
Analysis of DNA Methylation and Hydroxymethylation in the Genome of Crustacean Daphnia pulex

Abstract
:1. Introduction
2. Experimental Section
2.1. Preparation of Biological Samples
2.2. Preparation of Genomic DNA
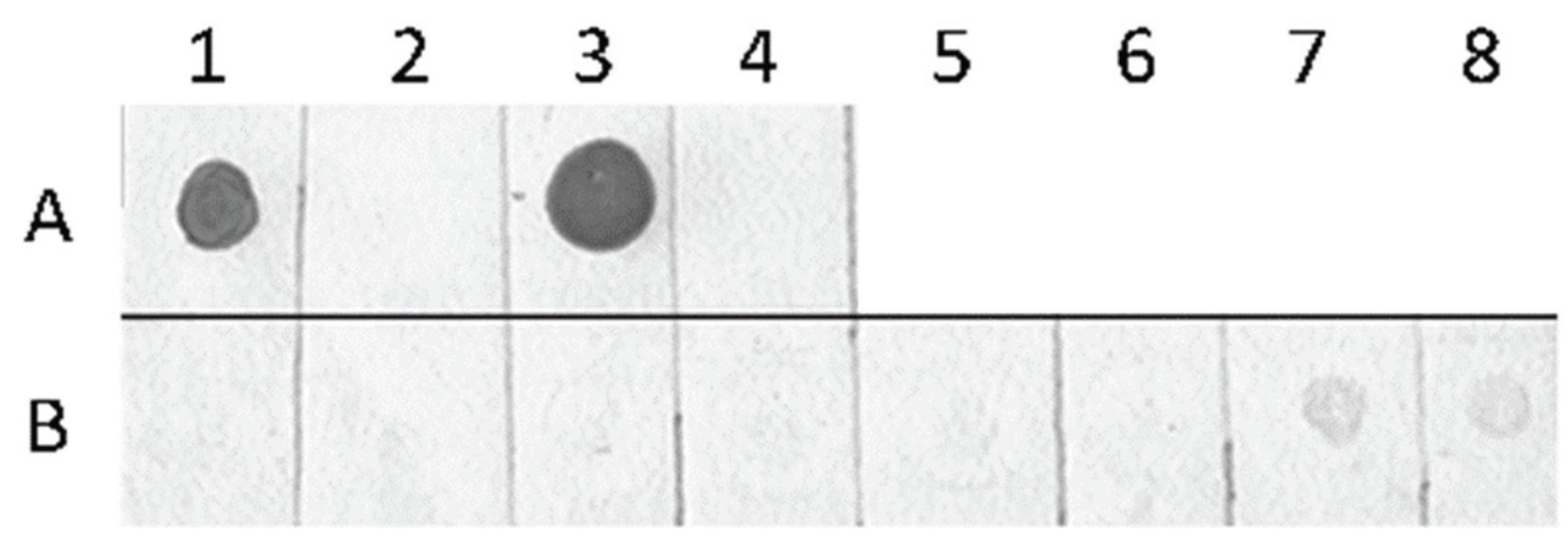
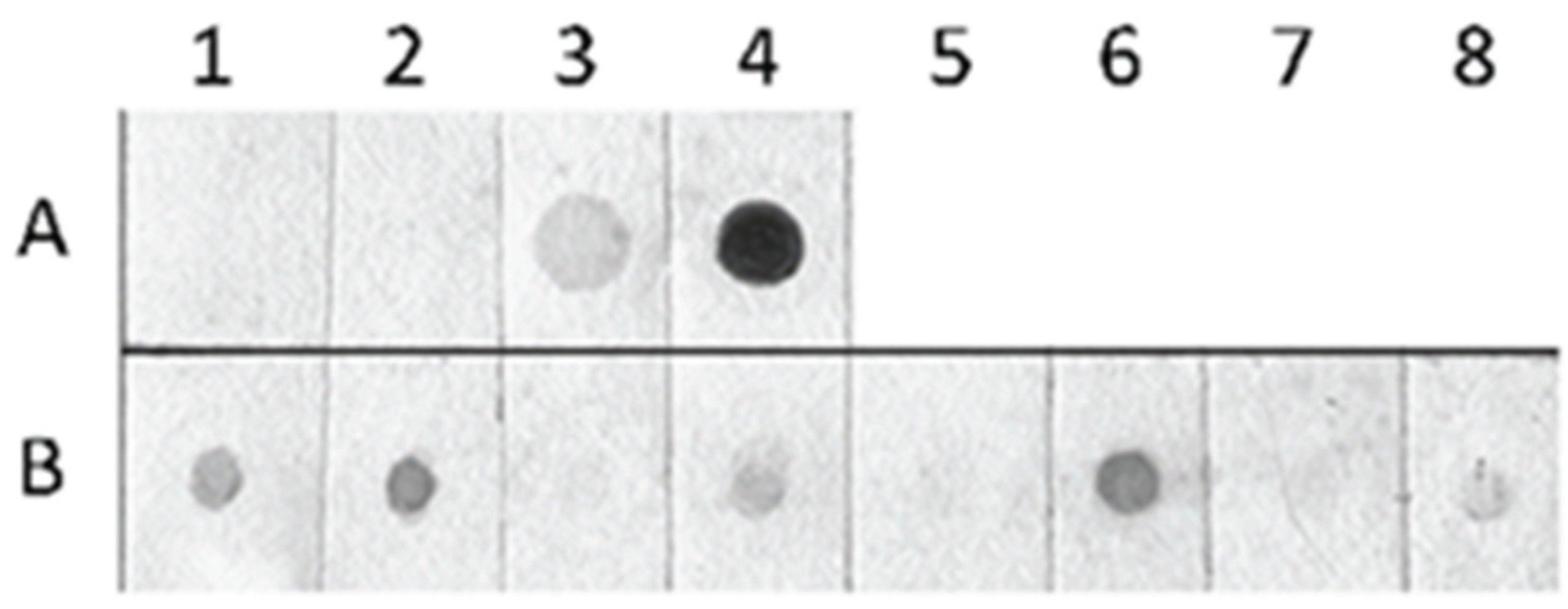
2.3. Detection of Global Genome Methylation and Hydroxymethylation Using Immuno-Dot Blot Analysis
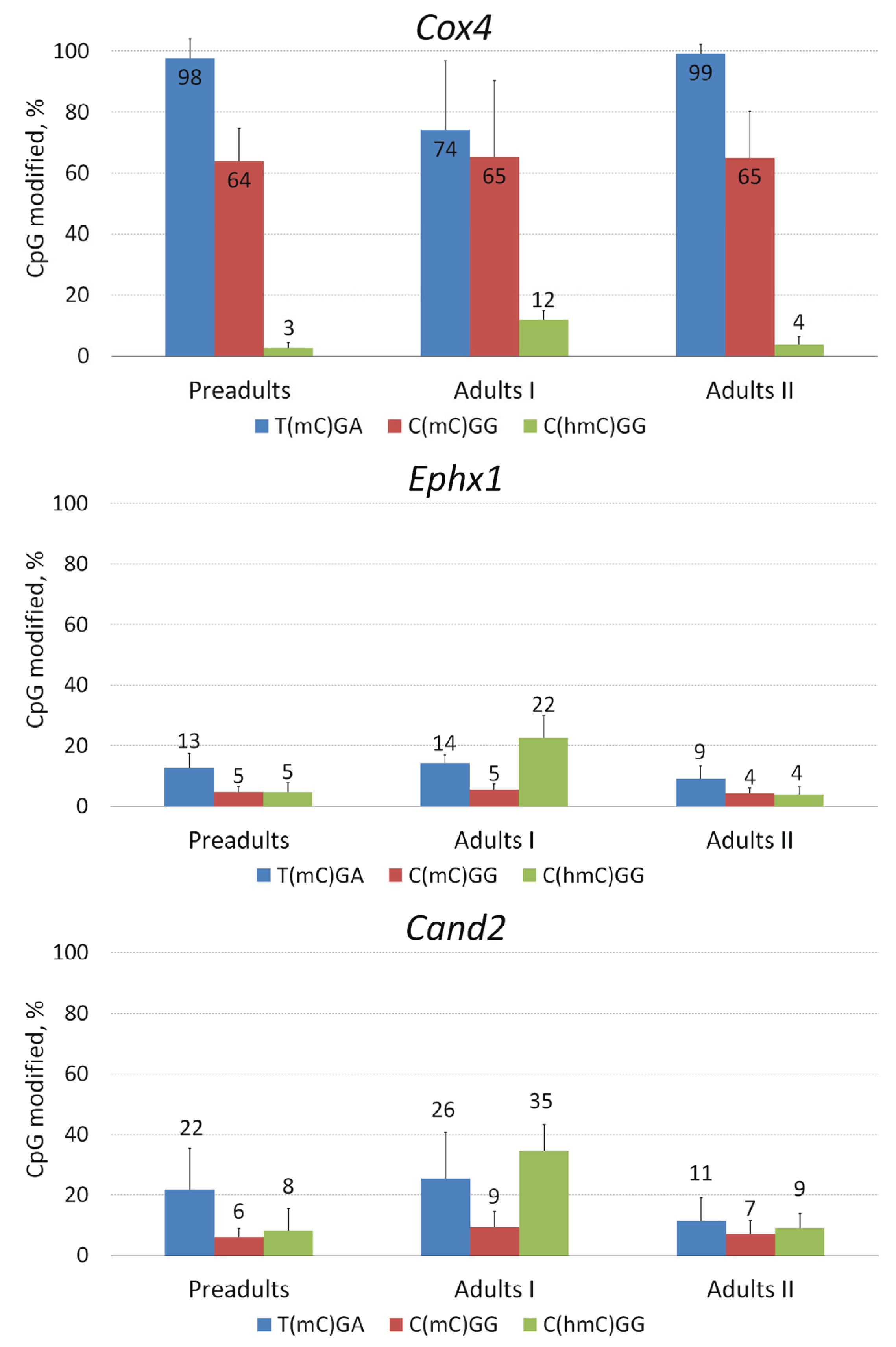
2.4. Local Methylation and Hydroxymethylation Analysis in Selected Genes of D. pulex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Symbol | Genomic Location | Length, bp | Number of Exons | Length of Exons, bp |
|---|---|---|---|---|---|
| Juvenile hormone epoxide hydrolase 1 | Ephx1 | scaffold_121: 159299–164657 | 5358 | 11 | 1598 |
| Cullin-associated Nedd8-dissociated 2 | Cand2 | scaffold_4446: 1087–2177 | 1090 | 4 | 882 |
| Cytochrome c oxidase, subunit IV | Cox4 | scaffold_23: 1334549–1335729 | 1180 | 4 | 780 |
| Gene | Primer | Primer Sequence |
|---|---|---|
| Cox4 | Forward | AGTTGGAGACCCAGTTAAAGC |
| Reverse | AGGTTTGGCAGAAAGATGCTC | |
| Cand2 | Forward | GAAATACTTGCACCGCCAGAG |
| Reverse | TACTCCTGCAGCATTTCCGTG | |
| Ephx1 | Forward | CTCAAAACCCAGTGGGGAGG |
| Reverse | TTGTCGGATTCTTGAGTCAGC |
2.5. Next Generation Sequencing
