
A Genome-Wide Identification and Analysis of the Basic Helix-Loop-Helix Transcription Factors in Brown Planthopper, Nilaparvata lugens

Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Rearing
2.2. bHLH Sequence Identification from N. lugens
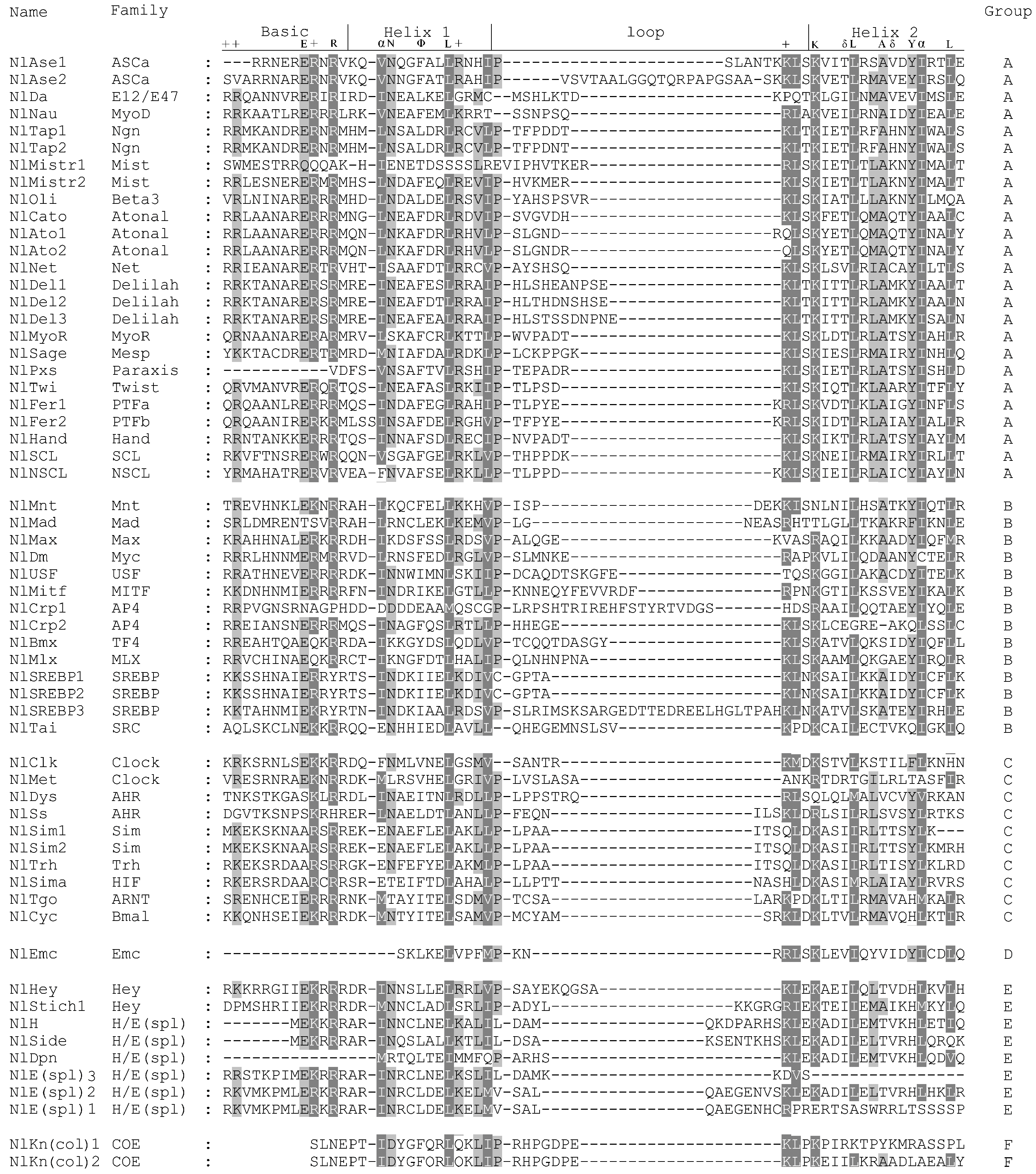
2.3. Multiple Sequence Alignments and Phylogenetic Analysis
2.4. Domain Prediction
2.5. Molecular Cloning
3. Results and Discussion
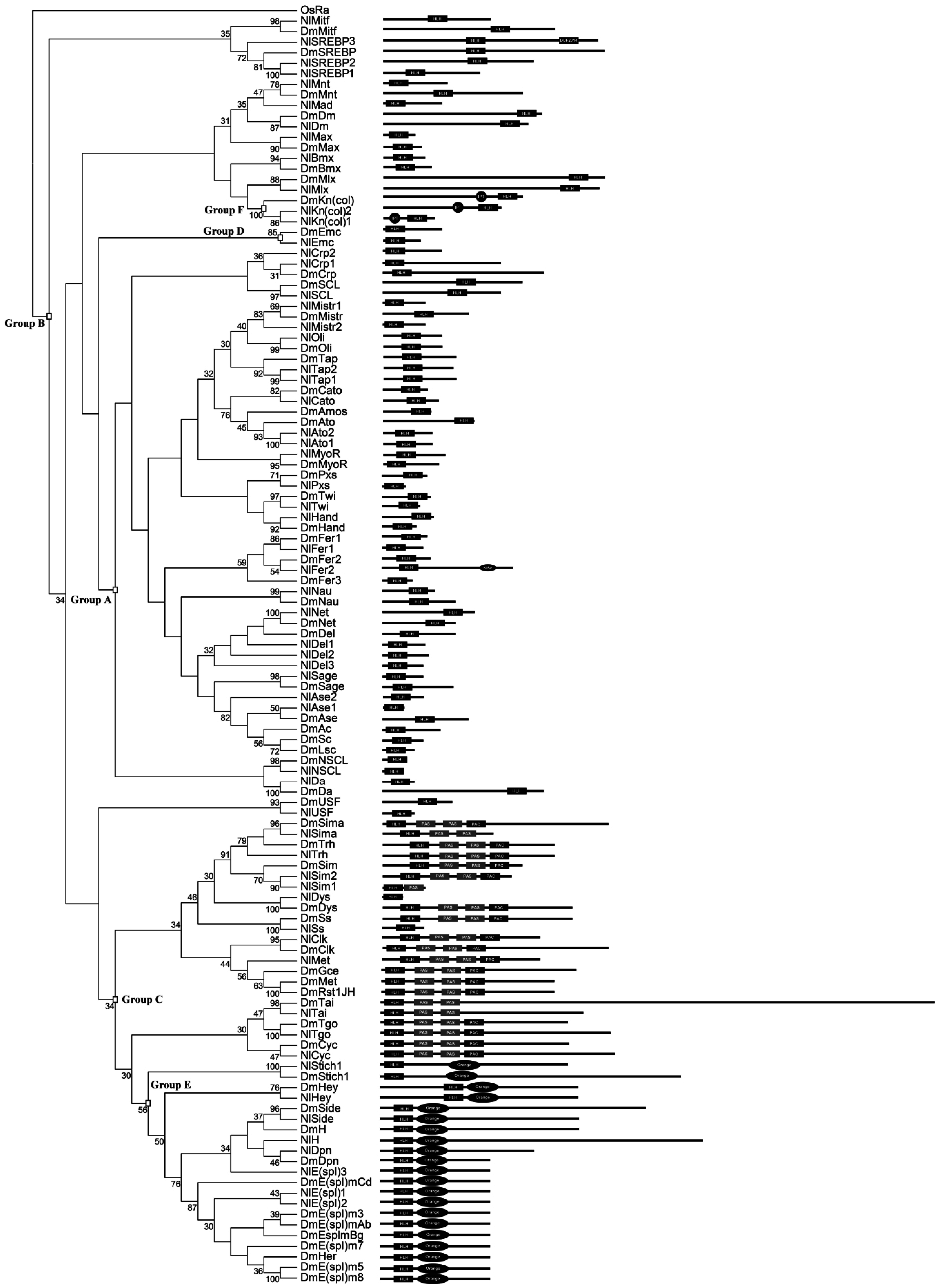
3.1. Identification of bHLH Members in N. lugens
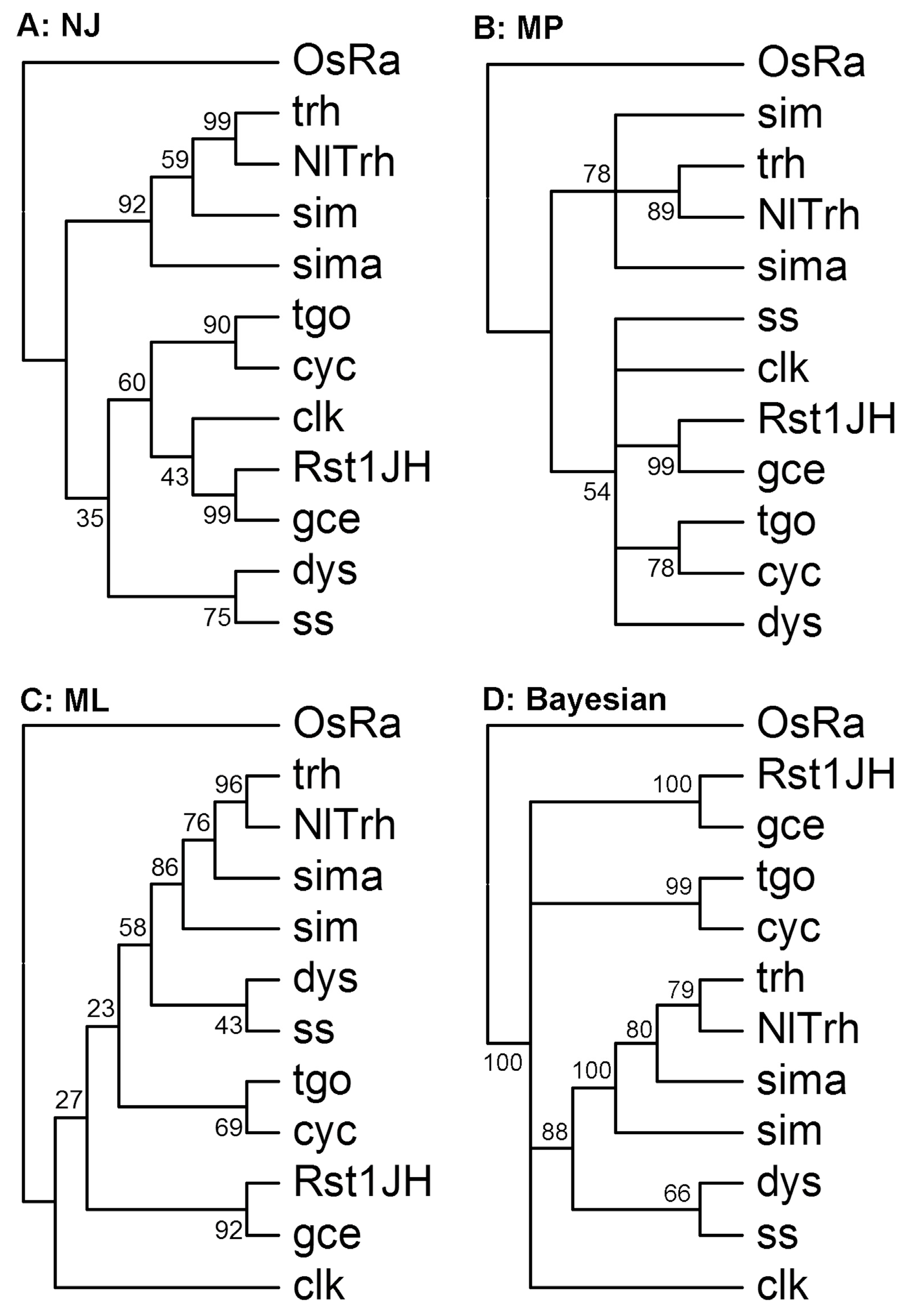
3.2. Identification of Orthologous Families
3.3. Genomic Distribution of N. lugens bHLH Genes
3.4. Intron–Exon Structure of N. lugens bHLH Genes
3.5. Molecular Cloning and Predicted Function of N. lugens bHLHs
3.6. The bHLH Repertoire of N. lugens and Other Insect Species
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Robinson, K.A.; Lopes, J.M. Survey and summary: Saccharomyces cerevisiae basic helix–loop–helix proteins regulate diverse biological processes. Nucleic Acids Res. 2000, 28, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Atchley, R.W.; Terhalle, W.; Dress, A. Positional dependence, cliques, and predictive motifs in the bHLH protein domain. J. Mol. Evolut. 1999, 48, 501–516. [Google Scholar] [CrossRef]
- Robinson, K.A.; Koepke, J.I.; Kharodawala, M.; Lopes, J.M. A network of yeast basic helix–loop–helix interactions. Nucleic Acids Res. 2000, 28, 4460–4466. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. Functional genomics: Recognizing DNA in the library. Nature 2000, 404, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Murre, C.; McCaw, P.S.; Baltimore, D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 1989, 56, 777–783. [Google Scholar] [CrossRef]
- Wang, X.H.; Wang, Y.; Zhang, D.B.; Liu, A.K.; Yao, Q.; Chen, K.P. A genome-wide identification of basic helix-loop-helix motifs in Pediculus humanus corporis (phthiraptera: Pediculidae). J. Insect Sci. 2014, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.-W.; Wang, Y.; Chen, K.-P.; Yao, Q.; Zhang, D.; Guo, M. The basic helix-loop-helix transcription factor family in the pea aphid, Acyrthosiphon pisum. J. Insect Sci. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-T.; Wang, Y.; Wang, X.-H.; Tao, X.-F.; Yao, Q.; Chen, K.-P. A genome-wide identification and classification of basic helix-loop-helix genes in the jewel wasp, Nasonia vitripennis (hymenoptera: Pteromalidae). Genome 2014, 57, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Wang, Y.; Dang, C.; Zhang, D.; Song, H.; Yao, Q.; Chen, K. A genome-wide identification and analysis of the basic helix-loop-helix transcription factors in the ponerine ant, Harpegnathos saltator. BMC Evolut. Biol. 2012, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, K.; Yao, Q.; Wang, W.; Zhu, Z. The basic helix-loop-helix transcription factor family in the honey bee, Apis mellifera. J. Insect Sci. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Bitra, K.; Tan, A.; Dowling, A.; Palli, S.R. Functional characterization of pas and hes family bHLHtranscription factors during the metamorphosis of the red flour beetle, Tribolium castaneum. Gene 2009, 448, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.-Y.; Meng, Q.-W.; Lü, F.-G.; Guo, W.-C.; Ahmat, T.; Li, G.-Q. The basic helix–loop–helix transcription factors in the colorado potato beetle Leptinotarsa decemlineata. J. Asia Pac. Entomol. 2015, 18, 197–203. