip has recently added another eight new loci associated with T2D in the European population and 2 novel loci not previously reported in populations of European descent [62].
Recently, a number of GWAS and meta-analysis studies have also been performed in non-European cohorts, adding several new loci to the list of genome-wide significant associations [63,64,65,66,67,68,69,70]. Interestingly, it seems like most associations found in one ethnic group also show some evidence of association in populations with other ethnicities. Association of the same variants in more than one population adds to the robustness of the association. KCNQ1, for instance, was reported simultaneously as a T2D risk allele in two studies based on East Asian populations and further replicated in the European population as well [71,72]. Similarly, variants in UBE2E2, C2CD4A-C2CD4B, ANK1, GRK5, RASGRP1, PAX4 and others were discovered in the East Asian population and replicated in Caucasians with variants in PPARγ, KCNJ11, TCF2, TCF7L2, CDKAL1, CDKN2A-CDKN2B, IDE-KIF11-HHEX, IGF2BP2, whereas variants in MTNR1B, SLC30A8, KCNQ1, CDC123, GLIS3, HNF1B, DUSP9 and others were identified in Caucasian populations and were also replicated in East Asian populations [63,64,65,66,67,68,69,70,73]. In addition, there are also some population specific risk variants whose association with T2D risk in other populations is yet to be ascertained. For instance, variants in GLIS3, PEPD, ITM2-R3HDML-HNF4A, KCNK16, MAEA, GCC1-PAX4, PSMD6, and ZFAND were discovered in populations of East Asian descent, SLC16A11 in the Mexican population, and TMEM163 in chr 2 in a North Indian population [63,74,75,76].
Trans-ethnic GWAS have been highly effective in the discovery of complex trait loci, and provide valuable insights into the genetic architecture of T2D across populations of diverse ancestry [70]. In addition, they enable fine-mapping by taking advantage of differences in genomic linkage disequilibrium across ethnically diverse populations, and facilitate prioritization of candidate genes [77]. However, allele frequencies can vastly vary between diverse populations and in terms of effects, and need to be taken into consideration while performing such meta-analyses. A method which takes into consideration the expected similarity in allelic effects between the most closely related populations, while allowing for heterogeneity between more diverse ethnic groups has been proposed to allow for this [78].
In total, GWAS have provided ~153 variants for T2D mapping to >120 loci (Table 1, Figure 2 and Figure 3) as well as numerous loci for glucose or insulin-related traits (Table 2) and more are likely to come.

{kind=link}
{kind=link}
{kind=link}
| N | T2D Risk SNP | Gene/Nearest Gene | Gene Location | Chr | RA | OA | OR | TRAIT | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs17106184 | FAF1 | intron | 1 | G | A | 1.10 | T2D | [70] |
| 2 | rs2296172 | MACF1 | coding - missense | 1 | G | A | 1.10 | 2D | [79] |
| 3 | rs10923931 | NOTCH2 | intron | 1 | T | G | 1.13 | T2D | [4,80] |
| 4 | rs340874 | PROX1 | intergenic | 1 | C | T | 1.07 | Fasting glucose/HOMA B/T2D | [81] |
| 5 | rs243021 | BCL11A | intergenic | 2 | A | G | 1.08 | T2D | [25] |
| 6 | rs243088 | BCL11A | intergenic | 2 | T | A | 1.07 | T2D | [62] |
| 7 | rs2975760 | CAPN10 | intron | 2 | C | T | 1.17 | T2D | [48,82] |
| 8 | rs3792267 | CAPN10 | intron | 2 | G | A | 1.17 | T2D | [48,82] |
| 9 | rs7607980 | COBLL1 | coding-missense | 2 | T | C | 1.14 | T2D | [79] |
| 10 | rs560887 | G6PC2/ABCB11 | intron | 2 | T | C | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 11 | rs780094 | GCKR | intron | 2 | C | T | 1.06 | T2D/Fasting glucose/beta-cell function/triglycerides/fasting insulin | [81] |
| 12 | rs3923113 | GRB14 | intergenic | 2 | A | C | 1.07 | T2D | [62,65] |
| 13 | rs13389219 | GRB14 | intergenic | 2 | C | T | 1.07 | T2D | [62] |
| 14 | rs2943641 | IRS1 | intergenic | 2 | C | T | 1.19 | Fasting glucose/T2D/HOMAB, HOMA IR/AUC ins/AUC ratio/ISI | [83] |
| 15 | rs7578326 | KIAA1486/IRS1 | intron of uncharacterized LOC646736 | 2 | A | G | 1.11 | T2D | [25] |
| 16 | rs7593730 | RBMS1/ITGB6 | intronic | 2 | C | T | 1.11 | T2D | [84] |
| 17 | rs7560163 | RND3 | intergenic | 2 | G | C | 1.33 | T2D | [67] |
| 18 | rs7578597 | THADA | coding-missense | 2 | T | C | 1.15 | T2D | [4,80] |
| 19 | rs10200833 | THADA | intron | 2 | G | C | 1.06 | T2D | [80,85] |
| 20 | rs6723108 | TMEM163 | intergenic | 2 | T | G | 1.31 | Decreased fasting plasma insulin/HOMA-IR/T2D | [74] |
| 21 | rs998451 | TMEM163 | intron | 2 | G | A | 1.56 | Decreased fasting plasma insulin/HOMA-IR/T2D | [74] |
| 22 | rs4607103 | ADAMTS9-AS2 | intron | 3 | C | T | 1.09 | T2D | [4,80] |
| 23 | rs6795735 | ADAMTS9-AS2 | intron | 3 | C | T | 1.09 | T2D | [4,80] |
| 24 | rs11708067 | ADCY5 | intron | 3 | A | G | 1.12 | T2D/2hr glucose/HOMA B | [81,86] |
| 25 | rs2877716 | ADCY5 | intron | 3 | C | T | 1.12 | 2 h insulin adjusted for 2 h glucose/2 h glucose/T2D | [81,86] |
| 26 | rs11071657 | FAM148B | intergenic | 3 | A | G | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 27 | rs4402960 | IGF2BP2 | intron | 3 | T | G | 1.11 | T2D | [59] |
| 28 | rs1470579 | IGF2BP2 | intron | 3 | C | A | 1.15 | T2D | [26,59,72,87] |
| 29 | rs6808574 | LPP | intergenic | 3 | C | T | 1.07 | T2D | [70] |
| 30 | rs1801282 | PPARG | coding-missense | 3 | C | G | 1.09 | T2D | [59] |
| 31 | rs13081389 | PPARG | intergenic | 3 | A | G | 1.24 | T2D | [25,53,59,87] |
| 32 | rs17036160 | PPARG | intron | 3 | C | T | 1.11 | T2D | [85] |
| 33 | rs1797912 | PPARG | intron | 3 | A | C | 1.06 | T2D | [85] |
| 34 | rs831571 | PSMD6 | intergenic | 3 | C | T | 1.09 | T2D | [63] |
| 35 | rs7647305 | SFRS10 | intergenic | 3 | C | T | 1.08 | BMI/obesity T2D | [88] |
| 36 | rs16861329 | ST6GAL1 | intron | 3 | G | A | 1.09 | T2D | [65] |
| 37 | rs6780569 | UBE2E2 | intergenic | 3 | G | A | 1.21 | T2D | [73] |
| 38 | rs6815464 | MAEA | intron | 4 | C | G | 1.13 | T2D | [63] |
| 39 | rs7656416 | MAEA | intron | 4 | C | T | 1.15 | T2D | [63,64] |
| 40 | rs6813195 | TMEM154 | intergenic | 4 | C | T | 1.08 | T2D | [70] |
| 41 | rs10010131 | WFS1 | intron | 4 | G | A | 1.14 | T2D | [4,89] |
| 42 | rs4689388 | WFS1 | nearGene-5 | 4 | T | C | 1.16 | T2D | [83] |
| 43 | rs6446482 | WFS1 | intron | 4 | G | C | 1.11 | T2D | [25,89,90] |
| 44 | rs1801214 | WFS1 | coding-missense | 4 | T | C | 1.13 | T2D | [25,89,90] |
| 45 | rs459193 | ANKRD55 | intergenic | 5 | G | A | 1.08 | T2D | [62] |
| 46 | rs702634 | ARL15 | intron | 5 | A | G | 1.06 | T2D | [70] |
| 47 | rs4457053 | ZBED3 | intron of ZBED3-AS1 | 5 | G | A | 1.08 | T2D | [25] |
| 48 | rs1048886 | C6orf57 | coding-missense | 6 | G | A | 1.54 | T2D | [91] |
| 49 | rs7754840 | CDKAL1 | intron | 6 | C | G | 1.17 | T2D | [25,27,59,60,66,73,92,93] |
| 50 | rs7756992 | CDKAL1 | intron | 6 | G | A | 1.20 | T2D | [27] |
| 51 | rs2206734 | CDKAL1 | intron | 6 | T | C | 1.20 | T2D | [25,27,59,60,66,73,92,93] |
| 52 | rs4712523 | CDKAL1 | intron | 6 | G | A | 1.27 | T2D | [25,27,59,60,66,73,83,92,93] |
| 53 | rs10946398 | CDKAL1 | intron | 6 | C | A | 1.12 | T2D | [25,27,59,60,66,73,92,93] |
| 54 | rs7766070 | CDKAL1 | intron | 6 | A | C | 1.23 | T2D | [25,27,59,60,66,73,92,93] |
| 55 | rs2244020 (rs9266650) | HLA-B | intergenic | 6 | G | A | 1.09 | T2D | [94] |
| 56 | rs1535500 | KCNK16 | coding-missense | 6 | T | G | 1.08 | T2D | [63] |
| 57 | rs3130501 | POU5F1-TCF19 | nearGene-5 | 6 | G | A | 1.07 | T2D | [70] |
| 58 | rs9505118 | SSR1-RREB1 | intron | 6 | A | G | 1.06 | T2D | [70] |
| 59 | rs9470794 | ZFAND3 | intron | 6 | C | T | 1.12 | T2D | [63] |
| 60 | rs17168486 | DGKB | intergenic | 7 | T | C | 1.15 | T2D | [62] |
| 61 | rs2191349 | DGKB/TMEM195 | intergenic | 7 | T | G | 1.06 | Fasting glucose, Homa B/T2D | [81] |
| 62 | rs6467136 | GCC1-PAX4 | intergenic | 7 | G | A | 1.11 | T2D | [63] |
| 63 | rs4607517 | GCK | intergenic | 7 | A | G | 1.07 | Fasting glucose/T2D/HOMA B | [81] |
| 64 | rs864745 | JAZF1 | intron | 7 | T | C | 1.10 | T2D | [4,80] |
| 65 | rs849134 | JAZF1 | intron | 7 | A | G | 1.13 | T2D | [25,80] |
| 66 | rs12113122 | JAZF1 | intron | 7 | G | C | 1.55 | T2D | [85] |
| 67 | rs972283 | KLF14 | intergenic | 7 | G | A | 1.07 | Reduced insulin sensitivity T2D | [25] |
| 68 | rs516946 | ANK1 | intron | 8 | C | T | 1.09 | T2D | [62] |
| 69 | rs515071 | ANK1 | intron | 8 | G | A | 1.18 | T2D Reduced beta-cell function | [62,64] |
| 70 | rs13266634 | SLC30A8 | coding-missense | 8 | C | T | 1.19 | T2D | [58] |
| 71 | rs11558471 | SLC30A8 | UTR-3 | 8 | A | G | 1.15 | Fasting glucose, HOMA B T2D | [25,58,59,81,87,92,93] |
| 72 | rs3802177 | SLC30A8 | UTR-3 | 8 | G | A | 1.26 | T2D | [25,58,59,81,87,92,93] |
| 73 | rs896854 | TP53INP1 | intron | 8 | T | C | 1.06 | T2D | [25] |
| 74 | rs10965250 | CDKN2A/2B | intergenic | 9 | G | A | 1.20 | T2D | [25,27,59,60,66,73,92,93] |
| 75 | rs2383208 | CDKN2A/2B | intergenic | 9 | A | G | 1.19 | T2D | [25,27,59,60,66,73,92,93] |
| 76 | rs7018475 | CDKN2A/2B | intergenic | 9 | G | T | 1.35 | T2D | [25,27,59,60,66,73,92,93] |
