
Recovery from the DNA Replication Checkpoint


Department of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN 55455, USA
*
Author to whom correspondence should be addressed.
Genes 2016, 7(11), 94; https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110094
Submission received: 26 September 2016
/
Revised: 20 October 2016
/
Accepted: 23 October 2016
/
Published: 28 October 2016
(This article belongs to the Special Issue DNA Replication Controls)
{kind=link}
Abstract
:Checkpoint recovery is integral to a successful checkpoint response. Checkpoint pathways monitor progress during cell division so that in the event of an error, the checkpoint is activated to block the cell cycle and activate repair pathways. Intrinsic to this process is that once repair has been achieved, the checkpoint signaling pathway is inactivated and cell cycle progression resumes. We use the term “checkpoint recovery” to describe the pathways responsible for the inactivation of checkpoint signaling and cell cycle re-entry after the initial stress has been alleviated. The DNA replication or S-phase checkpoint monitors the integrity of DNA synthesis. When replication stress is encountered, replication forks are stalled, and the checkpoint signaling pathway is activated. Central to recovery from the S-phase checkpoint is the restart of stalled replication forks. If checkpoint recovery fails, stalled forks may become unstable and lead to DNA breaks or unusual DNA structures that are difficult to resolve, causing genomic instability. Alternatively, if cell cycle resumption mechanisms become uncoupled from checkpoint inactivation, cells with under-replicated DNA might proceed through the cell cycle, also diminishing genomic stability. In this review, we discuss the molecular mechanisms that contribute to inactivation of the S-phase checkpoint signaling pathway and the restart of replication forks during recovery from replication stress.
1. Introduction
The DNA replication or S-phase checkpoint monitors the integrity of DNA synthesis. Perturbations in DNA synthesis—such as a scarcity of free nucleotides or damaged DNA—leads to replication fork stalling and activation of the checkpoint pathway [1,2]. The replication checkpoint promotes cell viability by mediating a transcriptional response [3], stabilizing replication forks [1,2,4,5], suppressing firing of origins of replication [6,7], and stalling DNA synthesis [6,8]. Difficult to replicate DNA regions, such as repetitive sequences or fragile sites, can also stall forks and lead to activation of the checkpoint. Thus, even an S-phase in ideal environmental conditions can lead to multiple activations of the checkpoint, although these activations may be local rather than global.
Recovery from checkpoint activation is key to a successful checkpoint mechanism. During checkpoint recovery, the checkpoint signaling pathway is inactivated, and cell cycle progression is resumed. As the S-phase checkpoint is sensitive to perturbations even under favorable conditions, it is likely that recovery from checkpoint initiation is critical in each and every cell division. It is clear that failure to activate the S-phase checkpoint has deleterious consequences. Stalled forks may become unstable and lead to DNA breaks or unusual DNA structures that are difficult to resolve, leading to genomic instability. Similarly, if checkpoint recovery mechanisms fail, stalled forks can persist, increasing the likelihood of DNA damage. Alternatively, if cell cycle resumption mechanisms become uncoupled from checkpoint inactivation, cells with under-replicated DNA would proceed through the cell cycle, also impacting genomic stability.