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, K.; Yao, Q.; Wang, W.; Zhu, Z. The basic helix-loop-helix transcription factor family in Bombyx mori. Dev. Genes Evolut. 2007, 217, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.B.; Wang, Y.; Liu, A.K.; Wang, X.H.; Dang, C.W.; Yao, Q.; Chen, K.P. Phylogenetic analyses of vector mosquito basic helix-loop-helix transcription factors. Insect Mol. Biol. 2013, 22, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Simionato, E.; Ledent, V.; Richards, G.; Thomas-Chollier, M.; Kerner, P.; Coornaert, D.; Degnan, B.M.; Vervoort, M. Origin and diversification of the basic helix-loop-helix gene family in metazoans: Insights from comparative genomics. BMC Evolut. Biol. 2007, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gyoja, F. A genome-wide survey of bHLH transcription factors in the Placozoan Trichoplax adhaerens reveals the ancient repertoire of this gene family in metazoan. Gene 2014, 542, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.W.; Barbel, S.; Jan, L.Y.; Jan, Y.N. A genomewide survey of basic helix-loop-helix factors in Drosophila. Proc. Natl. Acad. Sci. USA 2000, 97, 10436–11041. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Dolde, C.; Gillison, M.L.; Kato, G.J. Discrimination between related DNA sites by a single amino acid residue of Myc-related basic-helix-loop-helix proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Atchley, W.R.; Fitch, W.M. A natural classification of the basic helix–loop–helix class of transcription factors. Proc. Natl. Acad. Sci. USA 1997, 94, 5172–5176. [Google Scholar] [CrossRef] [PubMed]
- Ledent, V.; Vervoort, M. The basic helix-loop-helix protein family: Comparative genomics and phylogenetic analysis. Genome Res. 2001, 11, 754–770. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhou, X.; Zhang, C.-X.; Yu, L.-L.; Fan, H.-W.; Wang, Z.; Xu, H.-J.; Xi, Y.; Zhu, Z.-R.; Zhou, W.-W.; et al. Genomes of the rice pest brown planthopper and its endosymbionts reveal complex complementary contributions for host adaptation. Genome Biol. 2014, 15, 521. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-L.; Wan, P.-J.; Wang, W.-X.; Lai, F.-X.; Fu, Q. Ran involved in the development and reproduction is a potential target for rna-interference-based pest management in Nilaparvata lugens. PLoS ONE 2015, 10, e0142142. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Xu, X.; Liang, Y.; Tian, H.; Pan, Z.; Jin, S.; Wang, N.; Zhang, W. The insect ecdysone receptor is a good potential target for RNAi-based pest control. Int. J. Biol. Sci. 2014, 10, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yao, Y.; Wang, B. Methoprene-tolerant (Met) and krüpple-homologue 1 (Kr-h1) are required for ovariole development and egg maturation in the brown plant hopper. Sci. Rep. 2015, 5, 18064. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Chen, X.; Liu, W.-T.; Zhang, X.-Y.; Chen, M.-X.; Zhou, Q. Nutritional signaling regulates vitellogenin synthesis and egg development through juvenile hormone in Nilaparvata lugens (stål). Int. J. Mol. Sci. 2016, 17, 269. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Chen, X.; Liu, W.T.; Zhou, Q. TOR pathway-mediated juvenile hormone synthesis regulates nutrient-dependent female reproduction in Nilaparvata lugens (stål). Int. J. Mol. Sci. 2016, 17, 438. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef] [PubMed]
