
Hypermethylation of the VTRNA1-3 Promoter is Associated with Poor Outcome in Lower Risk Myelodysplastic Syndrome Patients

Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Healthy Donors
2.2. Cell Culture and Drug Treatments
2.3. DNA Extraction and Bisulfite Conversion
2.4. RNA Extraction and Reverse Transcriptase Quantitative PCR (RT-qPCR)
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. DNA Methylation Analyses
2.6.1. Bisulfite-Sequencing
2.6.2. Methylation-Sensitive Single-Nucleotide Primer Extension (MS-SNuPE)
2.6.3. Pyrosequencing
2.7. Survival Statistics
3. Results
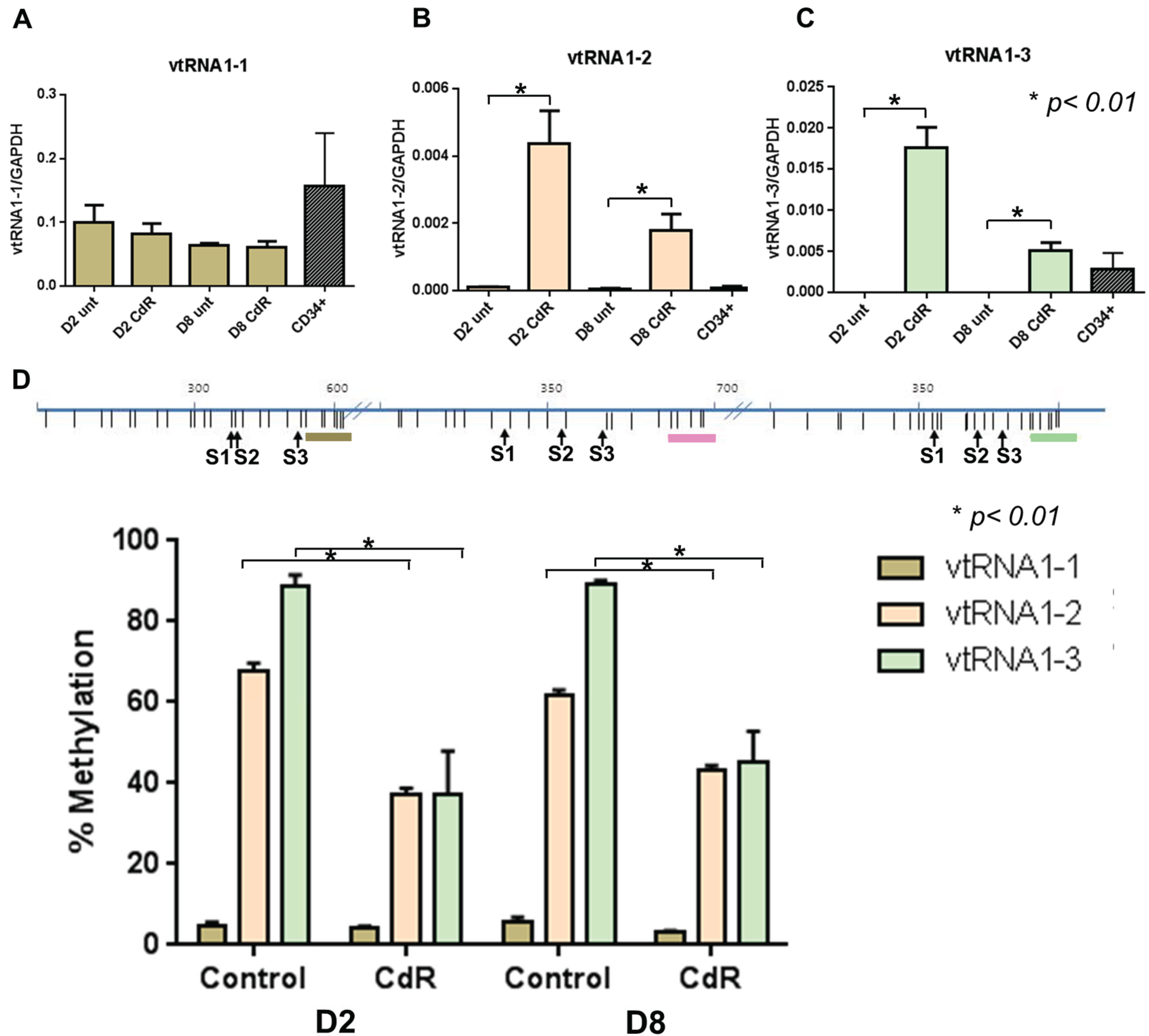
3.1. VTRNAs Are Regulated by DNA Methylation

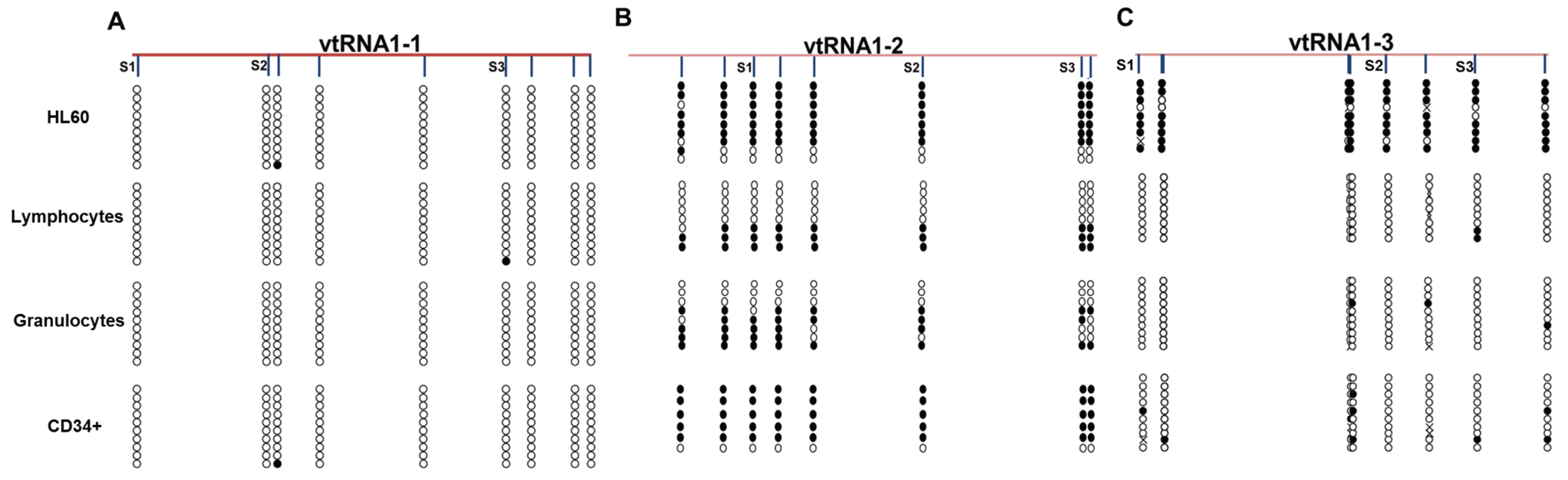
3.2. VTRNAs Are Differentially Methylated in Healthy Donor Cell Populations

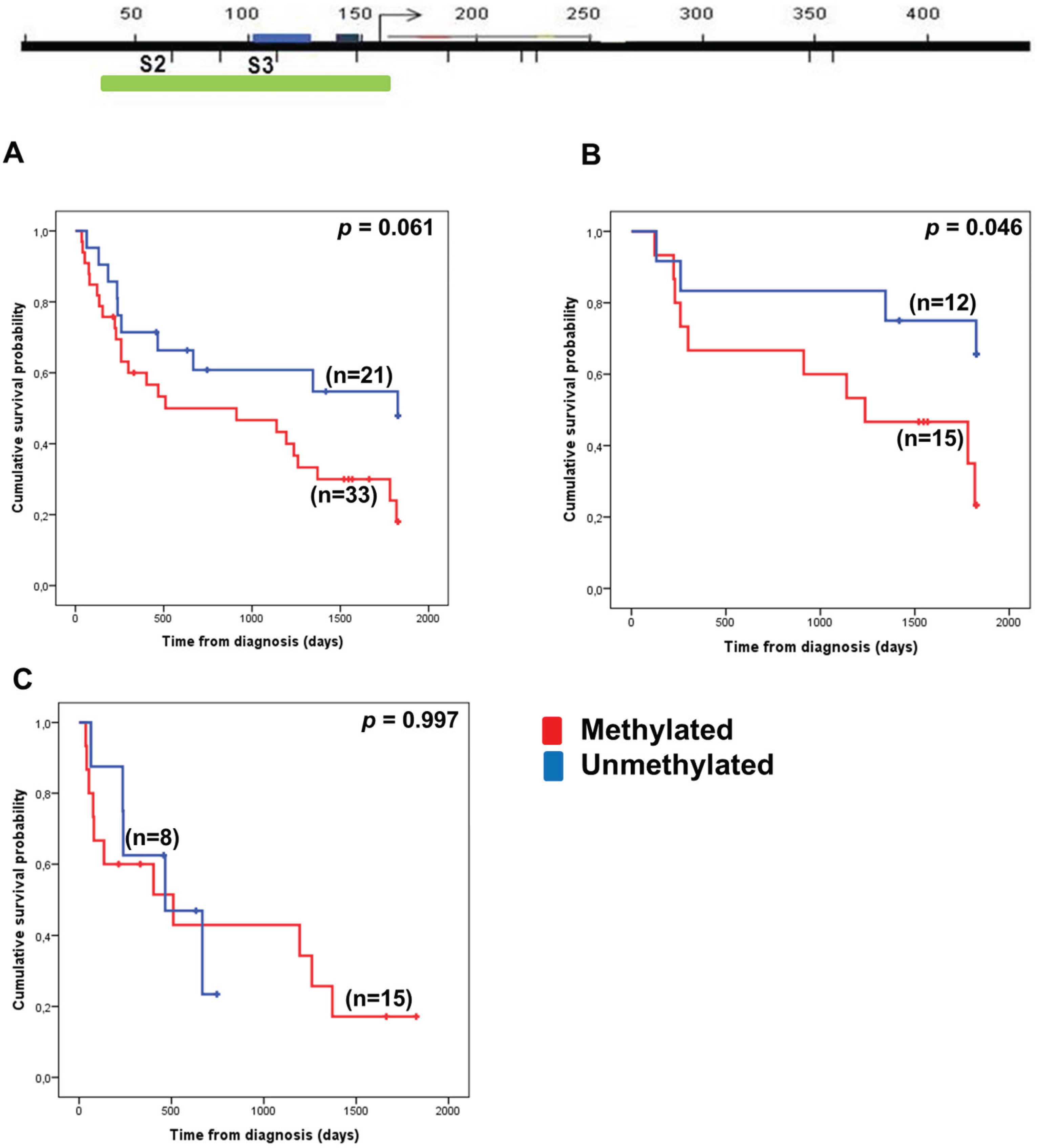
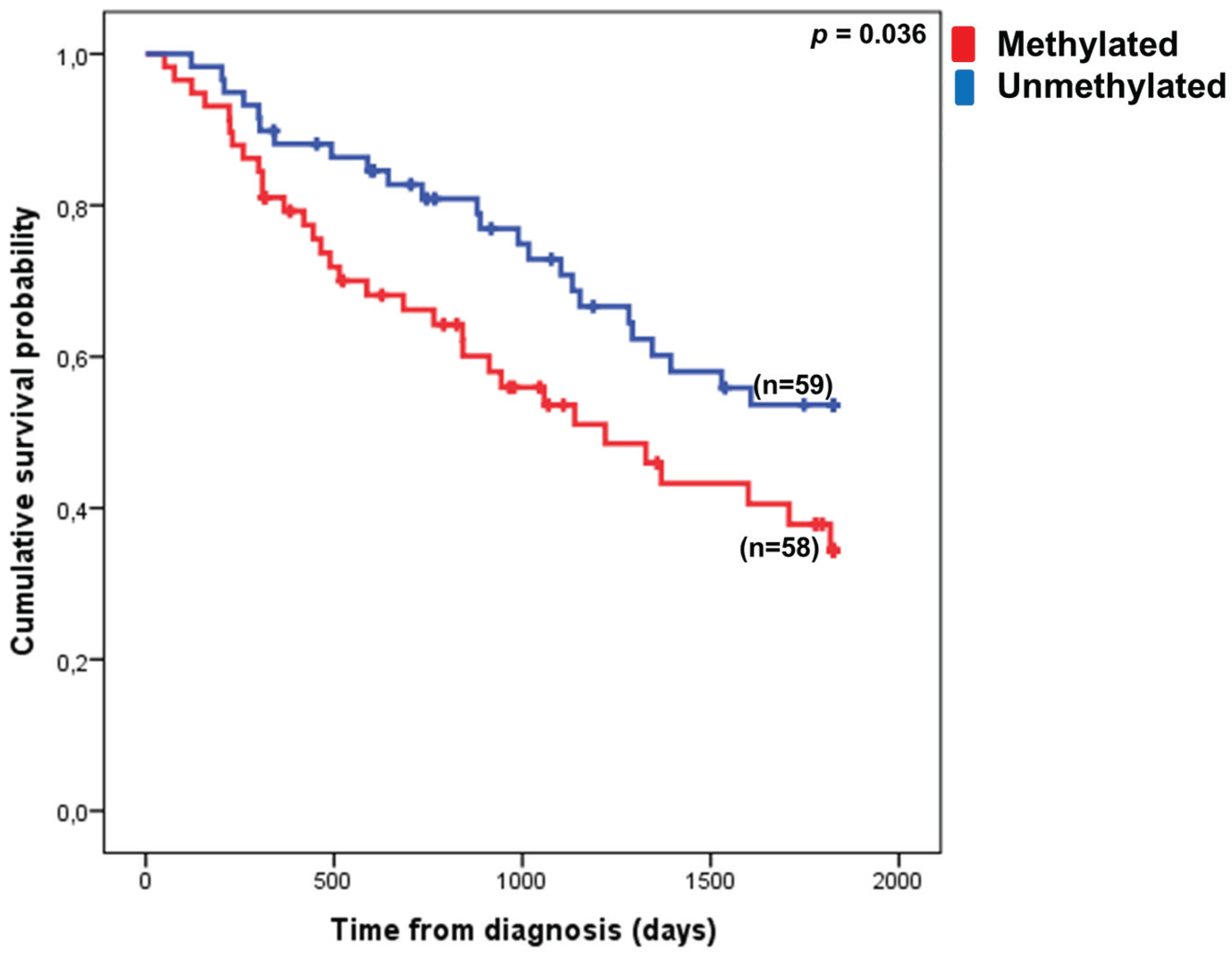
3.3. VTRNA1-3 Promoter Methylation Associates with Poor Survival in Lower Risk MDS Patients



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | % | ||
|---|---|---|---|
| Age | <65 | 20 | 17.1 |
| ≥65 | 97 | 82.9 | |
| Sex | Male | 59 | 54.1 |
| Female | 50 | 45.9 | |
| IPSS score | 0 | 61 | 60.3 |
| 0.5–1 | 50 | 39.7 |
| Covariate | Log-Rank Analysis | Multiple Cox Regression Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Coefficient | HR | 95% CI | p-value | Coefficient | HR | 95% CI | p-value | |
| Age (≥65 years) | 1.286 | 3.617 | 1.305–10.030 | 0.008 | 1.506 | 4.508 | 1.373–14.801 | 0.013 |
| Sex | −0.002 | 0.998 | 0.578–1.723 | 0.995 | −0.268 | 0.765 | 0.416–1.405 | 0.388 |
| VTRNA1-3 methylation | 0.562 | 1.755 | 1.031–2.985 | 0.037 | 0.360 | 1.434 | 0.801–2.566 | 0.225 |
| IPSS score (0 vs. 0.5–1) | 0.369 | 1.446 | 0.843–2.479 | 0.178 | 0.022 | 1.022 | 0.510–2.046 | 0.951 |