2. Activation of Checkpoint Signaling
Initiation of the S-phase checkpoint response is dependent on a signaling cascade that is remarkably conserved in eukaryotes. This review will highlight mechanisms identified in simple eukaryotes such as budding yeast and point out distinctions observed in higher eukaryotes, including humans. Both stalled replication forks and DNA damage are recognized by sensor complexes, which activate a kinase cascade to prevent cell cycle progression (for review, see [9,10]). In budding yeast, sensing of DNA damage or stalled replication forksSeSeSasdfasdfasieuyghwq relies on the Rad24-dependent loading of the heterotrimeric Rad17-Mec3-Ddc1 (9-1-1 complex in fission yeast and humans) sliding clamp onto DNA [11,12,13]. This leads to Mec1 kinase (ATR in humans) activation, followed by the downstream phosphorylation and activation of the primary signaling kinase Rad53 [14,15]. In higher eukaryotes, ATR activation primarily leads to Chk1 kinase activation during the S-phase checkpoint, rather than Rad53 homolog Chk2 [16,17,18,19]. Mec1-dependent activation of Rad53 requires the adaptor Mrc1 (Claspin in humans), which forms a complex to stabilize replication forks at sites of replication stress [1,2,4,5]. Several other proteins function to promote Rad53 activation, including Rad9, Csm3, and Tof1 [4,20,21]. Csm3 and Tof1 form a complex with Mrc1 at replication forks [22], whereas Rad9 typically functions during the DNA damage checkpoint response, but it can substitute for Mrc1 under specific conditions [4,21]. Sgs1 helicase—a member of the RecQ helicase family and yeast ortholog of the human Bloom Syndrome protein BLM—is important for recruiting Rad53 to stalled forks and in maintaining association of DNA polymerases α and ε with the replication fork during checkpoint activation [23,24,25]. Mrc1 appears to also have a role during DNA replication in the absence of replication stress. Mrc1 is loaded onto replication origins and travels with the replisome complex at the replication fork [5,26,27].
While substantial progress has been made in identifying factors, pathways, and molecular events central to checkpoint activation, our understanding of checkpoint recovery is much more limited in comparison. Recovery from replication stress occurs after the original damage or defect is repaired, thus triggering checkpoint inactivation and a return to progression through the cell cycle [28,29].
3. Checkpoint Signaling Inactivation
Checkpoint initiation programs must be counteracted to achieve recovery and re-entry into the cell cycle. S-phase checkpoint recovery has to accomplish two key steps: inactivation of checkpoint signaling and resumption of DNA replication.
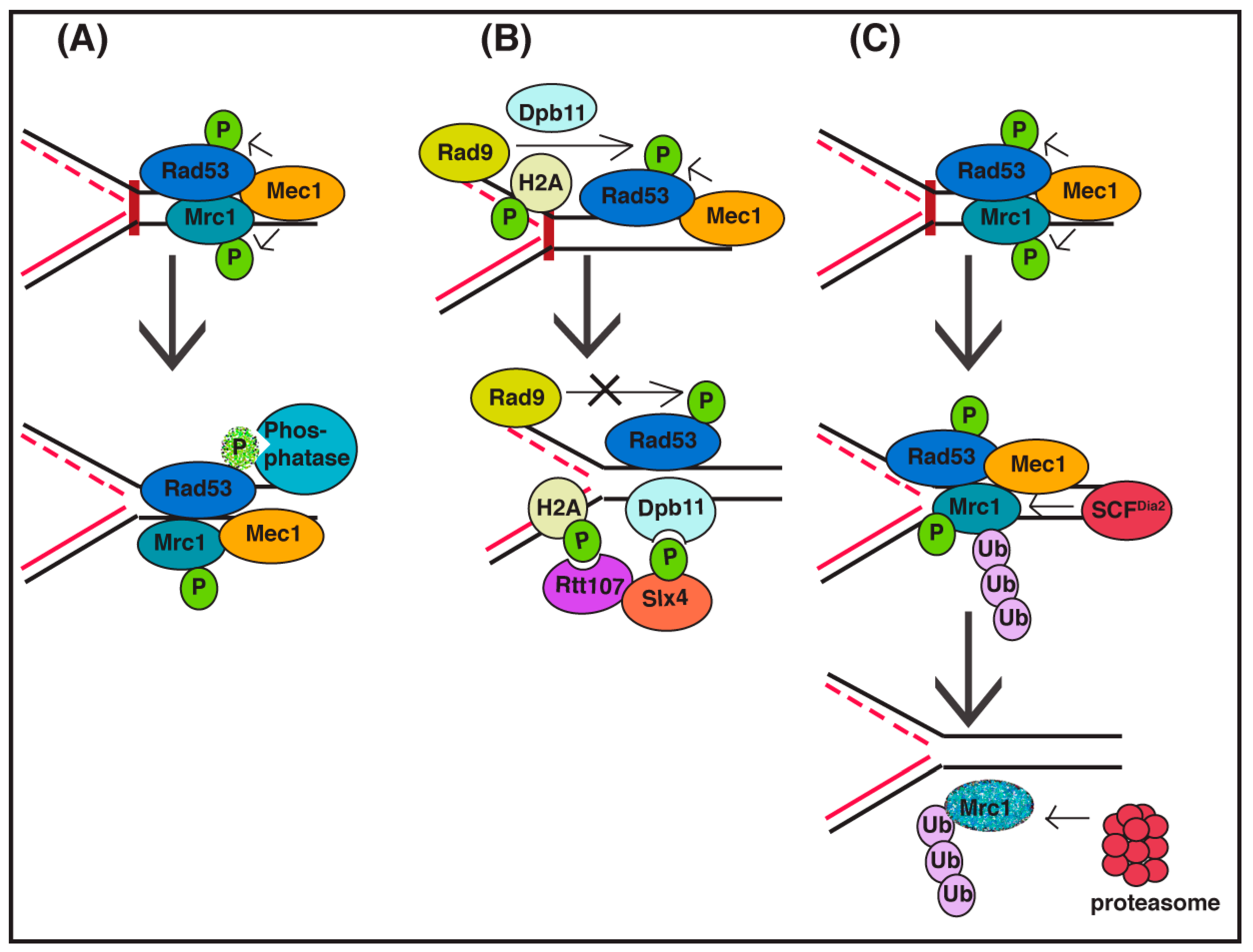