- Slater, G.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.-J.; Yang, L.; Wang, W.-X.; Fan, J.-M.; Fu, Q.; Li, G.-Q. Constructing the major biosynthesis pathways for amino acids in the brown planthopper, Nilaparvata lugens stål (hemiptera: Delphacidae), based on the transcriptome data. Insect Mol. Biol. 2014, 23, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. Smart: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Doerks, T.; Copley, R.R.; Schultz, J.; Ponting, C.P.; Bork, P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002, 12, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.; Cerutti, L.; de Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. Prosite, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010, 38, D161–D166. [Google Scholar] [CrossRef] [PubMed]
- Kafri, R.; Springer, M.; Pilpel, Y. Genetic redundancy: New tricks for old genes. Cell 2009, 136, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Ledent, V.; Paquet, O.; Vervoort, M. Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biol. 2002, 3, 1–18. [Google Scholar] [CrossRef]
- Ponting, C.P.; Aravind, L. Pas: A multifunctional domain family comes to light. Curr. Biol. 1997, 7, R674–R677. [Google Scholar] [CrossRef]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Davis, R.L.; Turner, D.L. Vertebrate hairy and enhancer of split related proteins: Transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene 2001, 20, 8342–8357. [Google Scholar] [CrossRef] [PubMed]
- Sharpton, T.J.; Neafsey, D.E.; Galagan, J.E.; Taylor, J.W. Mechanisms of intron gain and loss in Cryptococcus. Genome Biol. 2008, 9, R24. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.A. On the origin of RNA splicing and introns. Cell 1985, 42, 397–400. [Google Scholar] [CrossRef]
- Crick, F. Split genes and rna splicing. Science 1979, 204, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.H. How were introns inserted into nuclear genes? Trends Genet. TIG 1989, 5, 213–216. [Google Scholar] [CrossRef]
- Hankeln, T.; Friedl, H.; Ebersberger, I.; Martin, J.; Schmidt, E.R. A variable intron distribution in globin genes of Chironomus: Evidence for recent intron gain. Gene 1997, 205, 151–160. [Google Scholar] [CrossRef]
- Li, W.; Tucker, A.E.; Sung, W.; Thomas, W.K.; Lynch, M. Extensive, recent intron gains in Daphnia populations. Science 2009, 326, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Catania, F.; Lynch, M. Where do introns come from? PLoS Biol. 2008, 6, e283. [Google Scholar] [CrossRef] [PubMed]
- Irimia, M.; Rukov, J.L.; Penny, D.; Vinther, J.; Garcia-Fernandez, J.; Roy, S.W. Origin of introns by ‘intronization’ of exonic sequences. Trends Genet. 2008, 24, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Fink, G.R. Pseudogenes in yeast? Cell 1987, 49, 5–6. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. The pattern of intron loss. Proc. Natl. Acad. Sci. USA 2005, 102, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.-Y.; Qin, X.; Yu, B.; Chen, L.-B.; Wang, Z.-C.; Zhang, C.-X. Genomic insights into the serine protease gene family and expression profile analysis in the planthopper, Nilaparvata lugens. BMC Genom. 2014, 15, 507. [Google Scholar] [CrossRef] [PubMed]