2.6. NGS Data Analysis
2.6.1. Initial Processing of Reads
2.6.2. Assembly of Genome
- Reads that matched to draft D. pulex genome [11] were collected using Kraken 0.10.5 [22]. The overlapping 3' ends of reads were merged using FLASH v1.2.11 [23]. The assembly was performed using Spades 3.5.0 [24]. Bacterial contigs were filtered out using Kraken 0.10.5 and the remaining contigs were used for the subsequent step.
- The overlapping 3ʹ ends of reads were merged using FLASH v1.2.11. Then they were assembled using Spades 3.5.0 together with the contigs from the first step. Contigs that matched to draft D. pulex genome [11] were collected using Kraken 0.10.5. Out of these contigs the bacterial ones were discarded using second run of filtering with Kraken 0.10.5.
2.6.3. Detection of Genes and Their Functional Annotation
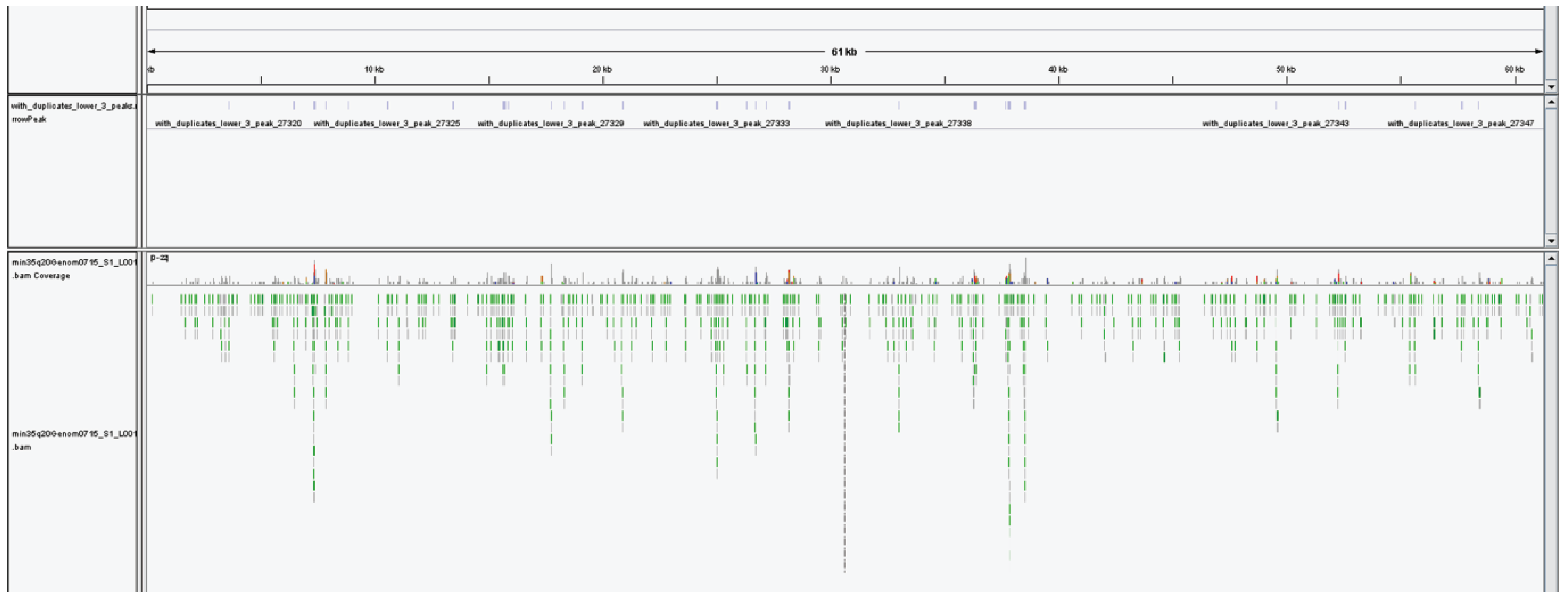
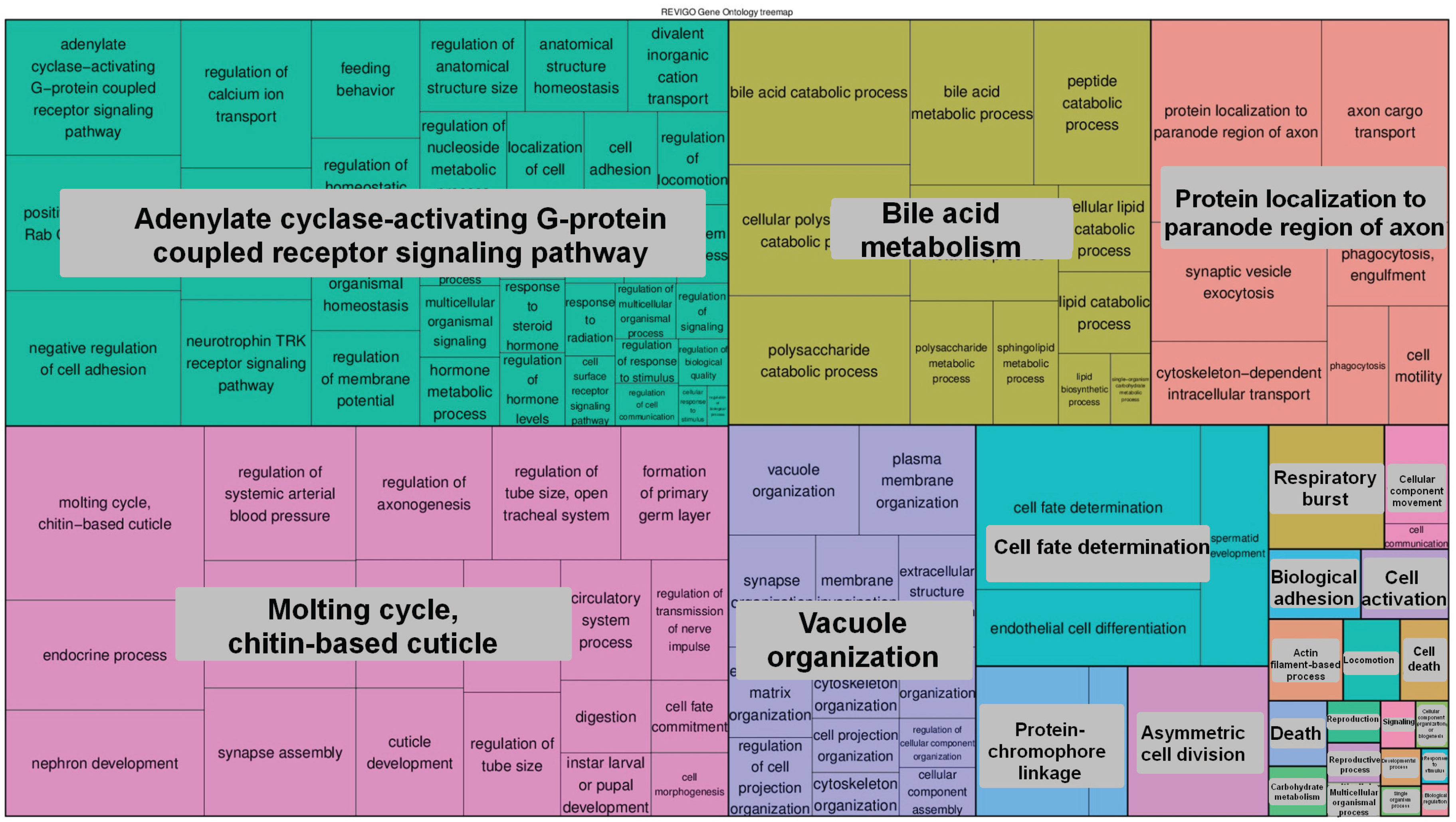
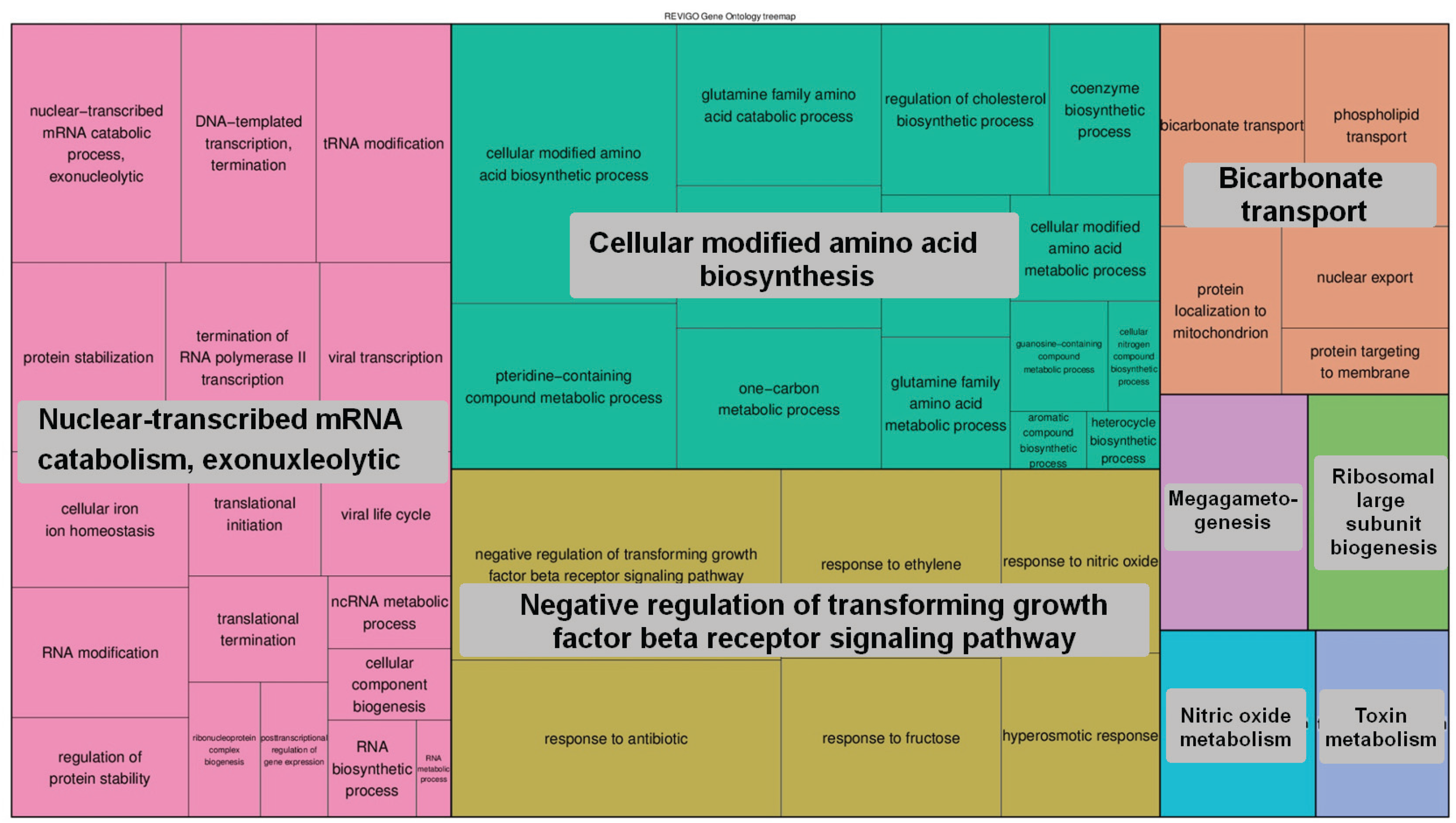
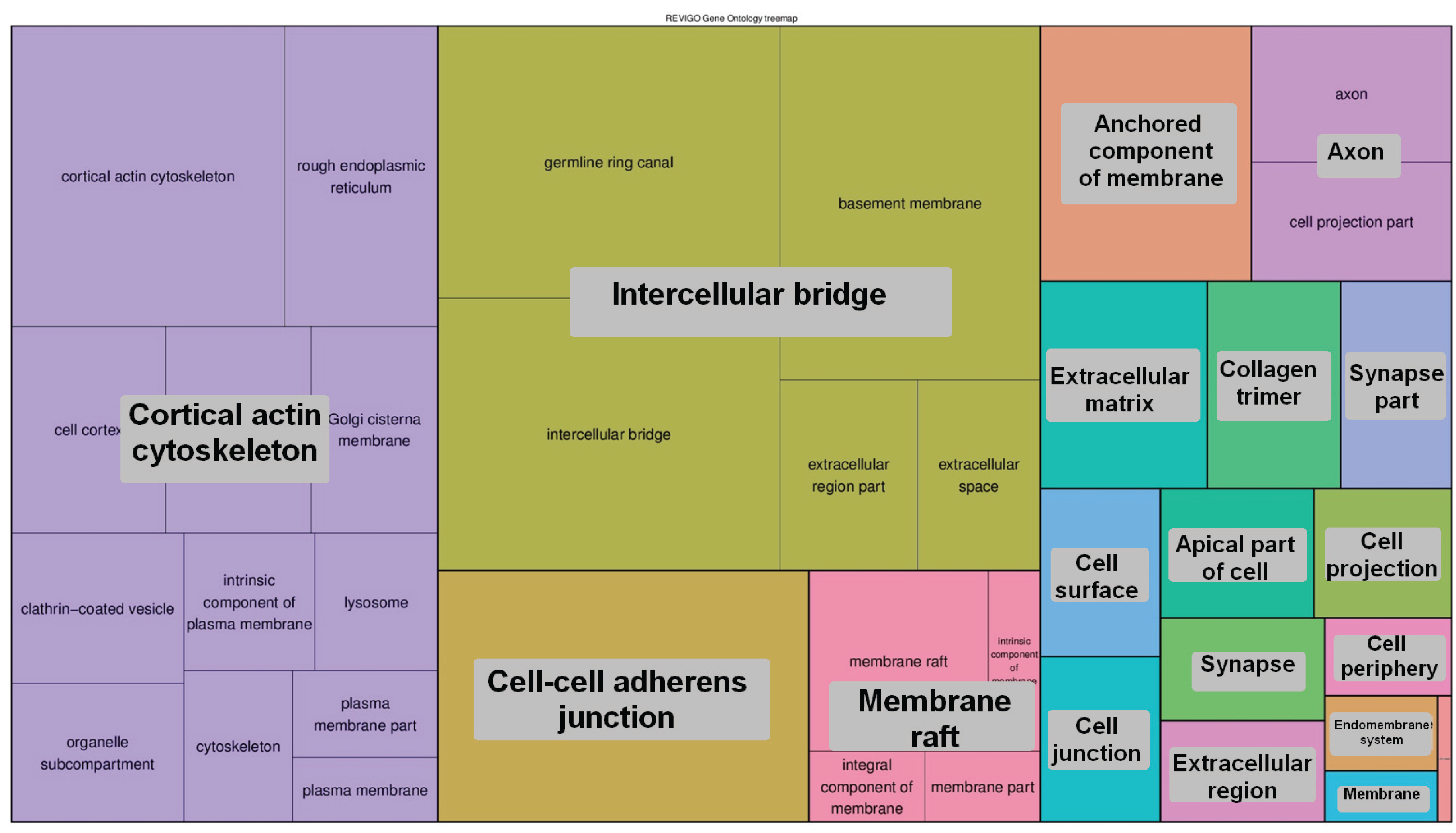
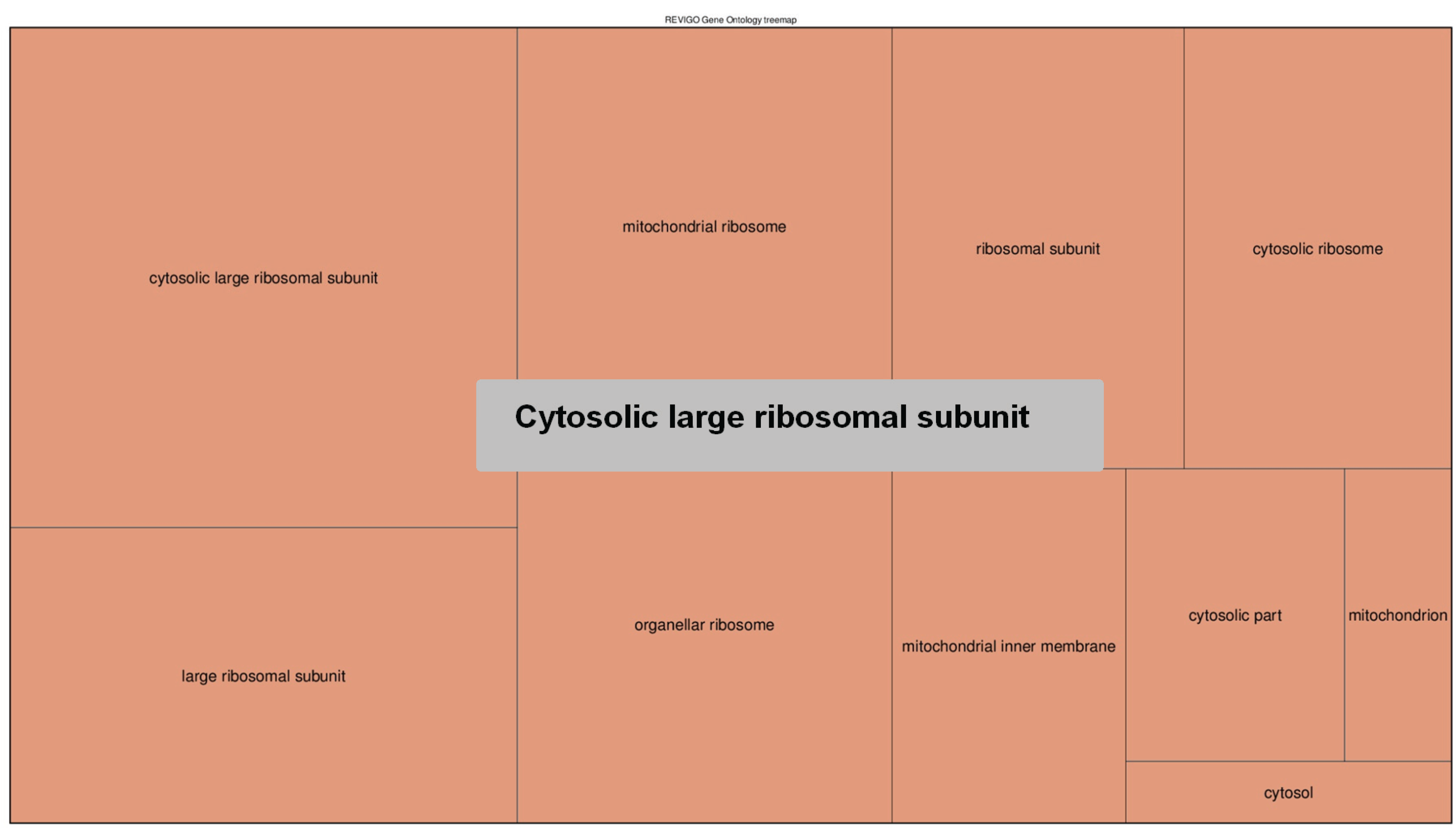
2.6.4. The 5-hmC Peak Calling and Analysis
3. Results and Discussion



| Assembled Genome | Reference Genome [11] | |||
|---|---|---|---|---|
| Assembly | Contigs | Contigs used for peak calling and annotation | Contigs | Scaffolds |