| 77 | rs564398 | CDKN2A/2B | intergenic | 9 | T | C | 1.12 | T2D | [25,27,59,60,66,73,92,93] |
| 78 | rs10757282 | CDKN2A/2B | intergenic | 9 | C | T | 1.14 | T2D | [25,27,59,60,66,73,92,93] |
| 79 | rs10811661 | CDKN2B | intergenic | 9 | T | C | 1.20 | T2D | [25,59,60,66,68,80,87,92] |
| 80 | rs7034200 | GLIS3 | intron | 9 | A | C | 1.03 | Fasting glucose/T2D/HOMA B | [81] |
| 81 | rs7041847 | GLIS3 | intron | 9 | A | G | 1.10 | T2D | [63,66] |
| 82 | rs10814916 | GLIS3 | intron | 9 | C | A | 1.11 | T2D | [63,66,81] |
| 83 | rs17584499 | PTPRD | intron | 9 | T | C | 1.57 | T2D | [95] |
| 84 | rs2796441 | TLE1 | intergenic | 9 | G | A | 1.07 | T2D | [62] |
| 85 | rs13292136 | TLE4 (CHCHD9) | intergenic | 9 | C | T | 1.11 | T2D | [25] |
| 86 | rs553668 | ADRA2A | UTR-3 | 10 | A | G | 1.42 | T2D | [96] |
| 87 | rs10885122 | ADRA2A | intergenic | 10 | G | T | 1.04 | Fasting glucose/HOMA B/T2D | [81] |
| 88 | rs12779790 | CDC123, CAMK1D | intergenic | 10 | G | A | 1.11 | T2D | [4,80] |
| 89 | rs11257655 | CDC123/CAMK1D | intergenic | 10 | C | T | 1.15 | T2D | [66,69,80] |
| 90 | rs10906115 | CDC123/CAMK1D | intergenic | 10 | A | G | 1.13 | T2D | [66,69,80] |
| 91 | rs10886471 | GRK5 | intron | 10 | C | T | 1.12 | T2D | [66] |
| 92 | rs5015480 | HHEX | intergenic | 10 | C | T | 1.13 | T2D | [25,58,59,92,93] |
| 93 | rs1111875 | HHEX/IDE | intergenic | 10 | C | T | 1.13 | T2D | [59] |
| 94 | rs7903146 | TCF7L2 | intronic/promoter | 10 | T | C | 1.35 | T2D, fasting glucose,.2 h glucose | [51] |
| 95 | rs4506565 | TCF7L2 | intron | 10 | T | A | 1.34 | Fasting glucose, HOMA B T2D | [25,27,51,58,59,60,61,80,87,92,93,97,98,99] |
| 96 | rs7901695 | TCF7L2 | intron | 10 | C | T | 1.37 | T2D | [25,27,51,58,59,60,61,80,87,92,93,97,98,99] |
| 97 | rs1802295 | VPS26A | UTR-3 | 10 | A | G | 1.08 | T2D | [65] |
| 98 | rs12571751 | ZMIZ1 | intron | 10 | A | G | 1.08 | T2D | [62] |
| 99 | rs11603334 | ARAP1 | UTR-5 | 11 | G | A | 1.13 | T2D fasting proinsulin levels/fasting glucose/ | [100] |
| 100 | rs1552224 | CENTD2 | intergenic | 11 | A | C | 1.14 | T2D | [25] |
| 101 | rs11605924 | CRY2 | intron | 11 | A | C | 1.04 | Fasting glucose/HOMA B/T2D | [81] |
| 102 | rs174550 | FADS1 | intron | 11 | T | C | 1.04 | Fasting glucose/T2D/HOMA B | [81] |
| 103 | rs2334499 | HCCA2 | intergenic | 11 | T | C | 1.35 | T2D | [101] |
| 104 | rs3842770 | INS-IGF2 | intron | 11 | A | G | 1.18 | T2D - African American | [94] |
| 105 | rs5219 | KCNJ11 | coding-missense | 11 | T | C | 1.14 | T2D | [54,59,60,87,99] |
| 106 | rs5215 | KCNJ11 | coding-missense | 11 | C | T | 1.14 | T2D | [54,59,60,87,99] |
| 107 | rs2237895 | KCNQ1 | intron | 11 | C | T | 1.45 | T2D | [71] |
| 108 | rs231362 | KCNQ1 | intron | 11 | G | A | 1.08 | T2D | [25] |
| 109 | rs163184 | KCNQ1 | intron | 11 | G | T | 1.22 | T2D | [62,71] |
| 110 | rs2237892 | KCNQ1 | intron | 11 | C | T | 1.25 | Reduced beta-cell function T2D | [25,71,72,92,95] |
| 111 | rs10501320 | MADD | intron | 11 | G | C | 1.01 | T2D fasting proinsulin levels/fasting glucose | [100] |
| 112 | rs10830963 | MTNR1B | intron | 11 | G | C | 1.09 | T2D | [102] |
| 113 | rs1387153 | MTNR1B | intergenic | 11 | T | C | 1.09 | Reduced beta-cell function T2D | [25,95,102] |
| 114 | rs7138803 | BCDIN3D/FAIM2 | intergenic | 12 | A | G | 1.11 | BMI/obesity T2D | [88,103] |
| 115 | rs11063069 | CCND2 | intergenic | 12 | G | A | 1.12 | T2D | [62] |
| 116 | rs1153188 | DCD | intergenic | 12 | A | T | 1.08 | T2D | [80] |
| 117 | rs1531343 | HMGA2 | intron of pseudogene | 12 | C | G | 1.10 | T2D | [25] |
| 118 | rs9668162 | HMGA2 | intron | 12 | G | C | 1.26 | T2D | [85] |
| 119 | rs7305618 | HNF1A | intergenic | 12 | C | T | 1.14 | T2D | [25,68] |
| 120 | rs35767 | IGF1 | nearGene-5 | 12 | G | A | 1.04 | Fasting insulin/T2D/HOMA IR | [81] |
| 121 | rs10842994 | KLHDC5 | intergenic | 12 | C | T | 1.10 | T2D | [62] |
| 122 | rs4275659 | MPHOSPH9 | intron | 12 | C | T | 1.06 | T2D | [70] |
| 123 | rs7957197 | OASL/TCF1/HNF1A | intron of OASL | 12 | T | A | 1.07 | T2D | [25] |
| 124 | rs7961581 | TSPAN8, LGR5 | intergenic | 12 | C | T | 1.09 | T2D | [4,80] |
| 125 | rs9552911 | SGCG | intron | 13 | G | A | 1.63 | T2D | [104] |
| 126 | rs1359790 | SPRY2 | intergenic | 13 | G | A | 1.15 | T2D | [69] |
| 127 | rs2028299 | AP3S2 | UTR-3 | 15 | C | A | 1.10 | T2D | [65] |
| 128 | rs7172432 | C2CD4A/B | intergenic | 15 | A | G | 1.14 | Reduced beta-cell function, T2D | [73] |
| 129 | rs7178572 | HMG20A | intergenic | 15 | A | G | 1.09 | lean T2D | [65,93] |
| 130 | rs7177055 | HMG20A | intergenic | 15 | A | G | 1.08 | T2D | [62] |
| 131 | rs8042680 | PRC1 | intron | 15 | A | C | 1.07 | T2D | [25] |
| 132 | rs7403531 | RASGRP1 | intron | 15 | T | C | 1.10 | T2D | [66] |
| 133 | rs4502156 | VPS13C/C2CD4A/B | intergenic | 15 | T | C | 1.07 | fasting proinsulin levels T2D | [100] |
| 134 | rs11634397 | ZFAND6 | intergenic | 15 | G | A | 1.06 | T2D | [25] |
| 135 | rs7202877 | BCAR1 | intergenic | 16 | T | G | 1.12 | T2D | [62] |
| 136 | rs8050136 | FTO | intron | 16 | A | C | 1.17 | Increased BMI, reduced insulin sensitivity, T2D | [25,60,61,80,87,93,99,105] |
| 137 | rs9939609 | FTO | intron | 16 | A | T | 1.25 | T2D (obese) | [25,60,61,80,87,93,99,105] |
| 138 | rs11642841 | FTO | intron | 16 | A | C | 1.13 | T2D | [25,60,61,80,87,93,99,105] |
| 139 | rs4430796 | HNF1B | intron | 17 | G | A | 1.19 | Reduced beta-cell function T2D | [66,106,66,107,108] |
| 140 | rs7501939 | HNF1B | intron | 17 | T | C | 1.09 | T2D | [106] |
| 141 | rs391300 | SRR | intron | 17 | G | A | 1.28 | T2D | [95] |
| 142 | rs4523957 | SRR | nearGene-5 | 17 | T | 1.27 | T2D | [95] | |
| 143 | rs8090011 | LAMA1 | intron | 18 | G | C | 1.13 | lean T2D | [93] |
| 144 | rs17782313 | MC4R | intergenic | 18 | C | T | 1.06 | BMI/T2D | [88,103] |
| 145 | rs12970134 | MC4R | intergenic | 18 | A | G | 1.08 | T2D/BMI/waist circumference/insulin resistance | [62,109] |
| 146 | rs3794991 | GATAD2A/CILP2 | intron, intergenic | 19 | T | C | 1.12 | T2D | [62,85] |
| 147 | rs8108269 | GIPR | intergenic | 19 | G | T | 1.05 | T2D | [62] |
| 148 | rs3786897 | PEPD | intron | 19 | A | G | 1.10 | T2D | [63] |
| 149 | rs10401969 | SUGP1/CILP2 | intron | 19 | C | T | 1.13 | T2D | [62,85] |
| 150 | rs6017317 | FITM2-R3HDML-HNF4A | intergenic | 20 | G | T | 1.09 | T2D | [63] |
| 151 | rs4812829 | HNF4A | intron | 20 | A | G | 1.09 | T2D | [65] |
| 152 | rs5945326 | DUSP9 | intergenic | X | A | G | 1.27 | T2D | [25] |
| 153 | rs12010175 | FAM58A | intron | X | G | A | 1.21 | T2D | [66] |
| N | SNPs | Gene/Nearest Gene | Gene Location | Chr | Effect Allele | Other Allele | Effect | TRAIT | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | rs9727115 | SNX7 | intron | 1 | G | A | 0.0133 | fasting proinsulin levels adjusted for fasting glucose | [100] |
| 2 | rs2785980 | LYPLAL1 | intergenic | 1 | T | C | 0.017 | fasting insulin | [110] |
| 3 | rs4675095 | IRS1 | intron | 2 | A | T | −0.006/−002 | fasting glucose/HOMA-IR | [81] |
| 4 | rs2943634 | IRS1 | intergenic | 2 | C | A | 0.025 | fasting insulin, CAD | [110] |
| 5 | rs1371614 | DPYSL5 | intron | 2 | T | C | 0.022 | Fasting glucose | [110] |
| 6 | rs11920090 | SLC2A2 | intron | 3 | T | A | 0.02 | Fasting glucose/HOMA B/HBA1C | [81] |
| 7 | rs17046216 | MSMO1 | intron | 4 | A | T | 0.18; 0.19 | Fasting Insulin; insulin resistance | [111] |
| 8 | rs4691380 | PDGFC | intron | 4 | C | T | 0.021 | fasting insulin | [110] |
| 9 | rs6235 | PCSK1 | coding-missense | 5 | G | C | 0.0394/−0.014 | fasting proinsulin levels/Fasting glucose | [100] |
| 10 | rs13179048 | PCSK1 | intergenic | 5 | C | A | 0.018 | Fasting glucose | [110] |
| 11 | rs4646949 | TAF11 | nearGene-3 | 6 | T | G | 0.020 | fasting insulin | [110] |
| 12 | rs6943153 | GRB10 | intron | 7 | C | T | 0.0154 | FG, FI | [26] |
| 13 | rs4841132 | PPP1R3B | intergenic | 8 | A | G | 0.030 | Fasting glucose | [110] |
| 14 | rs7077836 | TCERG1L | intergenic | 10 | T | C | 0.28; 0.34 | Fasting Insulin ; insulin resistance | [111] |
| 15 | rs7944584 | MADD | intron | 11 | A | T | 0.021 | Fasting proinsulin/Fasting glucose/Homa B | [81] |
| 16 | rs10838687 | MADD | intron | 11 | T | G | 0.0253 | fasting proinsulin levels | [100] |
| 17 | rs1483121 | OR4S1 | intergenic | 11 | G | A | 0.015 | Fasting glucose | [110] |
| 18 | rs2074356 | HECTD4/C12orf51 | intron | 12 | 1-h plasma glucose | [112] | |||
| 19 | rs2293941 | PDX1 - AS1 | intron | 13 | A | G | 0.016 | Fasting glucose | [110] |
| 20 | rs17271305 | VPS13C | intron | 15 | G | A | 0.07 | 2hr glucose/2-h insulin, adjusted for 2-h glucose | [86] |
| 21 | rs1549318 | LARP6 | intergenic | 15 | T | C | 0.0192 | fasting proinsulin levels | [100] |
| 22 | rs4790333 | SGSM2 | intron | 17 | T | C | 0.0154 | fasting proinsulin levels | [100] |
| 23 | rs10423928 | GIPR | intron | 19 | A | T | 2 h glucose/Insulinogenic index/AUCins/gluc/2-h insulin, adjusted for 2 h glucose/T2D | [86] | |
| 24 | rs6048205 | FOXA2/LINC00261 | intergenic/nearGene-5 | 20 | A | G | 0.029 | Fasting glucose | [110] |
Figure 2.