- Simionato, E.; Kerner, P.; Dray, N.; Le Gouar, M.; Ledent, V.; Arendt, D.; Vervoort, M. Atonal- and achaete-scute-related genes in the annelid Platynereis dumerilii: Insights into the evolution of neural basic-helix-loop-helix genes. BMC Evol. Biol. 2008, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, J.; de Taffin, M.; Crozatier, M.; Vincent, A.; Dubois, L. Combinatorial coding of Drosophila muscle shape by Collier and Nautilus. Dev. Biol. 2012, 363, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.T.; Liu, Y.; Ayyanathan, K.; Benner, C.; Jiang, Y.; Prokop, J.W.; Paz, H.; Wang, D.; Li, H.-R.; Fu, X.-D.; et al. An evolutionarily conserved DNA architecture determines target specificity of the twist family bHLH transcription factors. Genes Dev. 2015, 29, 603–616. [Google Scholar] [CrossRef] [PubMed]
- García-Bellido, A.; de Celis, J.F. The complex tale of the achaete-scute complex: A paradigmatic case in the analysis of gene organization and function during development. Genetics 2009, 182, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manetopoulos, C.; Hansson, A.; Karlsson, J.; Jönsson, J.-I.; Axelson, H. The LIM-only protein LMO4 modulates the transcriptional activity of HEN1. Biochem. Biophys. Res. Commun. 2003, 307, 891–899. [Google Scholar] [CrossRef]
- Tanaka-Matakatsu, M.; Miller, J.; Borger, D.; Tang, W.J.; Du, W. Daughterless homodimer synergizes with eyeless to induce atonal expression and retinal neuron differentiation. Dev. Biol. 2014, 392, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Nachman, A.; Halachmi, N.; Matia, N.; Manzur, D.; Salzberg, A. Deconstructing the complexity of regulating common properties in different cell types: Lessons from the delilah gene. Dev. Biol. 2015, 403, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Osanai, K.; Ohyoshi, T.; Wang, H.-B.; Iwanaga, M.; Kawasaki, H. Ecdysteroid promotes cell cycle progression in the Bombyx wing disc through activation of c-myc. Insect Biochem. Mol. Biol. 2016, 70, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harmansa, S.; Hamaratoglu, F.; Affolter, M.; Caussinus, E. Dpp spreading is required for medial but not for lateral wing disc growth. Nature 2015, 527, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.J.; Wong, R.H.F.; Pandya, N.; Sul, H.S. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. J. Biol. Chem. 2007, 282, 5453–5467. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, F.; Shen, J.; Rosbash, M. PDF and cAMP enhance PER stability in Drosophila clock neurons. Proc. Natl. Acad. Sci. USA. 2014, 111, E1284–E1290. [Google Scholar] [CrossRef] [PubMed]
- Crane, B.R.; Young, M.W. Interactive features of proteins composing eukaryotic circadian clocks. Annu. Rev. Biochem. 2014, 83, 191–219. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Krasnow, M. Development of the Drosophila tracheal system. Dev. Drosoph. Melanogaster 1993, 1, 609–685.69. [Google Scholar]
- Wilk, R.; Weizman, I.; Shilo, B.Z. Trachealess encodes a bhlh-pas protein that is an inducer of tracheal cell fates in drosophila. Genes Dev. 1996, 10, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Long, S.K.; Fulkerson, E.; Breese, R.; Hernandez, G.; Davis, C.; Melton, M.A.; Chandran, R.R.; Butler, N.; Jiang, L.; Estes, P. A comparison of midline and tracheal gene regulation during Drosophila development. PLoS ONE 2014, 9, e85518. [Google Scholar] [CrossRef] [PubMed]
- Lerner, I.; Bartok, O.; Wolfson, V.; Menet, J.S.; Weissbein, U.; Afik, S.; Haimovich, D.; Gafni, C.; Friedman, N.; Rosbash, M.; et al. Clk post-transcriptional control denoises circadian transcription both temporally and spatially. Nat. Commun. 2015, 6, 7056. [Google Scholar] [CrossRef] [PubMed]