| # contigs | 17338 | 5572 | 19008 | 5191 |
| Total length | 85410271 | 66080228 | 158604366 | 197261574 |
| GC (%) | 40.74 | 41.08 | 40.76 | 40.77 |
| N50 | 10601 | 13511 | 49250 | 642089 |
| # N's per 100 kbp | 0 | 0 | 1.33 | 19582.65 |

| Group | Total_Bases | Tag_Count | Tags/Kb |
|---|---|---|---|
| CDS_Exons | 17597397 | 10651 | 0.61 |
| Introns | 17376020 | 4652 | 0.27 |
| TSS_up_1 kb | 12615965 | 1012 | 0.08 |
| TSS_up_5 kb | 35083848 | 2304 | 0.07 |
| TSS_up_10 kb | 57615402 | 2585 | 0.04 |
| TES_down_1 kb | 11356574 | 1191 | 0.10 |
| TES_down_5 kb | 31789833 | 2351 | 0.07 |
| TES_down_10 kb | 53166077 | 2506 | 0.05 |




4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stross, R.G.; Hill, R.G. Diapause induction in Daphnia requires two stimuli. Science 1965, 150, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, G.R.; Hughes, R.N. The effect of food availability, female-density and photoperiod on ephippia production in Daphnia magna Straus (Crustacea, Cladocera). Freshw. Biol. 1983, 13, 37–46. [Google Scholar] [CrossRef]
- Caceres, C.E. Interspecific variation in the abundance, production, and emergence of Daphnia diapausing eggs. Ecology 1998, 79, 1699–1710. [Google Scholar] [CrossRef]