| Blast count (<5% vs. 5%–10%) | 1.098 | 2.999 | 1.397–6.437 | 0.003 | 1.310 | 3.705 | 1.574–8.722 | 0.003 |
| Cytogenetics (good vs. intermediate/poor) | 0.854 | 2.350 | 1.236–4.465 | 0.007 | 1.195 | 3.303 | 1.662–6.564 | 0.001 |
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Myelodysplastic syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Elias, H.K.; Schinke, C.; Bhattacharyya, S.; Will, B.; Verma, A.; Steidl, U. Stem cell origin of myelodysplastic syndromes. Oncogene 2013, 33, 1–12. [Google Scholar]
- Woll, P.S.; Kjällquist, U.; Chowdhury, O.; Doolittle, H.; Wedge, D.C.; Thongjuea, S.; Erlandsson, R.; Ngara, M.; Anderson, K.; Deng, Q.; et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell 2014, 25, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P. A Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dunbar, A.; Gondek, L.P.; Mohan, S.; Rataul, M.; O’Keefe, C.; Sekeres, M.; Saunthararajah, Y.; Maciejewski, J.P. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009, 113, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P. Epigenetic changes in the myelodysplastic syndrome. Hematol. Oncol. Clin. North Am. 2010, 24, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar, S.C. Genetic and epigenetic pathways in myelodysplastic syndromes: A brief overview. Adv. Biol. Regul. 2014, 58, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Grønbæk, K.; Hother, C.; Jones, P.A. Epigenetic changes in cancer. APMIS 2007, 115, 1039–1059. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Issa, J.-P.J.