- Jaumouillé, E.; Machado Almeida, P.; Stähli, P.; Koch, R.; Nagoshi, E. Transcriptional regulation via nuclear receptor crosstalk required for the Drosophila circadian clock. Curr. Biol. 2015, 25, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Jindra, M.; Uhlirova, M.; Charles, J.-P.; Smykal, V.; Hill, R.J. Genetic evidence for function of the bHLH-PAS protein Gce/Met as a juvenile hormone receptor. PLoS Genet. 2015, 11, e1005394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitra, K.; Palli, S.R. Bhlh transcription factors: Potential target sites for insecticide development. In Advanced Technologies for Managing Insect Pests; Ishaaya, I., Palli, R.S., Horowitz, R.A., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 13–30. [Google Scholar]
- Cheng, Y.J.; Tsai, J.W.; Hsieh, K.C.; Yang, Y.C.; Chen, Y.J.; Huang, M.S.; Yuan, S.S. Id1 promotes lung cancer cell proliferation and tumor growth through akt-related pathway. Cancer Lett. 2011, 307, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.T.; Shin, S.W.; Dou, W.; Roy, S.; Zhao, B.; Hou, Y.; Wang, X.L.; Zou, Z.; Girke, T.; Raikhel, A.S. Hairy and groucho mediate the action of juvenile hormone receptor methoprene-tolerant in gene repression. Proc. Natl. Acad. Sci. USA 2016, 113, E735–E743. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.; Wang, Y.; Zhang, D.; Yao, Q.; Chen, K. A genome-wide survey on basic helix-loop-helix transcription factors in giant panda. PLoS ONE 2011, 6, e26878. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Usui, T.; Satoh, D.; Moriyama, S.; Shimono, K.; Itoh, T.; Shirahige, K.; Uemura, T. Sensory-neuron subtype-specific transcriptional programs controlling dendrite morphogenesis: Genome-wide analysis of abrupt and knot/collier. Dev. Cell 2013, 27, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer (5′to 3′) | Reverse Primer (5′to 3′) | Amplicon Size (bp) |
|---|---|---|---|
| NlAse1 | CGTCATTCGCACTCGAGATGG | GGACATGGGCTGAACGTGGT | 473 |
| NlAse2 | TAACAAGCCCTCACGGAGCGT | GTGTACCTTGCGTTCCAGGA | 624 |
| NlDa | AAGTTGAGTTCTCAGCCACGGA | GGGCACTAACTAGTCGAGTGG | 735 |
| NlTap1 | TCACCGTCTGCATCGGACAG | CCATAGTAGGTCAACTCTTGGTG | 433 |
| NlTap2 | ATGTAACCGTCTGCATCGGA | CCACGTAATCAGGCGAACTC | 405 |
| NlMistr1 | GTCAAGTATGAGTGCCGACAG | CCTTGTTCCTCGTAGGGCGA | 246 |
| NlMistr2 | CGCCAAACGGAAATGTCTGC | GTGCCATAATGTAGTTCTTGGCT | 233 |
| NlOli | CTACAACAGTTGAGCGGACC | GGAATCGACATCGTTCCTTGAGC | 314 |
| NlCato | ACCGTCGTCAAGAAGCGT | ACGCTGCAGCATATCACAC | 189 |
| NlAto1 | AGTCGCCTCCACCGTTCTGCAA | ACTGTAGCAAGTCGTAGAGGGCA | 615 |
| NlAto2 | AGTCGCCTCCACCGTTCTGCAA | ACTGTAGCAAGTCGTAGAGGGCA | 615 |
| NlSage | GATAAATTGCCGCTATGCAAGC | TCAATGGTTGTAAGCAGTGGTG | 321 |
| NlPxs | ATTTCAGTGTGAATTCGGCAT | AAGAACATTAACCTGTTGTGAGTTG | 155 |
| NlTwi | GGCAAACACGACTTGACCAG | GGCGTTCTCTTACATTCGCCA | 325 |
| NlFer1 | AGGCACTTCCTGGATGGCTACGT | GGGTCCACACCTTGGCGTACAT | 586 |
| NlFer2 | AGAATGCAGTACAAGCGGTC | CATCCTTCTGTTCTGAATGTGC | 379 |
| NlHand | AACGAGGTGCCTGTCATACG | TTTACCTGATTGGTGGCCCT | 302 |
| NlSCL | TGCTGAGGAAGGTGTTCACC | GCGTCGGATGCGTTACTCAA | 517 |
| NlDel1 | AAGCTACTCGTTGCGACCCAG | ACTCTGAAATGAGGCTGACGT | 602 |
| NlDel2 | TAACTCCGATTTGGCGTCGACC | TCAAAATCCACCGCTGACGT | 249 |
| NlDel3 | CCATGAACCCGCAGGTGCTA | GGGCGTCAACTTGTAGGGCT | 399 |
| NlMnt | TGAGATTAGGAACTCGCGAAGTG | CAATGAGTAGAATTGGAAGGGCT | 703 |
| NlMad | AATGGTTCCCCTGGGCAACGA | GAGTCGGCTGGTGGACATAGC | 505 |
| NlDm | GTATGAACCGCGACTGGCTCCA | AGACTGCGGGCCACCGTCTT | 547 |
| NlUSF | TAATTCCTGATTGCGCTCAGGAC | GATCTGAATTGGGTATGATGCCA | 252 |