- Wolf, H.G.; Carvalho, G.R. Testing eggs of lake-Daphnia II. In situ observations on the hatching of eggs and their contribution to population and community structure. Freshw. Biol. 1989, 22, 471–478. [Google Scholar] [CrossRef]
- Hairston, N.G., Jr.; Hansen, A.M.; Schaffner, W.R. The effect of diapause emergence on the seasonal dynamics of zooplankton assemblage. Freshw. Biol. 2000, 22, 471–478. [Google Scholar] [CrossRef]
- Hairston, N.G., Jr.; van Brunt, R.A.; Kearns, C.M.; Engstrom, D.R. Age and survivorship of diapausing eggs in a sediment egg bank. Ecology 1995, 76, 1706–1711. [Google Scholar] [CrossRef]
- Spanier, K.; Leese, F.; Mayer, C.; Colbourne, J.K.; Gilbert, D.; Pfrender, M.E.; Tollrian, R. Predator induced defences in Daphnia pulex: Selection and evaluation of internal reference genes for gene expression studies with real-time PCR. BMC Mol. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Arbačiauskas, K. Seasonal phenotypes of Daphnia: Post-diapause and directly developing offspring. J. Limnol. 2004, 63, S7–S15. [Google Scholar] [CrossRef]
- Harris, K.M.D.; Bartlett, N.J.; Lloyd, V.K. Daphnia as an emerging epigenetic model organism. Genet. Res. Intern. 2012. [Google Scholar] [CrossRef] [PubMed]