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Lubbert, M.; Suciu, S.; Baila, L.; Ruter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; Labar, B.; Germing, U.; Salih, H.R.; et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk Myelodysplastic Syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized phase III study of the European Organisation for Rese. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Treppendahl, M.B.; Kristensen, L.S.; Grønbæk, K. Predicting response to epigenetic therapy. J. Clin. Invest. 2014, 124, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 2002, 99, 4638–4641. [Google Scholar] [CrossRef] [PubMed]
- Pellagatti, A.; Boultwood, J. Recent advances in the 5q- syndrome. Mediterr. J. Hematol. Infect. Dis. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Meggendorfer, M.; Cruz, M.M.A.; Fabisch, J.; Klesse, S.; Köhler, L.; Göhring, G.; Ganster, C.; Shirneshan, K.; Gutermuth, A.; et al. Frequency and prognostic impact of casein kinase 1A1 mutations in MDS patients with deletion of chromosome 5q. Leukemia 2015, 29, 1–4. [Google Scholar] [CrossRef]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Treppendahl, M.B.; Qiu, X.; Søgaard, A.; Yang, X.; Nandrup-Bus, C.; Hother, C.; Andersen, M.K.; Kjeldsen, L.; Möllgaard, L.; Hellström-Lindberg, E.; et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood 2012, 119, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Song, Y.; Bi, N.; Shen, J.; Liu, W.; Fan, J.; Sun, G.; Tong, T.; He, J.; Shi, Y.; et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013, 73, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Park, J.L.; Lee, K.; Richardson, L.E.; Johnson, B.H.; Lee, H.S.; Lee, J.S.; Kim, S.B.; Kwon, O.H.; Song, K.S.; et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget 2014, 5, 3944–3955. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Lee, K.; Jang, H.-J.; Lee, G.K.; Park, J.