| NlCrp2 | CCGAATACCACATTCACTCG | ATCACTGAGCCAGGGTATGG | 248 |
| NlBmx | CCATCAAGAAGGGGTATGACTCG | GCCAACTGAAGACACATGCT | 378 |
| NlMlx | ACAAATCCTACTGGCAGCGA | TGATATGCACAGCTGCCGAG | 1176 |
| NlSREBP3 | GCAGATGGCCGGTCAACCTT | GTCCTTGGATCGCCTTTGCAG | 1207 |
| NlTai | TATGTCAGCACAGCAAGTGCCTT | ACTTCTAGGAGGAGATTGCCGAA | 271 |
| NlDys | AGGTTCGACACGAACAAGTC | GTAACTGCGAAAGACGCTGTC | 130 |
| NlSim1 | ACATCGACCAAGCGGAAGTTGCA | TGACACTGGTGTATCCAGCCGTG | 472 |
| NlSim2 | TCATCTACTCCAGACGCTGG | TGCATTCCTTTTCGCTAGGACG | 283 |
| NlTrh | CGTGATCGAAACTGCAAGGTTCG | TTACTTTGAGCGATTTGGCAGCT | 574 |
| NlSima | CACCTCGACAAGGCGTCCAT | CGTAGCCAAGAAACTCTTCCA | 1384 |
| NlTgo | CACTCGATGGACGGCAAGTT | TGGCTGTGCGTGGTAGAGTG | 810 |
| NlCyc | TACGCGATGTCTCGCAAGCTGGA | ACAATGTACTCGCGGTCCATCTG | 1194 |
| NlMet | TATCGGTTCCACTCCACAAA | AAGGGATCATTGTTGAAGCC | 440 |
| NlEmc | TGTGACTTGCAGTACGCTCTGGA | AGGCTTCCTGCGTGGAACAC | 196 |
| NlHey | TGGACTACCACAACATCGGCTT | TTCATCTGAGAGGAAACCTGGT | 607 |
| NlSide | GAGGACATGCTGATGGCCGTCAA | AGACATCTTCGTCCTTGTCGGCA | 568 |
| NlDpn | TACCTGGAAACGTTCTGCCAT | TGTTTCAGTAGATGGTTGAGGCT | 554 |
| NlH | AACTGGAGAAAGCGGACATCCTT | TGGGGATAGTGCCTCAACAA | 1681 |
| NlE(spl)1 | GAAGGCTGACATCCTCGAGC | CTACCACGGCCTCCAGACTG | 482 |
| NlE(spl)3 | GGAAAGCGACGAGGATTACTG | GCGATTTGAGTTCATTGAGGCA | 191 |
| NlKn(col)1 | TTGATCCCTCAGATGGCCTGTA | GCAAAACTGTTTCGACTTGTAGG | 266 |
| NlKn(col)2 | TATGTCTCCCTGAACGAGCCA | TATTTGGAAGACCCGACCAGTGG | 680 |
| No. | Gene Name | Family | Fruit Fly Homolog | Statistical Support | Gene ID | Evidence Support | |||
|---|---|---|---|---|---|---|---|---|---|
| NJ | MP | ML | Bayesian | ||||||
| 01 | NlAse1 | ASCa | ase | 99 | 97 | 87 | 99 | NA | EST |
| 02 | NlAse2 | ASCa | ase | 99 | 100 | 98 | 68 | NLU023528 | RT-PCR and EST |
| 03 | NlDa | E12/E17 | da | 100 | 100 | 100 | 100 | NLU002710 | RT-PCR and EST |
| 04 | NlNau | MyoD | nau | 99 | 99 | 95 | 100 | NLU022422 | RT-PCR and EST |
| 05 | NlTap1 | Ngn | tap | 97 | 91 | 91 | 100 | NLU007911 | RT-PCR and EST |
| 06 | NlTap2 | Ngn | tap | 97 | 92 | 91 | 100 | NLU023195 | RT-PCR and EST |
| 07 | NlMistr1 | Mist | Mistr | 96 | 89 | 94 | 100 | NLU012420 | RT-PCR and EST |
| 08 | NlMistr2 | Mist | Mistr | 100 | 98 | 98 | 100 | NLU027753 | RT-PCR and EST |
| 09 | NlOli | Beta3 | Oli | 100 | 100 | 100 | 100 | NLU011046 | RT-PCR and EST |
| 10 | NlCato | Atonal | cato | 37 | 97 | 78 | 98 | NLU013048 | RT-PCR and EST |
| 11 | NlAto1 | Atonal | ato | 99 | 88 | 92 | 98 | NLU020408 | RT-PCR and EST |
| 12 | NlAto2 | Atonal | ato | 98 | 86 | 92 | 98 | NLU012608 | RT-PCR and EST |
| 13 | NlNet | Net | net | 100 | 99 | 97 | 100 | NLU003697 | EST |
| 14 | NlMyoR | MyoRa | MyoR | 99 | 97 | 95 | 100 | NLU020439 | EST |
| 15 | NlSage | Mesp | sage | 100 | 100 | 96 | 100 | NLU017450 | RT-PCR and EST |
| 16 | NlPxs | Paraxis | Pxs | 88 | 77 | 80 | 100 | NA | RT-PCR and EST |
| 17 | NlTwi | Twist | twi | 98 | 94 | 79 | 100 | NLU023739 | RT-PCR and EST |
| 18 | NlFer1 | PTFa | Fer1 | 99 | 92 | 72 | 98 | NLU018740 | RT-PCR and EST |
| 19 | NlFer2 | PTFb | Fer2 | 99 | 94 | 65 | 92 | NLU001388 | RT-PCR and EST |
| 20 | NlHand | Hand | Hand | 98 | 93 | 65 | 97 | NLU005290 | RT-PCR and EST |
| 21 | NlSCL | SCL | SCL | 100 | 100 | 99 | 100 | NLU016321 | RT-PCR and EST |
| 22 | NlNSCL | NSCL | NSCL | 100 | 99 | 95 | 100 | NLU009115 | EST |
| 23 | NlDel1 | Delilah | del | 96 | 91 | 77 | 96 | NLU025535 | RT-PCR and EST |