- Mirbahai, L.; Chipman, J.K. Epigenetic memory of environmental organisms: A reflection oflifetime stressor exposures. Mutat. Res. 2014, 764, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K.; et al. The ecoresponsive genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Vandegehuchte, M.B.; Kyndt, T.; Vanholme, B.; Haegeman, A.; Gheysen, G.; Janssen, C.R. Occurrence of DNA methylation in Daphnia magna and influence of multigeneration Cd exposure. Environ. Intern. 2009, 35, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandegehuchte, M.B.; Lemiere, F.; Janssen, C.R. Quantitative DNA-methylation in Daphnia magna and effects of multigeneration Zn exposure. Comp. Biochem. Physiol. 2009, 150, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Glastad, K.M.; Hunt, B.G.; Yi, S.V.; Goodisman, M.A.D. DNA methylation in insects: On the brink of the epigenomic era. Insect Mol. Biol. 2011, 20, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Zhang, D.; de Souza, R.F.; Pukkila, P.J.; Rao, A.; Aravind, L. Lineage-specific expansions of TET/JBP genes and a new class of DNA transposons shape fungal genomic and epigenetic landscapes. PNAS 2014, 111, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Arbačiauskas, K; Gasiūnaitė, Z.R. Growth and fecundity of Daphnia after diapause and their impact on the development of a population. Hydrobiologia 1996, 320, 209–222. [Google Scholar]