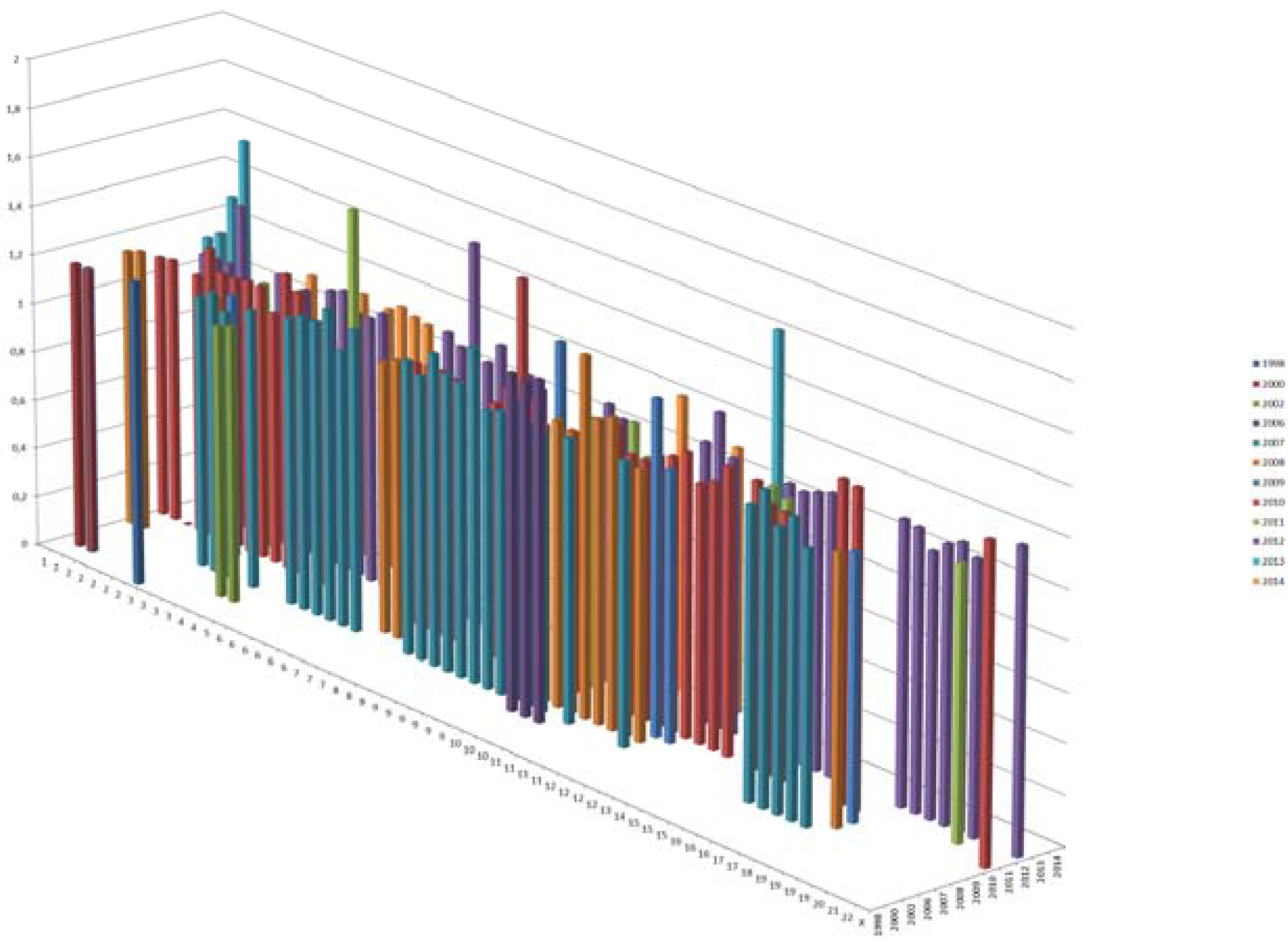
T2D risk variants. X axis shows the chromosomal location, Y shows the effect sizes and Z axis shows the year of discovery. Only 1 risk variant was reported in 1998; there were 2 in 2002, and today, we have a total of ~153 T2D variants.
Figure 2.
T2D risk variants. X axis shows the chromosomal location, Y shows the effect sizes and Z axis shows the year of discovery. Only 1 risk variant was reported in 1998; there were 2 in 2002, and today, we have a total of ~153 T2D variants.
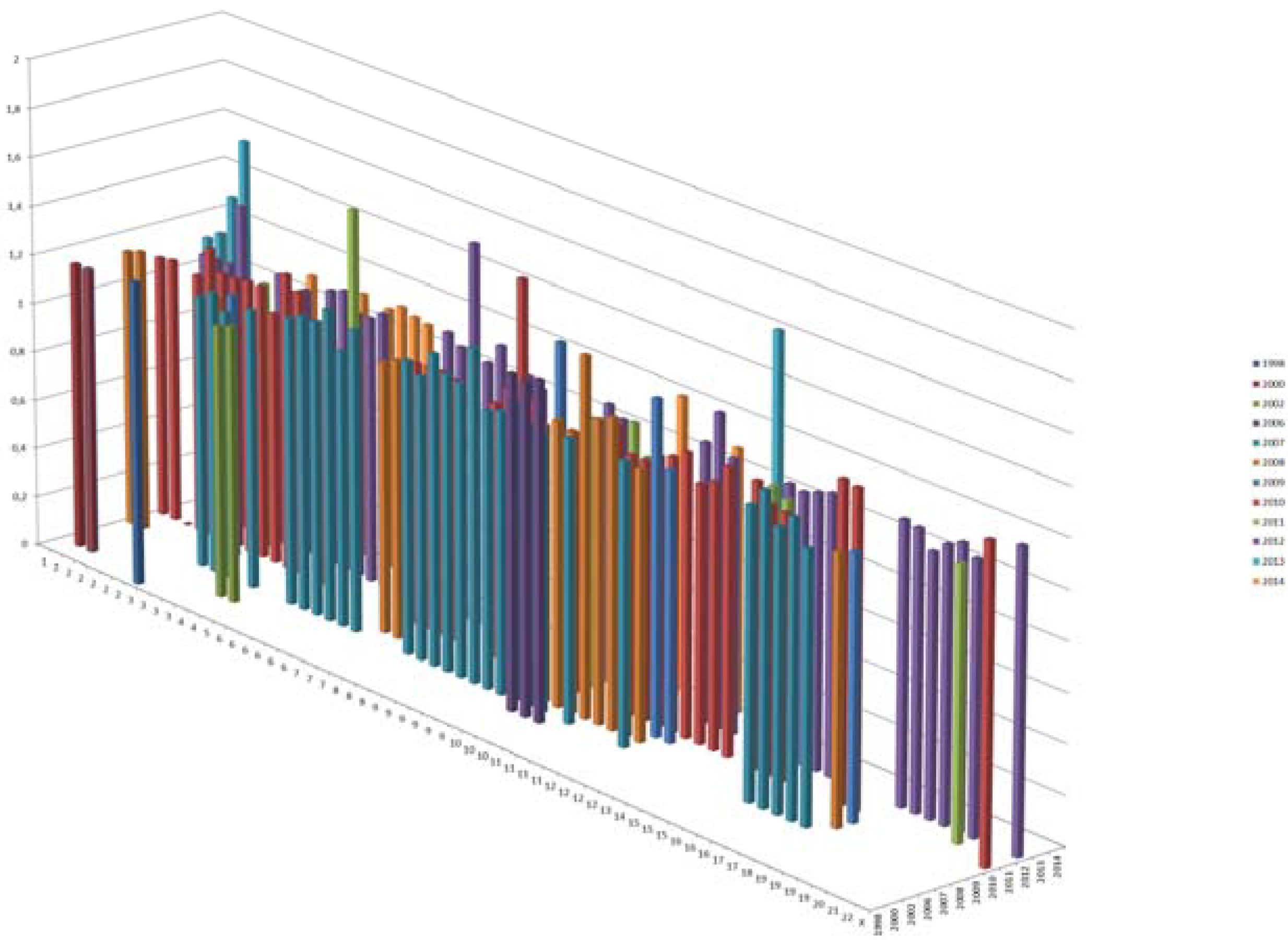
Figure 3.
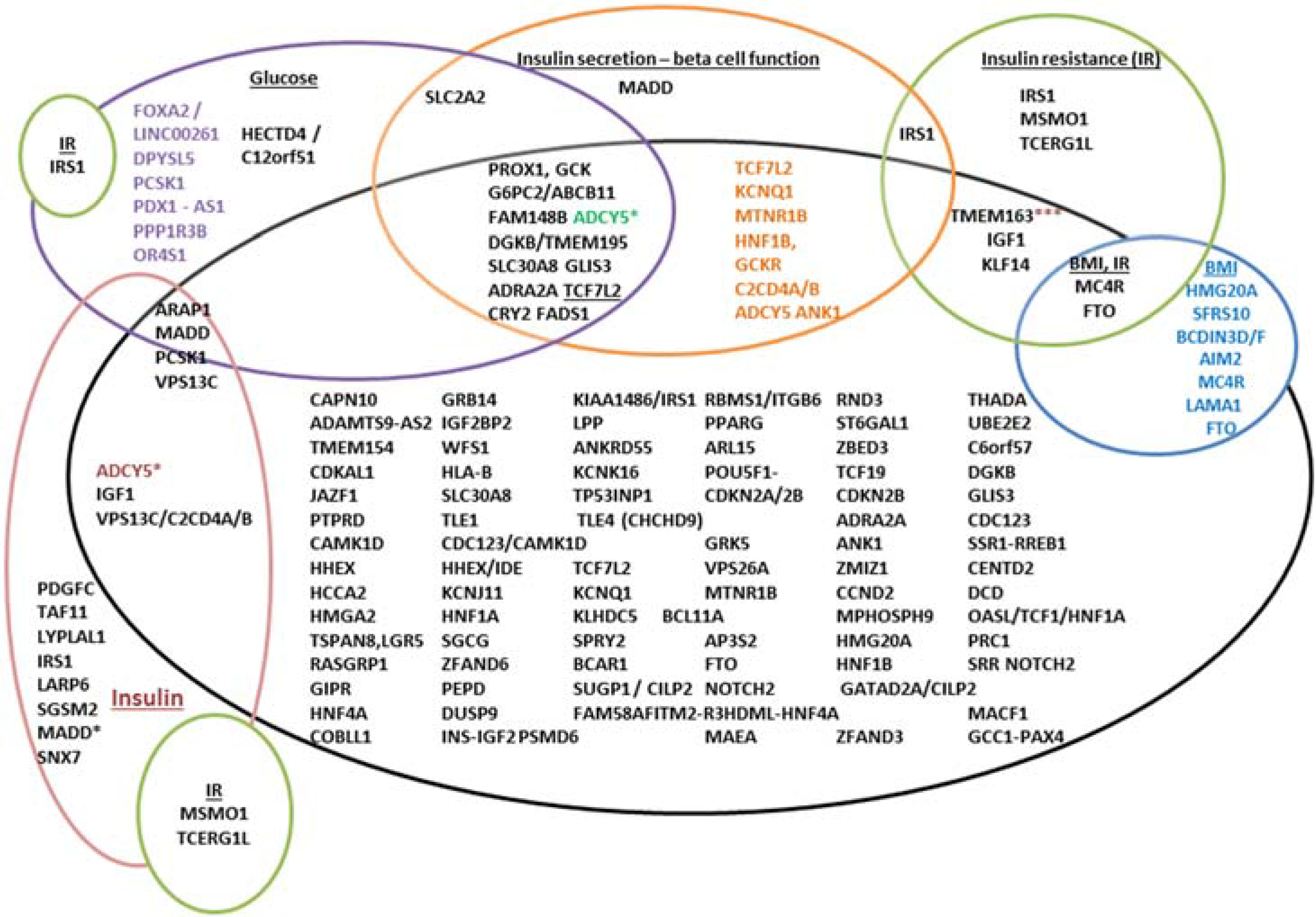
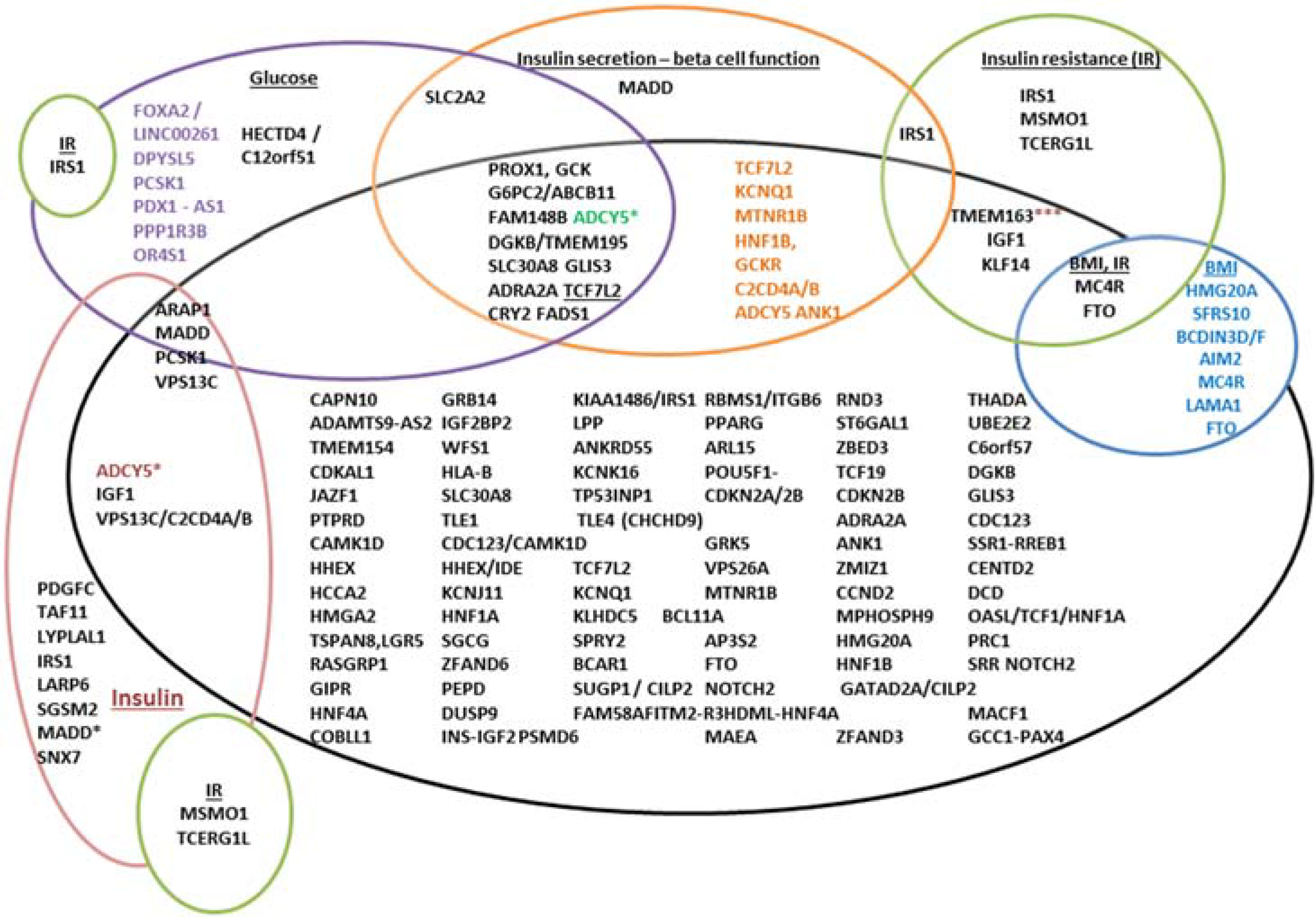
T2D and glycemic trait associated variants. The variants are represented by gene names here, which could indicate that the location is present either in the gene, or in the vicinity of the gene. The black circle represents T2D, and the gene names in black in this represent variants only associated with T2D. The overlapping circles indicate additional reporting associations for that variant for instance, TCF7L2, KCNQ1, MTNR1B etc., are associated with T2D and also with beta-cell dysfunction. An ADCY5 variant is associated with 2 h insulin adjusted for 2 h glucose; 2 h glucose/T2D (in brown) *** variants in TMEM163 are also associated with fasting insulin, TCF7L2—associated with fasting and 2 h glucose and MADD variants associated with fasting proinsulin, fasting glucose and HOMA-B.
Figure 3.
T2D and glycemic trait associated variants. The variants are represented by gene names here, which could indicate that the location is present either in the gene, or in the vicinity of the gene. The black circle represents T2D, and the gene names in black in this represent variants only associated with T2D. The overlapping circles indicate additional reporting associations for that variant for instance, TCF7L2, KCNQ1, MTNR1B etc., are associated with T2D and also with beta-cell dysfunction. An ADCY5 variant is associated with 2 h insulin adjusted for 2 h glucose; 2 h glucose/T2D (in brown) *** variants in TMEM163 are also associated with fasting insulin, TCF7L2—associated with fasting and 2 h glucose and MADD variants associated with fasting proinsulin, fasting glucose and HOMA-B.
4.1.4. Rare Variants
Rare variants are more recent and therefore more likely to be those that arose lately in an extended pedigree. Of course, natural selection removes the more deleterious variants before they reach a high frequency; risk alleles for diseases should thus be enriched at lower frequencies. The idea that there are unique rare variant combinations in families and their role in disease etiology is referred to as “clan genomics” [113]. Current data suggests that the combined effects of rare and common variants contribute in varying degrees to disease causation, even linkage; in fact, rare alleles may explain the majority of heritability [114]. Next-generation sequencing provides even denser coverage of genetic variation rendering detection of causal rare variants more feasible. Whole genome sequencing (WGS) of 2,630 Icelanders and imputation into 11,114 Icelandic cases and 267,140 controls followed by testing in Danish and Iranian samples revealed variants in PAM and PDX1 as risk of T2D [115] (Table 3). Array-based genotyping and exome sequencing on a small founder population from Greenland revealed a nonsense p.Arg684Ter variant (allele frequency of 17%) in TBC1D4 associated with higher concentrations of 2 h glucose and serum insulin [116]. Whole-exome sequencing in a Latino population revealed a rare missense variant in HNF1A (c.1522G > A [p.E508K]) was associated with type 2 diabetes prevalence [117]. Additionally, rare variants associated with glycemic traits were discovered through exome sequencing of 9,717 individuals from the METSIM study, Finland [118] (Table 3).
| N | SNPs | GENE/Nearest Gene | Gene Location | Chr | Refs. |
|---|---|---|---|---|---|
| 1 | rs35658696 | PAM | coding-missense | 5 | [118] |
| 2 | rs78408340 | PAM | coding-missense | 5 | [118] |
| 3 | rs36046591 | PPIP5K2 | coding-missense | 5 | [118] |
| 4 | p.Lys34Serfs*50 | SLC30A8 | coding-missense | 8 | [119] |
| 5 | p.Arg138* | SLC30A8 | coding-missense | 8 | [119] |
| 6 | rs3824420 | KANK1 | coding-missense | 9 | [118] |
| 7 | rs505922 | ABO | Intronic | 9 | [118] |
| 8 | rs60980157 | GPSM1 | coding-missense | 9 | [118] |
| 9 | p.Leu5Val (20) | ATG13 | coding-missense | 11 | [118] |
| 10 | p.Ile131Val (1) | ATG13 | coding-missense | 11 | [118] |
| 11 | p.Gln249Pro (3) | ATG13 | coding-missense | 11 | [118] |
| 12 | p.Arg392Trp (1) | ATG13 | coding-missense | 11 | [118] |
| 13 | p.Leu427Gln (3) | ATG13 | coding-missense | 11 | [118] |
| 14 | p.Gly434Arg (488) | ATG13 | coding-missense | 11 | [118] |
| 15 | p.X406Gly (200) | ATG13 | coding-missense | 11 | [118] |
| 16 | rs35233100 | MADD | coding-missense | 11 | [118] |
| 17 | p.Arg279Cys (324) | TBC1D30 | coding-missense | 12 | [118] |