| 24 | NlDel2 | Delilah | del | 94 | 87 | 78 | 93 | NLU027494 | RT-PCR and EST |
| 25 | NlDel3 | Delilah | del | 94 | 90 | 77 | 95 | NLU005401 | RT-PCR and EST |
| 26 | NlMnt | Mnt | Mnt | 96 | 88 | 90 | 100 | NLU002070 | RT-PCR and EST |
| 27 | NlMad* | Mnt | ApMad | 100 | 100 | 100 | 100 | NLU010490 | RT-PCR and EST |
| 28 | NlMax | Max | Max | 99 | 97 | 97 | 100 | NA | EST |
| 29 | NlDm | Myc | dm | 81 | 82 | 97 | 100 | NLU025779 | RT-PCR and EST |
| 30 | NlUSF | USF | USF | 91 | 68 | 94 | 100 | NLU023467 | RT-PCR and EST |
| 31 | NlMitif | MITF | Mitif | 100 | 100 | 100 | 100 | NLU017474 | EST |
| 32 | NlCrp1 | AP4 | Crp | 83 | 44 | 50 | 88 | NLU016559 | EST |
| 33 | NlCrp2 | AP4 | Crp | 98 | 96 | 88 | 100 | NLU011530 | RT-PCR and EST |
| 34 | NlBmx | TF4 | bmx | 99 | 81 | 98 | 95 | NA | RT-PCR and EST |
| 35 | NlMlx | MLX | MLX | 100 | 97 | 97 | 100 | NLU009394 | RT-PCR and EST |
| 36 | NlSREBP1 | SREBP | SREBP | 94 | 73 | 81 | 100 | NLU005608 | EST |
| 37 | NlSREBP2 | SREBP | SREBP | 96 | 73 | 81 | 100 | NLU006435 | EST |
| 38 | NlSREBP3 | SREBP | SREBP | 90 | 59 | 72 | 98 | NLU021448 | RT-PCR and EST |
| 39 | NlTai | SRC | tai | 93 | 99 | 100 | 100 | NLU023056 | RT-PCR and EST |
| 40 | NlClk | Clock | clk | 100 | 100 | 98 | 100 | NLU027428 | EST |
| 41 | NlDys | AHR | dys | 100 | 100 | 100 | 100 | NA | RT-PCR and EST |
| 42 | NlSs | AHR | ss | 100 | 100 | 100 | 100 | NLU022623 | EST |
| 43 | NlSim1 | Sim | sim | 87 | 79 | 71 | 78 | NLU022755 | RT-PCR and EST |
| 44 | NlSim2 | Sim | sim | 93 | 83 | 72 | 74 | NLU008712 | RT-PCR and EST |
| 45 | NlTrh | Trh | trh | 99 | 89 | 96 | 84 | NLU009957 | RT-PCR and EST |
| 46 | NlSima | HIF | sima | 79 | 87 | 96 | 100 | NLU019462 | RT-PCR and EST |
| 47 | NlTgo | ARNT | tgo | 100 | 100 | 100 | 100 | NLU026318 | RT-PCR and EST |
| 48 | NlCyc | Bmal | cyc | 97 | 88 | 55 | 86 | NA | RT-PCR and EST |
| 49 | NlMet | Met | Met | 77 | 68 | 77 | 95 | NA | RT-PCR and EST |
| 50 | NlEmc | Emc | emc | 93 | 92 | 88 | 100 | NLU011228 | RT-PCR and EST |
| 51 | NlHey | Hey | Hey | 96 | 89 | 84 | 92 | NLU027503 | RT-PCR and EST |
| 52 | NlStich1 | Hey | Stich1 | 100 | 100 | 100 | 100 | NLU010132 | EST |
| 53 | NlSide | H/E(spl) | side | 97 | 89 | 95 | 100 | NLU019226 | RT-PCR and EST |
| 54 | NlDpn | H/E(spl) | dpn | 61 | n/m | 21 | n/m | NLU021732 | RT-PCR and EST |
| 55 | NlH * | H/E(spl) | ?-ApH | 93 | 93 | 55 | 67 | NLU017783 | RT-PCR and EST |
| 56 | NlE(spl)1 * | H/E(spl) | ?-ApHES1 | 93 | 65 | 62 | 62 | NLU012936 | RT-PCR and EST |
| 57 | NlE(spl)2 * | H/E(spl) | ? | n/m | n/m | n/m | n/m | NLU007850 | EST |
| 58 | NlE(spl)3 * | H/E(spl) | ?-ApHES1 | 99 | 96 | 85 | 89 | NLU021733 | RT-PCR and EST |
| 59 | NlKn(col)1 | COE | Kn(col) | 100 | 100 | 100 | 100 | NLU001955 | RT-PCR and EST |
| 60 | NlKn(col)2 | COE | Kn(col) | 100 | 100 | 100 | 100 | NLU011325 | RT-PCR and EST |
| Group | Family name | N.l. | A.p. | N.v. | H.s. | A.m. | T.c. | L.d. | B.m. | D.m. | A.a. | A.g. | C.q. | P.h. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | ASCa | 2 | 0 | 2 | 2 | 2 | 3 | 1 | 4 | 4 | 4 | 2 | 4 | 2 |
| A | ASCb | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| A | MyoD | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | E12/E17 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Ngn | 2 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 2 | 2 | 1 |
| A | NeuroD | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| A | Atonal | 3 | 3 | 3 | 3 | 3 | 3 | 2 | 1 | 3 | 5 | 4 | 5 | 3 |