- Arbačiauskas, K. Life-history traits of exephippial and parthenogenetically derived daphnids: Indicators of different life-history strategies. Arch. Hydrobiol. Spec. Issues Adv. Limnol. 1998, 52, 339–358. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB J. 2011. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Field, L.M.; Lyko, F.; Mandrioli, M.; Prantera, G. DNA methylation in insects. Insect Mol. Biol. 2004, 13, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Nong, G.; Preston, J.F.; Ben-Ami, F.; Ebert, D. Comparative metagenomics of Daphnia symbionts. BMC Genomics 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, S.; Zhou, C.; Aoki, T.; Sato, N.; Chiba, T.; Tanaka, K.; Yoshida, S.; Nabeshima, Y.; Nabeshima, Y.; Tamura, T.A. TBP-interacting protein 120B (TIP120B)/cullin-associated and neddylation-dissociated 2 (CAND2) inhibits SCF-dependent ubiquitination of myogenin and accelerates myogenic differentiation. J. Biol. Chem. 2007, 282, 9017–9028. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Niklison-Chirou, M.V.; Inoue, S.; Tomasini, R.; Harris, I.S.; Marino, A.; Federici, M.; Dinsdale, D.; Knight, R.A.; Melino, G.; et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012, 26, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Ge, X.; Wang, W.; Yu, J.; Hu, S. A gene catalogue for post-diapause development of an anhydrobiotic arthropod Artemia franciscana. BMC Genomics 2009. [Google Scholar] [CrossRef] [PubMed]