| 18 | p.Pro746Leu (427) | TBC1D30 | coding-missense | 12 | [118] |
| 19 | c.1522G > A [p.E508K] | HNF1A | coding-missense | 12 | [117] |
| 20 | rs76895963 | CCND2 | intergenic | 12 | [115] |
| 21 | rs75615236 | CCND2 | intergenic | 12 | [115] |
| 22 | rs150781447 | TBC1D30 | coding-missense | 12 | [118] |
| 23 | rs2650000 | HNF1A | Intergenic | 12 | [118] |
| 24 | Chr. 13: g.27396636delT | PDX1 | coding-missense | 13 | [119] |
| 25 | p.Tyr416Cys (78) | SGSM2 | coding-missense | 17 | [118] |
| 26 | p.Thr789Pro (3) | SGSM2 | coding-missense | 17 | [118] |
| 27 | p.Val996Ile (236) | SGSM2 | coding-missense | 17 | [118] |
| 28 | rs61741902 | SGSM2 | coding-missense | 17 | [118] |
4.1.5. Structural Variants
Most studies to date have been limited to SNP leaving structural polymorphisms relatively unexplored. However, since common structural variants are likely to be tagged by surrounding SNPs, they are unlikely to explain a large proportion of missing heritability. A recent study identified a common copy number variation (CNV), CNVR5583.1 (TSPAN8), as associated with T2D [120]. This association could be convincingly replicated by previously typed SNPs that tag the CNV [80].
4.1.6. Protective Variants
The average T2D risk variant frequency is 54% in the general population, which raises the question: Is T2D the default condition? If so, then does carrying protective variants makes a difference in the disease susceptibility? Studies have been performed to address this question but included controls that, despite a clustering of risk factors for T2D, have escaped the disease. A rare (0.66%) loss of function mutation (R138X) was detected in the SLC30A8 gene in the Botnia region from Finland and subsequently replicated in a massive effort applying the Exome chip to >150,000 individuals from other European countries. Also, the DeCode group had identified another LoF mutation, a frameshift mutation which also was enriched in the non-diabetic Icelandic population. The SLC30A8 gene encodes the islet zinc transporter 8 with a putative effect on insulin secretion. Notably, a common variant in the same gene increases susceptibility to T2D, whereas autoantibodies to T1D predispose to T1D.
Collectively, carriers of these protein-truncating mutations have a 65% lower risk of T2D [119]. Other studies based on Icelandic, Danish and Iranian populations identified a low frequency variant in CCND2 that reduced T2D risk by half [115]. Moreover, variants in TCF2 were found to be protective against T2D [106]. It is also likely that the more recent variants that are only a few generations old segregate in families and could be detected though sequencing in families.
4.1.7. The Genetic Architecture of T2D
Genetic architecture of a complex phenotype is defined by the number, frequencies and effect sizes of causal alleles. Many hypotheses have been proposed to define the genetic architecture of T2D: one hypothesis suggested that the unexplained heritability lies in a large number of common variants with low additive effects and that the disease represents the extremes of a normal distribution [121]. Another proposed that rare alleles might be responsible for effects observed with common variants (synthetic associations) and explain a majority of the heritability [113,122,123]. One argument against common and more so against rare variants is that they would have been removed from the population by natural selection [47]; however, this is not a valid argument for a disease like T2D whereby the penetrance of the genetic effect depends strongly on interactions with the environment, especially since this environment has changed in recent years and the genetic risk variants could have been neutral or even beneficial before the introduction of the Westernized lifestyle.
While extreme models are excluded by the present data based on epidemiological, linkage and GWAS, models wherein rare variants explain a little (<25%) or a lot (>80%) of heritability remain consistent [124]. Large next-generation sequencing studies in families will hopefully answer the questions about the role of rare variants in complex diseases. There is already substantial evidence for parent-of-origin effects on T2D risk, and studies are ongoing to explore this further. Structural polymorphisms and microRNAs add a further layer of complexity and have not yet been exhaustively studied.
The genetic architecture could also be influenced by gene-gene interactions (epistasis) where rare, variants with high penetrance, could act jointly with common alleles to increase risk of disease. The extent of allelic heterogeneity seems to be less pronounced for the common form of T2D than for monogenic forms like MODY. Moreover, there could be differences in the genetic predisposition to T2D due to phenotypic heterogeneity within T2D cases. For instance, lean T2D cases are likely to carry a disproportionately high load of type 2 diabetes risk alleles [93].