| A | Mist | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| A | Beta3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Oligo | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| A | Net | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Delilah | 3 | 1 | 0 | 0 | 0 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Mesp | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Twist | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | Paraxis | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| A | MyoRa | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | MyoRb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| A | Hand | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | PTFa | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 |
| A | PTFb | 1 | 2 | 2 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 |
| A | SCL | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | NSCL | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | SRC | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | Figα | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| B | Myc | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | Mad | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| B | Mnt | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | Max | 1 | 3 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | USF | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | MITF | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 |
| B | SREBP | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| B | AP4 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | MLX | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| B | TF4 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| C | Clock | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | 2 | 2 | 2 |
| C | ARNT | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| C | Bmal | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 |
| C | AHR | 2 | 2 | 2 | 3 | 2 | 1 | 2 | 3 | 2 | 2 | 2 | 2 | 2 |
| C | Sim | 2 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 2 | 1 | 1 |
| C | Trh | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| C | HIF | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| D | Emc | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 |
| E | Hey | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 | 2 |
| E | H/E(spl) | 6 | 6 | 4 | 6 | 6 | 6 | 8 | 5 | 11 | 4 | 4 | 4 | 8 |
| F | COE | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 60 | 54 | 48 | 57 | 51 | 50 | 49 | 52 | 59 | 55 | 55 | 57 | 55 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, P.-J.; Yuan, S.-Y.; Wang, W.-X.; Chen, X.; Lai, F.-X.; Fu, Q. A Genome-Wide Identification and Analysis of the Basic Helix-Loop-Helix Transcription Factors in Brown Planthopper, Nilaparvata lugens. Genes 2016, 7, 100. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110100
Wan P-J, Yuan S-Y, Wang W-X, Chen X, Lai F-X, Fu Q. A Genome-Wide Identification and Analysis of the Basic Helix-Loop-Helix Transcription Factors in Brown Planthopper, Nilaparvata lugens. Genes. 2016; 7(11):100. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110100
Chicago/Turabian StyleWan, Pin-Jun, San-Yue Yuan, Wei-Xia Wang, Xu Chen, Feng-Xiang Lai, and Qiang Fu. 2016. "A Genome-Wide Identification and Analysis of the Basic Helix-Loop-Helix Transcription Factors in Brown Planthopper, Nilaparvata lugens" Genes 7, no. 11: 100. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110100