- Brisson, J. Aphid wing dimorphisms: Linking environmental and genetic control of trait variation. Phil. Transact. R. Soc. Ser. B 2010, 365, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Nijhout, H.F.; Riddiford, L.M.; Mirth, C.; Shingleton, A.W.; Suzuki, Y.; Callier, V. The developmental control of size in insects. WIREs Dev. Biol. 2014, 3, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Benzie, J.A.H. Cladocera: The Genus Daphnia (Including Daphniopsis); Backhuys Publishers: Leiden, The Netherland, 2005. [Google Scholar]
- Duggan, I.C.; Robinson, K.V.; Burns, C.W.; Banks, J.C.; Hogg, I.D. Identifying incertebrate invasionsusing morphological and molecular analyse: North American Daphnia “pulex” in New Zealand fresh water. Aquat. Invasions 2012, 7, 585–590. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Li, X.; Yan, L.; Tan, Y.; Li, R.; Zhao, Y.; Wang, Y.; Xie, J.; Zhang, Y.; Song, C.; et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, L.; Helfand, B.T.; Jang, T.L.; Sharma, V.; Kozlowski, J.; Kuzel, T.M.; Zhu, L.J.; Yang, X.J.; Javonovic, B.; et al. TGF-β regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS ONE 2011, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Szulwach, K.E.; Su, Y.; Li, Y.; Yao, B.; Xu, Z.; Shin, J.H.; Xie, B.; Gao, Y.; Ming, G.L.; et al. Genome-wide antagonism between 5-hydroxymethylcytosine and DNA methylation in the adult mouse brain. Front. Biol. 2014, 9, 66–74. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strepetkaitė, D.; Alzbutas, G.; Astromskas, E.; Lagunavičius, A.; Sabaliauskaitė, R.; Arbačiauskas, K.; Lazutka, J. Analysis of DNA Methylation and Hydroxymethylation in the Genome of Crustacean Daphnia pulex. Genes 2016, 7, 1. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7010001
Strepetkaitė D, Alzbutas G, Astromskas E, Lagunavičius A, Sabaliauskaitė R, Arbačiauskas K, Lazutka J. Analysis of DNA Methylation and Hydroxymethylation in the Genome of Crustacean Daphnia pulex. Genes. 2016; 7(1):1. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7010001
Chicago/Turabian StyleStrepetkaitė, Dovilė, Gediminas Alzbutas, Eimantas Astromskas, Arūnas Lagunavičius, Rasa Sabaliauskaitė, Kęstutis Arbačiauskas, and Juozas Lazutka. 2016. "Analysis of DNA Methylation and Hydroxymethylation in the Genome of Crustacean Daphnia pulex" Genes 7, no. 1: 1. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7010001