4.2. Functions of Associated Genes
Most identified diabetes loci have not been mechanistically tied to the disease. While loci are commonly referred to by the names of genes located close to them, only a few are close to strong biological candidates, e.g., the melatonin receptor (MTNR1B) and the insulin receptor substrate-1 (IRS1). For others, like TCF7L2 and GIPR, the evidence is quite strong that an intronic SNP is the causal SNP. Melatonin receptor 1B (MTNR1B) has been associated with both fasting glucose and T2D risk [102,125,126]. Melatonin works as a chronobiotic factor, adjusting the timing of the biological clock. Its receptors are present in the pancreas and melatonin is proposed to contribute to the nocturnal lowering of insulin in humans. The MTNR1B risk genotype is associated with impaired early insulin release to both oral and intravenous glucose and insulin secretion deteriorates over time in the risk allele carriers [125]. The proposed mechanism by which MTNR1B polymorphism could predispose to T2D involves altered expression of MTNR1B in pancreatic β-cells leading to decreased cAMP/cGMP concentrations via G proteins and, thereby, impaired insulin secretion.
The insulin receptor substrate 1 (IRS1) gene encodes a protein that mediates insulin’s control of various cellular processes by transmitting signals from the insulin receptor to intracellular signaling pathways. The C allele of rs2943641 has been shown to be associated with insulin resistance and increased risk of diabetes. The genetic variant causes reduced basal levels of IRS1 protein and decreased insulin induction of IRS1-associated phosphatidylinositol-3-hydroxykinase activity in human skeletal muscle biopsies [83].
TCF7L2 is a transcription factor playing an important role in the Wnt signaling pathway. The risk allele is associated with decreased insulinogenic index and lower disposition index, suggesting a reduced capacity for insulin secretion in relation to insulin sensitivity. Since it was identified as a diabetes gene, it has been shown to be important for several vital functions in the pancreatic islet, including pancreas development, determination of beta-cell mass, maintenance of the secretory function of mature beta cells, and regulation of insulin production and processing [127,128].
The incretin hormone GIP (glucose-dependent insulintropic polypeptide) promotes pancreatic β-cell function by potentiating insulin secretion and β-cell proliferation. The GIP receptor (GIPR) locus showed association to postprandial insulin levels in a meta-analysis performed by the MAGIC consortium but was surprisingly not associated with risk of diabetes in the DIAGRAM+ study [25,86]. The reason seems to be that the same variant results in decreased BMI, which neutralizes the effect of the SNP on risk of T2D. GIP influences expression of the inflammatory cytokine OPN in islets, which in turn has protective effects on β-cell proliferation and potentially apoptosis [129].
Many of the other identified loci can be sub-grouped based on their association with other phenotypes with a key role in T2D etiology. Exploration of the effects of T2D-associated variants on glucose and insulin traits in non-diabetic populations has shown that most of the known loci act through an effect on insulin secretion rather than insulin resistance (Table 1) [25,130,131,132].
Fasting glucose-raising alleles of the MADD, GIPR, GCK, FADS, DGKB, PROX1, TCF7L2, SLC30A8 and C2CD4B loci have all been associated with either abnormal insulin processing or secretion, whereas GCKR and IGF1 are associated with OGTT-based disposition indices and β-cell function [131]. The DIAGRAM+ consortium observed that three loci (TCF7L2, ARAP1 and CDKAL1) were associated with reduced fasting insulin also suggestive of β-cell dysfunction, whereas the T2D risk alleles at PPARG, FTO, IRS1 and KLF14 were associated with higher fasting insulin, indicating a primary effect on insulin action [25].
Translational Studies
The ADRA2A (adrenergic receptor alpha 2) locus was recently identified as a T2D risk locus after first having been positionally mapped in congenic GK rats where it was associated with impaired insulin granule docking and reduced β-cell exocytosis [130]. Human carriers of the ADRA2A risk variant (rs553668) have reduced fasting insulin and decreased insulin secretion as a consequence of increased expression of the ADRA2A receptor in pancreatic islets. It is well known that epinephrine excess can suppress insulin secretion and cause diabetes. The α2AAR antagonist yohimbine enhances insulin release in vitro in islets from organ donors carrying the risk allele to levels similar to those in non-risk carriers. A randomized clinical study was performed blocking α2AAR pharmacologically to increase insulin secretion T2D patients with the rs553668 risk allele. Yohimbine administration enhanced 30 min insulin, corrected insulin response and disposition index in the risk group, making secretion similar to patients carrying the low-risk allele. Insulin secretion defect in patients carrying the ADRA2A risk genotype could be corrected by α2AAR antagonism [133]. This demonstrated the potential application of genetic risk variants to guide therapeutic interventions that target the underlying pathophysiology: one step closer to individualized medicine.
5. Pitfalls
5.1. Heritability Estimates
Heritability parameters facilitate understanding the genetic architecture of complex traits such as T2D. However, the >80 loci identified explain less than 20% of the heritability of T2D. There are many possible explanations for the missing heritability, including assumptions made about the genetic architecture of the disease and the definitions of heritability. The estimations of heritability explained assumes that only additive affects determine disease risk and that the risk follows the liability threshold model, i.e., that the genetic and environmental effects sum up to form a normal distribution of liability and that disease arises in individuals surpassing a certain threshold in the distribution [134]. If these assumptions are not true, the estimate of heritability explained will not be correct.
Intrauterine effects can also affect heritability estimates because monozygotic twins are often monochorionic which results in growth retardation compared to dizygotic twins, and low birth weight is associated with increased risk of T2D later in life. Furthermore, there could be other explanations to the “missing heritability” problem. However, one should keep in mind that heritability can only be estimated from the most recent generations for which information on affection status is available, whereas most of the variants studied thus far are ancestral variants hundreds of generations old. We do not know whether these ancestral variants (that have modest effects and have escaped purifying selection) can really explain the diabetes epidemic we see in the most recent generations or whether this can be ascribed to rare variants with stronger effects.
Moreover, heritability estimates are based on “top” SNPs from GWAS associations; novel methods have been proposed which take into account the (i) scale 0–1 scale as opposed to liability (2) ascertainment bias and (3) quality control of the GWAS SNPs. Estimating the proportion of variance explained by all SNPs in GWAS as opposed to only the most significantly associated SNPs could result in a more detailed estimate of heritability [135]. Applying an approach that considers all SNPs on the chip could in fact explain a much larger proportion of the “narrow-sense” heritability (>50%) supporting the existence of numerous yet unidentified loci with smaller effects [25,136,137].
5.2. Parent of Origin Effects
The risk of T2D in offspring is greater if the mother has T2D compared to if the father is affected in contrast to T1D, where the risk of T1D in offspring is greater if the father is affected [138,139]. Sex-specific parental effects have been reported for insulin response to the oral glucose load, with male offspring of diabetic mothers showing the lowest insulin values, as well as influencing HDL concentrations [138]. One potential explanation for this could be preferential parental specific transmissions of risk alleles to offspring, which is often associated with DNA methylation and imprinting. Epigenetic modifications have the potential to be stable and heritable across cell divisions and [140,141] manifest as parent-of-origin effects. A large-scale family-based study on the Icelandic population determined that variants in KCNQ1 and KLF14 show stronger effects on T2D when the risk allele is transmitted from the mother than from the father [101,142] and was replicated in later studies [143] including our own.
The conflict hypothesis suggests that imprinting arose due to a genomic tug-of-war between mothers and fathers over the use of maternal resources in the fetus. The paternal imprinting maximizes the utilization of intrauterine resources to the offspring which would increase his evolutionary fitness whereas the maternal imprinting tries to minimize this in order to conserve it for her future offspring [144]. Conversely, the co-adaptation hypothesis suggests that imprinted genes coevolve to optimize parental care of offspring. While there is insufficient evidence to support either theory, the significant role of imprinting in defining paternal and maternal effects has nevertheless been consistently established [145].
The intrauterine environment plays a significant role in determining fetal programming. It has been shown that poor nutrition can affect fetal growth, can produce permanent changes in glucose-insulin metabolism, and often results in low birth weight [146] This can induce permanent changes in metabolism and affect chronic disease susceptibility as proposed by the DoHAD hypothesis [147]. If this intrauterine programming results in a reduced β-cell mass, it could predispose to diabetes later in life when the insulin requirements increase as a consequence of obesity resulting in insulin resistance. KCNQ1 could represent an example of fetal programming wherein the maternally expressed gene was mono-allelically expressed in fetal tissues and bi-allelically expressed in adult tissues [148].
Dissection of genetic parent-of-origin effects requires genotype data from families and only heterozygous parents are informative yielding reduced power for relatively rare variants. However, long-range phasing and imputation methods allow for predicting genotypes with a great likelihood, thus making this a valuable method to find “surrogate” parents even if DNA exists from only a few family members. When the paternal and maternal allele have effects in opposite directions—as, for instance in a situation where the maternal allele could confer risk while the paternal allele could be protective—such an association would be almost impossible to detect in a traditional case-control GWAS. However, some novel POE detection methods allow detection of imprinting effects from differences in the phenotypic variance of heterozygotes in very large case-control studies [149]. Parent-of-origin effects could explain a large portion of the missing heritability and must be taken into consideration in investigations of genetic T2D susceptibility.
5.3. Gene−Gene and Gene−Environment Interactions
Gene−gene interactions, or epistasis, have been suggested as a possible explanation for difficulties in replicating genetic association in complex diseases [150]. The standard statistical methods used in association studies are usually limited to analysis of single marker effects and thereby do not account for interactions between markers. Previous attempts to study epistasis in complex diseases have focused on interactions between candidate regions [151,152]. However, the recent abundance of GWAS data has made a comprehensive search across the genome more feasible. Some studies have attempted to account for epistasis in GWAS using a two-step approach in which significant SNPs are tested against each other or against all other SNPs in the study with variable results [153,154]. The main problem when studying epistasis is power, since interaction between loci with modest effects is difficult to detect without extremely large sample sizes. However, some studies have pointed at novel tests to increase power [155]. Thorough studies in diabetes addressing epistasis using this approach are missing. Furthermore, a recent paper by Eric Lander and co-workers provided compelling evidence that gene−gene interaction can also contribute to missing heritability by causing “phantom heritability” that inflates the estimated narrow sense heritability of the trait [156].
Gene−environment interactions are equally difficult to study but are likely to play an important role in T2D development. The epidemic of T2D only dates back 50 years, and it is quite obvious that during this period that only the environment, and not the genes, have changed. However, the genetic architecture determines our response to the environment. Genetic variants could affect specific metabolic processes to make an individual more susceptible to the harmful effects of a poor diet but also personality traits that make an individual more or less likely to over-consume and live a sedentary lifestyle. It will however be a formidable task to identify the environmental triggers for most of the genetic variants increasing susceptibility to diabetes as this will require very large studies with precise information on diet, exercise, energy expenditure, etc.
5.4. Epigenetics
The environment can also influence the expression of the genome, and ultimately the phenotype, via the epigenome. Even though the DNA sequence is not changed, the phenotype is altered by epigenetic modifications of gene expression by mechanisms including methylation of DNA, posttranslational modification of histones, or activation of microRNAs. Changes to the phenotype can be at the level of the cell, tissue, or whole organism.
It is tempting to speculate that environmental factors such as diet and exercise can change the level of DNA methylation and thereby cause changes in gene expression, but evidence that DNA methylation contributes to the increase in T2D is still lacking. Epigenetic mechanisms may however play a role in progression of the disease by inducing glucotoxicity in islets and predispose to diabetic complications [157]. Elevated glucose is a prerequisite to this condition and it is well established that cells can memorize changes in glucose concentrations. For example, two large studies, the UKPDS and DCCT studies, showed that an initial good metabolic control was associated with reduced frequency of diabetic complications decades later. The advanced “metabolic memory” hypothesis suggests that this is because glucose can induce histone modifications in endothelial cells that can be remembered long after [158].
5.5. Non-Coding RNAs−microRNAs
Non-coding RNAs have recently emerged as important regulators of gene expression and function. MicroRNAs (miRNAs) naturally regulate programs of gene expression. Altered miRNA function has been shown to contribute to human disease, and manipulation of specific miRNAs is now being explored as a novel therapeutic modalities [159]. The efficiency of miRNAs binding to target transcripts depends on both the sequence and the intra-molecular structure of the transcript. SNPs can contribute to alterations in the structure of regions flanking them, thereby influencing the accessibility for miRNA binding. Several studies have implicated miRNAs in diabetes and inflammation and common SNPs change the target sequence of miRNAs in several T2D susceptibility loci (http://miracle.igib.res.in/dbSMR) [160,161]. Other forms of non-coding RNAs, such as piRNAs (PIWI-interacting RNAs), snoRNAs (small nucleolar RNAs), lincRNAs (long intergenic non-coding RNAs), and lncRNAs (long non-coding RNAs), might also contribute to the development of diabetes. For example, the CDKN2A/B region on chromosome 9 is associated with T2D, as well as cardiovascular disease and a number of other disorders. This region harbors an lncRNA, ANRIL (non-protein coding CDKN2B-AS1 CDKN2B antisense RNA 1), which can potentially modify and explain some of these associations [162].