-L.; Kim, S.-Y.; Kim, S.-B.; Johnson, B.H.; Zo, J.I.; Lee, J.-S.; et al. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget 2014, 5, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, V.; Nakabayashi, K.; Vizoso, M.; Moran, S.; Iglesias-Platas, I.; Sugahara, N.; Simón, C.; Hata, K.; Esteller, M.; Court, F.; et al. Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer. Epigenetics 2014, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Johnson, B.H.; Lee, Y.S. A tumor surveillance model: A non-coding RNA senses neoplastic cells and its protein partner signals cell death. Int. J. Mol. Sci. 2012, 13, 13134–13139. [Google Scholar] [CrossRef] [PubMed]
- Van Zon, A.; Mossink, M.H.; Schoester, M.; Scheffer, G.L.; Scheper, R.J.; Sonneveld, P.; Wiemer, E.A. Multiple human vault RNAs. Expression and association with the vault complex. J. Biol. Chem. 2001, 276, 37715–37721. [Google Scholar]
- Amort, M.; Nachbauer, B.; Tuzlak, S.; Kieser, A.; Schepers, A.; Villunger, A.; Polacek, N. Expression of the vault RNA protects cells from undergoing apoptosis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.; Kvist, A.; Vallon-Christersson, J.; Medstrand, P.; Borg, A.; Rovira, C. The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nat. Cell Biol. 2009, 11, 1268–1271. [Google Scholar] [CrossRef] [PubMed]
- Nandy, C.; Mrázek, J.; Stoiber, H.; Grässer, F.A.; Hüttenhofer, A.; Polacek, N. Epstein-barr virus-induced expression of a novel human vault RNA. J. Mol. Biol. 2009, 388, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Kudo, M.; Takada, Y.; Matsugami, A.; Gopinath, S.C.B.; Kumar, P.K.R.; Katahira, M. Interactions between antitumor drugs and vault RNA. Nucleic Acids Symp. Ser. Oxf. 2008, 52, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.Y.; Eaton, S.A.; Young, P.E.; Lee, M.; Shuttleworth, R.; Humphreys, D.T.; Grau, G.E.; Combes, V.; Bebawy, M.; Gong, J.; et al. Glioma microvesicles carry selectively packaged coding and noncoding RNAs which alter gene expression in recipient cells. RNA Biol. 2013, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nolte-’t Hoen, E.N.M.; Buermans, H.P.J.; Waasdorp, M.; Stoorvogel, W.; Wauben, M.H.M.; AC’t Hoen, P. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012, 40, 9272–9285. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Hother, C.; Ralfkiær, U.M.; Søgaard, A.; Lu, Q.; Workman, C.T.; Liang, G.; Jones, P.A.; Grønbæk, K. Equitoxic doses of 5-azacytidine and 5-aza-2’deoxycytidine induce diverse immediate and overlapping heritable changes in the transcriptome. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Miranda, T.B.; Liang, G.; Berman, B.P.; Lin, J.C.; Tanay, A.; Jones, P.A. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol. Cell 2010, 39, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Gonzalgo, M.L.; Liang, G. Methylation-sensitive single-nucleotide primer extension (Ms-SNuPE) for quantitative measurement of DNA methylation. Nat. Protoc. 2007, 2, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Suzuki, H.; Yamamoto, E.; Imai, K.; Shinomura, Y. Emerging links between epigenetic alterations and dysregulation of noncoding RNAs in cancer. Tumour Biol. 2012, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Mufti, G.J. Excessive apoptosis in low risk myelodysplastic syndromes (MDS). Leuk. Lymphoma 2000, 40, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.-Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Lee, K.; Lee, K.S.; Kunkeaw, N.; Johnson, B.H.; Holthauzen, L.M.F.; Gong, B.; Leelayuwat, C.; Lee, Y.S. Characterization of the direct physical interaction of nc886, a cellular non-coding RNA, and PKR. FEBS Lett. 2012, 586, 3477–3484. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.; Henke, C.; Akman, B.; Li, H.; Rote, N.S.; Beckmann, M.W.; Zahnow, C.A.; Baylin, S.B.; Strick, R. Inhibiting DNA methylation causes an interferon response via double-stranded DNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Yau, H.L.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Pugh, T.J.; Jones, P.A.; et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helbo, A.S.; Treppendahl, M.; Aslan, D.; Dimopoulos, K.; Nandrup-Bus, C.; Holm, M.S.; Andersen, M.K.; Liang, G.; Kristensen, L.S.; Grønbæk, K. Hypermethylation of the VTRNA1-3 Promoter is Associated with Poor Outcome in Lower Risk Myelodysplastic Syndrome Patients. Genes 2015, 6, 977-990. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6040977
Helbo AS, Treppendahl M, Aslan D, Dimopoulos K, Nandrup-Bus C, Holm MS, Andersen MK, Liang G, Kristensen LS, Grønbæk K. Hypermethylation of the VTRNA1-3 Promoter is Associated with Poor Outcome in Lower Risk Myelodysplastic Syndrome Patients. Genes. 2015; 6(4):977-990. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6040977
Chicago/Turabian StyleHelbo, Alexandra Søgaard, Marianne Treppendahl, Derya Aslan, Konstantinos Dimopoulos, Cecilie Nandrup-Bus, Mette Skov Holm, Mette Klarskov Andersen, Gangning Liang, Lasse Sommer Kristensen, and Kirsten Grønbæk. 2015. "Hypermethylation of the VTRNA1-3 Promoter is Associated with Poor Outcome in Lower Risk Myelodysplastic Syndrome Patients" Genes 6, no. 4: 977-990. https://0-doi-org.brum.beds.ac.uk/10.3390/genes6040977