5.6. The T1D-T2D Paradox
T1D and T2D can be considered two extremes of the diabetes spectrum and share a few similarities in manifestation of underlying physiology including hyperglycemia, insulin deficiency and development of complications. However, the genetics of T1D and T2D vastly differ, with very few T2D susceptibility loci showing an association with T1D. Notable exceptions include the PPARG Pro12Ala variant, MTNR1B, HNF1A, GLIS3, 6q22.32 and novel loci near the MHC, which harbor the HLA class II genes associated with about half the T1D risk [70,163,164,165]. Based on these studies, the mechanisms underlying T1D and T2D appear to be intrinsically distinct. The distribution of T2D risk SNPs should be more random in a T1D patient; however, this does not seem to be the case. The strongest T2D−SNP in the TCF7L2 gene almost seems to protect from T1D. T1D risk variants for BCAR1, GLIS3 and RAD51L1 were protective for T2D whereas for those in C6orf173, COBL and C10orf59, the effects were coincident [62]. Also, it has been reported that APOC3 haplotypes increase risk of T1D; however, the same variants increase risk of T2D in lean carriers while having a protective effect in overweight carriers [166]. Common variants in SLC30A8 are associated with an increased risk of T2D, and rare variants with a protective effect [58,119]. Puzzlingly, SLC30A8 was also found to be a major autoantigen eliciting 60%–80% autoantibodies in new-onset T1D patients [167].
Curiously, gene variants associated with T1D underwent recent positive selection and have been increasing in prevalence. There is more selection in alleles increasing, rather than decreasing, susceptibility to T1D. This is indicative of an evolutionary benefit, wherein these variants were possibly protective against viruses and bacterial infections. However, no such link has been reported for type 2 diabetes risk variants, as could be expected from the thrifty genotype hypothesis [168]. In terms of genetics, T2D seems to have more in common with that of cancer rather than T1D [169]. It has been suggested there may rather be a specific yin−yang relationship between cancer and T2D, with too much cell proliferation resulting in cancer and insufficient proliferation of pancreatic islets resulting in T2D [169].
LADA (latent autoimmune diabetes in adults) is considered an intermediary form between T1D and T2D, also referred to as type 1.5 diabetes. There is much less information available as to the genetic basis of LADA compared to T1D and T2D. One way to understand this would be to assess to what extent LADA shares genetic similarities with T1D and T2D. The HLA locus, conferring 50% of the genetic susceptibility of T1D also shows similar associations with LADA, with a few differences. The T1D variant PTPN22 shows a weak association with LADA. Data on the INS class I variable number tandem repeats (VNTRs) has been inconclusive and associations have been reported with both T1D and T2D wherein the short tandem repeat is associated with T1D and the long with T2D. While previously several T1D-associated variants could be tested for association with LADA, it was not until the discovery of the common variant in the TCF7L2 gene being strongly associated with T2D that the genetic contribution of T2D to LADA could really be tested. This variant is clearly associated with LADA and T2D but not with T1D. This indicates that LADA is indeed a genetic admixture of T1D and T2D.
There are more lessons to be learned from differences between T1D and T2D rather than similarities. Elucidating the genetic heterogeneity of the spectrum of diabetes disorders will help us in understanding the mechanisms underlying the phenotypic heterogeneity of diabetes and would be a step towards individualized therapy.
5.7. Systems Biology
A restricted focus on the genome through GWAS provides limited insights into the molecular mechanisms driving disease and is akin to a snapshot of the genetics of the disease. To obtain an understanding of disease pathogenesis, it is important to analyze the GWAS data in the context of complementary follow-up analyses including DNA methylation and histone modifications, expression profiling under conditions relevant for the disease, related protein analysis, and analysis of genotype−phenotype associations. Global transcriptome profiling in relevant tissues such as pancreas has facilitated identification and cataloging of a wide array of transcription-based events in the pathogenesis of T2D [170]. For example, by combining GWAS information with metabolomics, it has been possible to identify strong associations between SNPs and metabolic reactions that otherwise would have been missed [171]. Network or pathway-based approaches, including enrichment in pre-defined pathways by, for example, KEGG [172] (http://www.genome.jp) and Gene Ontology (GO) (http://www.geneontology.org), have also been used to identify disease genes for various diseases. Thus, an integrative approach of several data types is likely to discover disease genes that would not be identified by the use of classical GWAS approaches. This was also illustrated in a recent study of human islets identifying novel candidate genes for T2D based upon expression differences and co-expression with known T2D genes as well as protein−protein interaction analyses [173,174]. Integration of GWAS data with such data could therefore facilitate a systems-based understanding of the pathogenic mechanisms.
6. Conclusions
The technical revolution in the field of genetics has allowed identification of numerous genetic variants that associate with T2D. The genetic landscape of T2D susceptibility is as yet incomplete, thus far only explaining a small proportion of the total heritability of diabetes. Many possibilities to dissect the architecture of T2D etiology have emerged in the form of large-scale genetic studies, meta-analyses and sequencing in families. It has already greatly contributed to our understanding of disease mechanisms by identifying pathways that could not be linked to diabetes by existing hypothetical models, even though many genetic findings are very recent and have yet to make their contribution to our knowledge about diabetes pathogenesis. However, one must bear in mind that diabetes is probably a much more diverse disease than the current subdivision into T1D and T2D implies and more precise subdivision into subgroups may both facilitate the investigation of T2D genetics and pave the way for more individualized treatment. A holistic systems biology approach will also be required to obtain a complete picture of how genetic variation leads to diabetes. The rapid technology development during the past years holds promises that this will be possible in a not-too-distant future.
Author Contributions
Rashmi B. Prasad and Leif Groop researched relevant literature, conceived, wrote and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Groop, L.; Lyssenko, V. Genetics of type 2 diabetes. An overview. Endocrinol. Nutr. 2009, 56 (Suppl. 4), 34–37. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Atlas. 2014. Available online: www.Idf.Org/diabetesatlas (accessed on 3 March 2014).
- WHO. WHO report. 2014. Available online: http://www.who.int/gho/publications/world_health_statistics/en/ (accessed on 3 March 2014).
- Lyssenko, V.; Jonsson, A.; Almgren, P.; Pulizzi, N.; Isomaa, B.; Tuomi, T.; Berglund, G.; Altshuler, D.; Nilsson, P.; Groop, L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 2008, 359, 2220–2232. [Google Scholar] [CrossRef] [PubMed]
- Groop, L. Pathogenesis of type 2 diabetes: The relative contribution of insulin resistance and impaired insulin secretion. Int. J. Clin. Pract. Suppl. 2000, 113, 3–13. [Google Scholar] [PubMed]
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Menconi, F.; Corathers, S.; Jacobson, E.M.; Tomer, Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: From epidemiology to mechanisms. Endocr. Rev. 2008, 29, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Dobreanu, M.; Raz, I. Prevention of type 1 diabetes: Today and tomorrow. Diabetes/Metab. Res. Rev. 2010, 26, 602–605. [Google Scholar] [CrossRef]
- Kyvik, K.O.; Green, A.; Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: A population based study of young Danish twins. BMJ 1995, 311, 913–917. [Google Scholar] [CrossRef]
- Pociot, F.; Norgaard, K.; Hobolth, N.; Andersen, O.; Nerup, J. A nationwide population-based study of the familial aggregation of type 1 (insulin-dependent) diabetes mellitus in Denmark. Danish study group of diabetes in childhood. Diabetologia 1993, 36, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Polychronakos, C.; Li, Q. Understanding type 1 diabetes through genetics: Advances and prospects. Nat. Rev. Genet. 2011, 12, 781–792. [Google Scholar] [CrossRef]
- Hakonarson, H.; Grant, S.F. Genome-wide association studies (GWAS): Impact on elucidating the aetiology of diabetes. Diabetes/Metab. Res. Rev. 2011, 27, 685–696. [Google Scholar] [CrossRef]
- Groop, L.; Pociot, F. Genetics of diabetes—Are we missing the genes or the disease? Mol. Cell. Endocrinol. 2014, 382, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Stankov, K.; Benc, D.; Draskovic, D. Genetic and epigenetic factors in etiology of diabetes mellitus type 1. Pediatrics 2013, 132, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, G.P.; Rewers, M. The epidemic of type 1 diabetes: What is it telling us? Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S. Genetic susceptibility of childhood type 1 diabetes mellitus in Japan. Pediatr. Endocrinol. Rev. 2012, 10 (Suppl. 1), 62–71. [Google Scholar] [PubMed]
- Thomas, I.H.; Pietropaolo, M. Type 1 diabetes: A genetic pandora’s box? Pediatr. Diabetes 2010, 11, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Tuomi, T.; Rowley, M.; Zimmet, P.; Mackay, I.R. Latent autoimmune diabetes in adults (LADA)—More than a name. Diabetologia 2006, 49, 1996–1998. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, W.V.; Kazlauskas, R.J. Molecular basis for enantioselectivity of lipase from pseudomonas cepacia toward primary alcohols. Modeling, kinetics, and chemical modification of tyr29 to increase or decrease enantioselectivity. J. Organ. Chem. 1999, 64, 2638–2647. [Google Scholar] [CrossRef]
- Tuomi, T.; Zimmet, P.Z.; Rowley, M.J.; Serjeantson, S.W.; Mackay, I.R. Persisting antibodies to glutamic acid decarboxylase in type 1 (insulin-dependent) diabetes mellitus are not associated with neuropathy. Diabetologia 1993, 36, 685. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Midthjell, K.; Grill, V. Influence of family history of diabetes on incidence and prevalence of latent autoimmune diabetes of the adult: Results from the nord-trondelag health study. Diabetes Care 2007, 30, 3040–3045. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, R.B. Mild familial diabetes with dominant inheritance. Q. J. Med. 1974, 43, 339–357. [Google Scholar] [PubMed]
- Voight, B.F.; Scott, L.J.; Steinthorsdottir, V.; Morris, A.P.; Dina, C.; Welch, R.P.; Zeggini, E.; Huth, C.; Aulchenko, Y.S.; Thorleifsson, G.; et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010, 42, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Lagou, V.; Welch, R.P.; Wheeler, E.; Montasser, M.E.; Luan, J.; Magi, R.; Strawbridge, R.J.; Rehnberg, E.; Gustafsson, S.; et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012, 44, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Turnbull, D.M.; Walker, M.; Hattersley, A.T. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243a>g mitochondrial point mutation. Diabet. Med. 2008, 25, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Van den Ouweland, J.M.; Lemkes, H.H.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.; van de Kamp, J.J.; Maassen, J.A. Mutation in mitochondrial tRNA(leu)(uur) gene in a large pedigree with maternally transmitted type ii diabetes mellitus and deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar]
- Pearson, E.R.; Flechtner, I.; Njolstad, P.R.; Malecki, M.T.; Flanagan, S.E.; Larkin, B.; Ashcroft, F.M.; Klimes, I.; Codner, E.; Iotova, V.; et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to kir6.2 mutations. N. Engl. J. Med. 2006, 355, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.M.; Black, M.H.; Xiang, A.H.; Allayee, H.; Lawrence, J.M.; Buchanan, T.A. Genetics of gestational diabetes mellitus and type 2 diabetes. Diabetes Care 2007, 30 (Suppl. 2), S134–S140. [Google Scholar] [CrossRef] [PubMed]
- Shaat, N.; Groop, L. Genetics of gestational diabetes mellitus. Curr. Med. Chem. 2007, 14, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, T.H.; Lim, S.; Choi, S.H.; Shin, H.D.; Lee, H.K.; Park, K.S.; Jang, H.C. Type 2 diabetes-associated genetic variants discovered in the recent genome-wide association studies are related to gestational diabetes mellitus in the Korean population. Diabetologia 2009, 52, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lauenborg, J.; Grarup, N.; Damm, P.; Borch-Johnsen, K.; Jorgensen, T.; Pedersen, O.; Hansen, T. Common type 2 diabetes risk gene variants associate with gestational diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, J.; Grant, A.M. The genetics of gestational diabetes mellitus: Evidence for relationship with type 2 diabetes mellitus. Genet. Med. 2008, 10, 240–250. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.B. Diabetes mellitus after GDM. Diabetes 1991, 40 (Suppl. 2), 131–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational diabetes and the incidence of type 2 diabetes: A systematic review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Young, B.C.; Ecker, J.L. Fetal macrosomia and shoulder dystocia in women with gestational diabetes: Risks amenable to treatment? Curr. Diabetes Rep. 2013, 13, 12–18. [Google Scholar] [CrossRef]
- Group, H.S.C.R.; Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Köbberling, J.; Tillil, H. Empirical risk figures for first-degree relatives of non-insulin dependent diabetics. In The Genetics of Diabetes Mellitus; Academic Press: London, UK, 1982; pp. 201–209. [Google Scholar]
- Kaprio, J.; Tuomilehto, J.; Koskenvuo, M.; Romanov, K.; Reunanen, A.; Eriksson, J.; Stengard, J.; Kesaniemi, Y.A. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992, 35, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Newman, B.; Selby, J.V.; King, M.C.; Slemenda, C.; Fabsitz, R.; Friedman, G.D. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia 1987, 30, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Kyvik, K.O.; Vaag, A.; Beck-Nielsen, H. Heritability of type ii (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance—A population-based twin study. Diabetologia 1999, 42, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Medici, F.; Hawa, M.; Ianari, A.; Pyke, D.A.; Leslie, R.D. Concordance rate for type ii diabetes mellitus in monozygotic twins: Actuarial analysis. Diabetologia 1999, 42, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of type 2 diabetes: The framingham offspring study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J. The double puzzle of diabetes. Nature 2003, 423, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Oda, N.; Cox, N.J.; Li, X.; Orho-Melander, M.; Hara, M.; Hinokio, Y.; Lindner, T.H.; Mashima, H.; Schwarz, P.E.; et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 2000, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, R.; Blangero, J.; Almasy, L.; Dyer, T.D.; Williams, K.L.; Leach, R.J.; O’Connell, P.; Stern, M.P. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am. J. Hum. Genet. 1999, 64, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Thorleifsson, G.; Benediktsson, R.; Sigurdsson, G.; Emilsson, V.; Einarsdottir, A.S.; Hjorleifsdottir, E.E.; Orlygsdottir, G.T.; Bjornsdottir, G.T.; Saemundsdottir, J.; et al. Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am. J. Hum. Genet. 2003, 73, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.F.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Lin, Y.; Zhang, Y.; Yang, J.; Zhang, Y.; Liu, H.; Zhang, B. Association between TCF7L2 gene polymorphisms and susceptibility to type 2 diabetes mellitus: A large human genome epidemiology (huge) review and meta-analysis. BMC Med. Genet. 2009, 10, e15. [Google Scholar] [CrossRef]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamaki, J.; Mykkanen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A pro12ala substitution in ppargamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hani, E.H.; Boutin, P.; Durand, E.; Inoue, H.; Permutt, M.A.; Velho, G.; Froguel, P. Missense mutations in the pancreatic islet beta cell inwardly rectifying k+ channel gene (kir6.2/bir): A meta-analysis suggests a role in the polygenic basis of type ii diabetes mellitus in Caucasians. Diabetologia 1998, 41, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell katp channel subunits kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.K.; Nielsen, E.M.; Ek, J.; Andersen, G.; Glumer, C.; Carstensen, B.; Mouritzen, P.; Drivsholm, T.; Borch-Johnsen, K.; Jorgensen, T.; et al. Analysis of separate and combined effects of common variation in KCNJ11 and PPARG on risk of type 2 diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Cummings, E.A.; Edghill, E.L.; Harries, L.W.; Scott, R.; Costa, T.; Temple, I.K.; Hattersley, A.T.; Ellard, S. Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 gene encoding the kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel. J. Clin. Endocrinol. Metab. 2004, 89, 3932–3935. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT; Lund University; Novartis Institutes of BioMedical Research; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segre, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat. Genet. 2012, 44, 67–72. [Google Scholar] [CrossRef]
- Imamura, M.; Maeda, S.; Yamauchi, T.; Hara, K.; Yasuda, K.; Morizono, T.; Takahashi, A.; Horikoshi, M.; Nakamura, M.; Fujita, H.; et al. A single-nucleotide polymorphism in ANK1 is associated with susceptibility to type 2 diabetes in Japanese populations. Hum. Mol. Genet. 2012, 21, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Kooner, J.S.; Saleheen, D.; Sim, X.; Sehmi, J.; Zhang, W.; Frossard, P.; Been, L.F.; Chia, K.S.; Dimas, A.S.; Hassanali, N.; et al. Genome-wide association study in individuals of south Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011, 43, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gan, W.; Lu, L.; Dong, X.; Han, X.; Hu, C.; Yang, Z.; Sun, L.; Bao, W.; Li, P.; et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese hans. Diabetes 2013, 62, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.D.; McDonough, C.W.; Hicks, P.J.; Roh, B.H.; Wing, M.R.; An, S.S.; Hester, J.M.; Cooke, J.N.; Bostrom, M.A.; Rudock, M.E.; et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLOS ONE 2012, 7, e29202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, E.J.; Below, J.E.; Krithika, S.; Valladares, A.; Barta, J.L.; Cox, N.J.; Hanis, C.L.; Wacher, N.; Garcia-Mena, J.; Hu, P.; et al. Genome-wide association study of type 2 diabetes in a sample from Mexico city and a meta-analysis of a Mexican-American sample from starr county, Texas. Diabetologia 2011, 54, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.O.; Long, J.; Cai, Q.; Qi, L.; Xiang, Y.B.; Cho, Y.S.; Tai, E.S.; Li, X.; Lin, X.; Chow, W.H.; et al. Identification of new genetic risk variants for type 2 diabetes. PLOS Genet. 2010, 6, e1001127. [Google Scholar] [CrossRef] [PubMed]
- DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium; Go, M.J.; Zhang, W.; Below, J.E.; Gaulton, K.J.; et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 2014, 46, 234–244. [Google Scholar]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jorgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Hara, K.; Maeda, S.; Yasuda, K.; Takahashi, A.; Horikoshi, M.; Nakamura, M.; Fujita, H.; Grarup, N.; Cauchi, S.; et al. A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat. Genet. 2010, 42, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Chauhan, G.; Dwivedi, O.P.; Mahajan, A.; Jaiswal, A.; Kaur, I.; Bandesh, K.; Singh, T.; Mathai, B.J.; Pandey, Y.; et al. Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes 2013, 62, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Consortium, S.T.D.; Williams, A.L.; Jacobs, S.B.; Moreno-Macias, H.; Huerta-Chagoya, A.; Churchhouse, C.; Marquez-Luna, C.; Garcia-Ortiz, H.; Gomez-Vazquez, M.J.; Burtt, N.P.; et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 2014, 506, 97–101. [Google Scholar] [PubMed]
- Sun, X.; Yu, W.; Hu, C. Genetics of type 2 diabetes: Insights into the pathogenesis and its clinical application. BioMed. Res. Int. 2014, 2014, 926713. [Google Scholar] [PubMed]
- Li, Y.R.; Keating, B.J. Trans-ethnic genome-wide association studies: Advantages and challenges of mapping in diverse populations. Genome Med. 2014, 6, e91. [Google Scholar] [CrossRef]
- Morris, A.P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 2011, 35, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Albrechtsen, A.; Grarup, N.; Li, Y.; Sparso, T.; Tian, G.; Cao, H.; Jiang, T.; Kim, S.Y.; Korneliussen, T.; Li, Q.; et al. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 2013, 56, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeggini, E.; Scott, L.J.; Saxena, R.; Voight, B.F.; Marchini, J.L.; Hu, T.; de Bakker, P.I.; Abecasis, G.R.; Almgren, P.; Andersen, G.; et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008, 40, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weedon, M.N.; Schwarz, P.E.; Horikawa, Y.; Iwasaki, N.; Illig, T.; Holle, R.; Rathmann, W.; Selisko, T.; Schulze, J.; Owen, K.R.; et al. Meta-analysis and a large association study confirm a role for calpain-10 variation in type 2 diabetes susceptibility. Am. J. Hum. Genet. 2003, 73, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Rung, J.; Cauchi, S.; Albrechtsen, A.; Shen, L.; Rocheleau, G.; Cavalcanti-Proenca, C.; Bacot, F.; Balkau, B.; Belisle, A.; Borch-Johnsen, K.; et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat. Genet. 2009, 41, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Cornelis, M.C.; Kraft, P.; Stanya, K.J.; Linda Kao, W.H.; Pankow, J.S.; Dupuis, J.; Florez, J.C.; Fox, C.S.; Pare, G.; et al. Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum. Mol. Genet. 2010, 19, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Elbers, C.C.; Guo, Y.; Peter, I.; Gaunt, T.R.; Mega, J.L.; Lanktree, M.B.; Tare, A.; Castillo, B.A.; Li, Y.R.; et al. Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am. J. Hum. Genet. 2012, 90, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.; Rayner, N.W.; Freathy, R.M.; et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Thorleifsson, G.; Walters, G.B.; Gudbjartsson, D.F.; Steinthorsdottir, V.; Sulem, P.; Helgadottir, A.; Styrkarsdottir, U.; Gretarsdottir, S.; Thorlacius, S.; Jonsdottir, I.; et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009, 41, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, M.S.; Weedon, M.N.; Fawcett, K.A.; Wasson, J.; Debenham, S.L.; Daly, A.; Lango, H.; Frayling, T.M.; Neumann, R.J.; Sherva, R.; et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat. Genet. 2007, 39, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Minton, J.A.; Hattersley, A.T.; Owen, K.; McCarthy, M.I.; Walker, M.; Latif, F.; Barrett, T.; Frayling, T.M. Association studies of genetic variation in the WFS1 gene and type 2 diabetes in U.K. Populations. Diabetes 2002, 51, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Sim, X.; Ong, R.T.; Suo, C.; Tay, W.T.; Liu, J.; Ng, D.P.; Boehnke, M.; Chia, K.S.; Wong, T.Y.; Seielstad, M.; et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLOS Genet. 2011, 7, e1001363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, F.; Serizawa, M.; Yamamoto, K.; Fujisawa, T.; Nakashima, E.; Ohnaka, K.; Ikegami, H.; Sugiyama, T.; Katsuya, T.; Miyagishi, M.; et al. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 2009, 58, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Voight, B.F.; Yengo, L.; Amin, N.; Dupuis, J.; Ganser, M.; Grallert, H.; Navarro, P.; Li, M.; Qi, L.; et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLOS Genet. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.C.; Shriner, D.; Chen, B.H.; Li, J.; Chen, W.M.; Guo, X.; Liu, J.; Bielinski, S.J.; Yanek, L.R.; Nalls, M.A.; et al. Meta-analysis of genome-wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLOS Genet. 2014, 10, e1004517. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.J.; Yang, C.F.; Chen, C.C.; Chuang, L.M.; Lu, C.H.; Chang, C.T.; Wang, T.Y.; Chen, R.H.; Shiu, C.F.; Liu, Y.M.; et al. A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLOS Genet. 2010, 6, e1000847. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.H.; Jokubka, R.; Tojjar, D.; Granhall, C.; Hansson, O.; Li, D.Q.; Nagaraj, V.; Reinbothe, T.M.; Tuncel, J.; Eliasson, L.; et al. Overexpression of alpha2a-adrenergic receptors contributes to type 2 diabetes. Science 2010, 327, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Gianniny, L.; Burtt, N.P.; Lyssenko, V.; Giuducci, C.; Sjogren, M.; Florez, J.C.; Almgren, P.; Isomaa, B.; Orho-Melander, M.; et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 2006, 55, 2890–2895. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.T.; Uimari, P.; Aalto, J.M.; Pirskanen, M.; Kaikkonen, J.; Todorova, B.; Hypponen, J.; Korhonen, V.P.; Asikainen, J.; Devine, C.; et al. Type 2 diabetes whole-genome association study in four populations: The diagen consortium. Am. J. Hum. Genet. 2007, 81, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Timpson, N.J.; Lindgren, C.M.; Weedon, M.N.; Randall, J.; Ouwehand, W.H.; Strachan, D.P.; Rayner, N.W.; Walker, M.; Hitman, G.A.; Doney, A.S.; et al. Adiposity-related heterogeneity in patterns of type 2 diabetes susceptibility observed in genome-wide association data. Diabetes 2009, 58, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Frigge, M.L.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet. 2009, 41, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Saleheen, D.; Been, L.F.; Garavito, M.L.; Braun, T.; Bjonnes, A.; Young, R.; Ho, W.K.; Rasheed, A.; Frossard, P.; et al. Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in sikhs of punjabi origin from India. Diabetes 2013, 62, 1746–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Steinthorsdottir, V.; Bergthorsson, J.T.; Thorleifsson, G.; Manolescu, A.; Rafnar, T.; Gudbjartsson, D.; Agnarsson, B.A.; Baker, A.; et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007, 39, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Winckler, W.; Burtt, N.P.; Holmkvist, J.; Cervin, C.; de Bakker, P.I.; Sun, M.; Almgren, P.; Tuomi, T.; Gaudet, D.; Hudson, T.J.; et al. Association of common variation in the hnf1alpha gene region with risk of type 2 diabetes. Diabetes 2005, 54, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Winckler, W.; Graham, R.R.; de Bakker, P.I.; Sun, M.; Almgren, P.; Tuomi, T.; Gaudet, D.; Hudson, T.J.; Ardlie, K.G.; Daly, M.J.; et al. Association testing of variants in the hepatocyte nuclear factor 4alpha gene with risk of type 2 diabetes in 7883 people. Diabetes 2005, 54, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Elliott, P.; Zabaneh, D.; Zhang, W.; Li, Y.; Froguel, P.; Balding, D.; Scott, J.; Kooner, J.S. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 2008, 40, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Bentley, A.; Adeyemo, A.; Shriner, D.; Zhou, J.; Doumatey, A.; Huang, H.; Ramos, E.; Erdos, M.; Gerry, N.; et al. Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum. Mol. Genet. 2012, 21, 4530–4536. [Google Scholar] [CrossRef] [PubMed]
- Go, M.J.; Hwang, J.Y.; Kim, Y.J.; Hee Oh, J.; Kim, Y.J.; Heon Kwak, S.; Soo Park, K.; Lee, J.; Kim, B.J.; Han, B.G.; et al. New susceptibility loci in myl2, C12orf51 and oas1 associated with 1-h plasma glucose as predisposing risk factors for type 2 diabetes in the Korean population. J. Hum. Genet. 2013, 58, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Belmont, J.W.; Boerwinkle, E.; Gibbs, R.A. Clan genomics and the complex architecture of human disease. Cell 2011, 147, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Thorleifsson, G.; Sulem, P.; Helgason, H.; Grarup, N.; Sigurdsson, A.; Helgadottir, H.T.; Johannsdottir, H.; Magnusson, O.T.; Gudjonsson, S.A.; et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 2014, 46, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Moltke, I.; Grarup, N.; Jorgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T.; et al. A common greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Consortium, S.T.D.; Estrada, K.; Aukrust, I.; Bjorkhaug, L.; Burtt, N.P.; Mercader, J.M.; Garcia-Ortiz, H.; Huerta-Chagoya, A.; Moreno-Macias, H.; Walford, G.; et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 2014, 311, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, J.R.; Jackson, A.U.; Fogarty, M.P.; Buchkovich, M.L.; Stancakova, A.; Stringham, H.M.; Sim, X.; Yang, L.; Fuchsberger, C.; Cederberg, H.; et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013, 45, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellcome Trust Case Control Consortium; Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; et al. Genome-wide association study of cnvs in 16,000 cases of eight common diseases and 3000 shared controls. Nature 2010, 464, 713–720. [Google Scholar]
- Plomin, R.; Haworth, C.M.; Davis, O.S. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef] [PubMed]
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J. What is complex about complex disorders? Genome Biol. 2012, 13, e237. [Google Scholar] [CrossRef]
- Agarwala, V.; Flannick, J.; Sunyaev, S.; Go, T.D.C.; Altshuler, D. Evaluating empirical bounds on complex disease genetic architecture. Nat. Genet. 2013, 45, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Nagorny, C.L.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spegel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet. 2009, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Bouatia-Naji, N.; Bonnefond, A.; Cavalcanti-Proenca, C.; Sparso, T.; Holmkvist, J.; Marchand, M.; Delplanque, J.; Lobbens, S.; Rocheleau, G.; Durand, E.; et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet. 2009, 41, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Park, S.Y.; Su, J.; Bailey, K.; Ottosson-Laakso, E.; Shcherbina, L.; Oskolkov, N.; Zhang, E.; Thevenin, T.; Fadista, J.; et al. TCF7L2 is a master regulator of insulin production and processing. Hum. Mol. Genet. 2014, 23, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjogren, M.; Ling, C.; Eriksson, K.F.; Lethagen, A.L.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Eliasson, L.; Kotova, O.; Pilgaard, K.; Wierup, N.; Salehi, A.; Wendt, A.; Jonsson, A.; de Marinis, Y.Z.; Berglund, L.M.; et al. Pleiotropic effects of gip on islet function involve osteopontin. Diabetes 2011, 60, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.H.; Braun, M.; Mahdi, T.; Andersson, S.A.; Travers, M.E.; Shigeto, M.; Zhang, E.; Almgren, P.; Ladenvall, C.; Axelsson, A.S.; et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes 2012, 61, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Langenberg, C.; Hivert, M.F.; Prokopenko, I.; Lyssenko, V.; Dupuis, J.; Magi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Grarup, N.; Andersen, G.; Krarup, N.T.; Albrechtsen, A.; Schmitz, O.; Jorgensen, T.; Borch-Johnsen, K.; Hansen, T.; Pedersen, O. Association testing of novel type 2 diabetes risk alleles in the JAZF1, CDC123/CAMK1D, TSPAN8, THADA, ADAMTS9, and NOTCH2 loci with insulin release, insulin sensitivity, and obesity in a population-based sample of 4516 glucose-tolerant middle-aged danes. Diabetes 2008, 57, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Axelsson, A.S.; Spegel, P.; Andersson, L.E.; Mulder, H.; Groop, L.C.; Renstrom, E.; Rosengren, A.H. Genotype-based treatment of type 2 diabetes with an alpha2a-adrenergic receptor antagonist. Sci. Transl. Med. 2014, 6, 257ra139. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Wray, N.R.; Goddard, M.E.; Visscher, P.M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 2011, 88, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Yang, J.; Goddard, M.E. A commentary on “common SNPs explain a large proportion of the heritability for human height” by yang et al. (2010). Twin Res. Hum. Genet. 2010, 13, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Groop, L.; Forsblom, C.; Lehtovirta, M.; Tuomi, T.; Karanko, S.; Nissen, M.; Ehrnstrom, B.O.; Forsen, B.; Isomaa, B.; Snickars, B.; et al. Metabolic consequences of a family history of niddm (the botnia study): Evidence for sex-specific parental effects. Diabetes 1996, 45, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X.; Sundquist, K.; Sundquist, J. Familial risks for type 2 diabetes in Sweden. Diabetes Care 2010, 33, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Whitelaw, E. Epigenetic germline inheritance. Curr. Opin. Genet. Dev. 2004, 14, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Tollefsbol, T.O. Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 2008, 10, 25–36. [Google Scholar] [PubMed]
- Small, K.S.; Hedman, A.K.; Grundberg, E.; Nica, A.C.; Thorleifsson, G.; Kong, A.; Thorsteindottir, U.; Shin, S.Y.; Richards, H.B.; Soranzo, N.; et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet. 2011, 43, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.L.; Guo, T.; Muller, Y.L.; Fleming, J.; Knowler, W.C.; Kobes, S.; Bogardus, C.; Baier, L.J. Strong parent-of-origin effects in the association of KCNQ1 variants with type 2 diabetes in American Indians. Diabetes 2013, 62, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. TIG 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Wolf, J.B.; Hager, R. A maternal-offspring coadaptation theory for the evolution of genomic imprinting. PLOS Biol. 2006, 4, e380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Travers, M.E.; Mackay, D.J.; Dekker Nitert, M.; Morris, A.P.; Lindgren, C.M.; Berry, A.; Johnson, P.R.; Hanley, N.; Groop, L.C.; McCarthy, M.I.; et al. Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets. Diabetes 2013, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Hoggart, C.J.; Venturini, G.; Mangino, M.; Gomez, F.; Ascari, G.; Zhao, J.H.; Teumer, A.; Winkler, T.W.; Tsernikova, N.; Luan, J.; et al. Novel approach identifies SNPs in slc2a10 and kcnk9 with evidence for parent-of-origin effect on body mass index. PLOS Genet. 2014, 10, e1004508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Wedig, G.C.; Jacobs, K.B.; Elston, R.C. Multilocus linkage tests based on affected relative pairs. Am. J. Hum. Genet. 2000, 66, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.J.; Frigge, M.; Nicolae, D.L.; Concannon, P.; Hanis, C.L.; Bell, G.I.; Kong, A. Loci on chromosomes 2 (niddm1) and 15 interact to increase susceptibility to diabetes in Mexican Americans. Nat. Genet. 1999, 21, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Gayan, J.; Marin, J.; Galan, J.J.; Saez, M.E.; Real, L.M.; Antunez, C.; Ruiz, A. Whole-genome conditional two-locus analysis identifies novel candidate genes for late-onset parkinson’s disease. Neurogenetics 2009, 10, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with il-23 and nf-kappab pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Marchini, J.; Morris, A.P.; Cardon, L.R. Two-stage two-locus models in genome-wide association. PLOS Genet. 2006, 2, e157. [Google Scholar] [CrossRef] [PubMed]
- Zuk, O.; Hechter, E.; Sunyaev, S.R.; Lander, E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 2012, 109, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Diabetic nephropathy—Emerging epigenetic mechanisms. Nat. Rev. Nephrol. 2014, 10, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.L.; McCray, P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valverde, S.L.; Taft, R.J.; Mattick, J.S. MicroRNAs in beta-cell biology, insulin resistance, diabetes and its complications. Diabetes 2011, 60, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, M.; Scaria, V.; Brahmachari, S.K. Dbsmr: A novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinform. 2009, 10, e108. [Google Scholar] [CrossRef]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the anril locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Eftychi, C.; Howson, J.M.; Barratt, B.J.; Vella, A.; Payne, F.; Smyth, D.J.; Twells, R.C.; Walker, N.M.; Rance, H.E.; Tuomilehto-Wolf, E.; et al. Analysis of the type 2 diabetes-associated single nucleotide polymorphisms in the genes IRS1, KCNJ11, and pparg2 in type 1 diabetes. Diabetes 2004, 53, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.M.; Howson, J.M.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Field, S.F.; Stevens, H.E.; Todd, J.A. No association of multiple type 2 diabetes loci with type 1 diabetes. Diabetologia 2009, 52, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.K.; Sterner, M.; Forsen, T.; Karajamaki, A.; Rolandsson, O.; Forsblom, C.; Groop, P.H.; Lahti, K.; Nilsson, P.M.; Groop, L.; et al. Type 2 diabetes susceptibility gene variants predispose to adult-onset autoimmune diabetes. Diabetologia 2014, 57, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, M.; van Herpt, T.W.; Dehghan, A.; Hofman, A.; Lieverse, A.G.; van Duijn, C.M.; Witteman, J.C.; Sijbrands, E.J. Association of an APOC3 promoter variant with type 2 diabetes risk and need for insulin treatment in lean persons. Diabetologia 2011, 54, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter znt8 (SLC30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [PubMed]
- Corona, E.; Dudley, J.T.; Butte, A.J. Extreme evolutionary disparities seen in positive selection across seven complex diseases. PLOS ONE 2010, 5, e12236. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Colhoun, H.; Florez, J.C. A genetic link between type 2 diabetes and prostate cancer. Diabetologia 2008, 51, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Fadista, J.; Vikman, P.; Laakso, E.O.; Mollet, I.G.; Esguerra, J.L.; Taneera, J.; Storm, P.; Osmark, P.; Ladenvall, C.; Prasad, R.B.; et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 13924–13929. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Shin, S.Y.; Petersen, A.K.; Mohney, R.P.; Meredith, D.; Wagele, B.; Altmaier, E.; CardioGram; Deloukas, P.; Erdmann, J.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Lang, S.; Sharma, A.; Fadista, J.; Zhou, Y.; Ahlqvist, E.; Jonsson, A.; Lyssenko, V.; Vikman, P.; Hansson, O.; et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012, 16, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Taneera, J.; Fadista, J.; Ahlqvist, E.; Zhang, M.; Wierup, N.; Renstrom, E.; Groop, L. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol. Cell. Endocrinol. 2013, 375, 35–42. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Prasad, R.B.; Groop, L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes 2015, 6, 87-123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010087
AMA Style
Prasad RB, Groop L. Genetics of Type 2 Diabetes—Pitfalls and Possibilities. Genes. 2015; 6(1):87-123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010087
Chicago/Turabian StylePrasad, Rashmi B., and Leif Groop. 2015. "Genetics of Type 2 Diabetes—Pitfalls and Possibilities" Genes 6, no. 1: 87-123. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6010087