One straightforward way to turn off a signaling pathway is by inhibiting key enzymatic steps in the pathway. There are S-phase checkpoint recovery mechanisms that involve deactivation of the checkpoint signaling pathway by interfering with Rad53 kinase activity (Figure 1A). Direct inactivation of the Rad53 signaling kinase by the action of PP2A-like phosphatase complex (Pph3/Psy2) has been observed to be important in S-phase checkpoint recovery [30,31,32,33]. Disruption of the Rad53 phosphatase complex leads to a defect in replication fork restart [34], suggesting that fork restart mechanisms are dependent on the inactivation of checkpoint signaling. Inhibition of a checkpoint kinase is also observed in the DNA damage checkpoint pathway, as PP2C-type phosphatases inhibit Rad53 signaling in this pathway [30,35]. As the initial checkpoint kinase activated during the S-phase checkpoint differs from the kinase activated during the DNA damage checkpoint, it follows that distinct Rad53 residues may need to be de-phosphorylated in each case [33]. Thus, the initiating event that triggers a specific checkpoint signaling pathway may determine (in part) the mechanism of inactivation during recovery.
Checkpoint signaling may also be affected by alterations of chromatin structure and changes to checkpoint protein complex architecture (Figure 1B). Dampening of checkpoint signaling can also be accomplished by Slx4-Rtt107 competition for Rad9 binding at sites of DNA lesions [36]. Slx4 and Rtt107 are scaffold proteins that can be recruited to stressed replication forks [37] as well as double-strand DNA breaks, uncapped telomeres, and other DNA lesions to displace Rad9 by competing for checkpoint-induced phosphosites on histone H2A, thus reducing the activation of Rad53 kinase and the checkpoint signaling pathway [38,39]. Indeed, evidence indicates that local action of the Slx4/Rtt107 complex at replication forks is complementary to the global activity of the Pph3/Psy2 Rad53 phosphatase [40]. In addition, chromatin remodeling factors Ino80 and Isw2—demonstrated to promote chromatin accessibility—attenuate S-phase checkpoint signaling and promote the recovery of stalled replication forks, although the mechanism by which this is accomplished in not known [41,42].
Protein degradation of checkpoint adaptor proteins is also important for S-phase checkpoint recovery, although whether this degradation is required solely for checkpoint signaling inactivation is not clear. Degradation of human Claspin (an adaptor for the Chk1 signaling kinase) is linked to checkpoint recovery after fork stalling caused by exposure to hydroxyurea, which limits free nucleotides [43,44,45]. Plk1-induced phosphorylation of Claspin triggers its degradation, facilitated by the SCFβTrCP ubiquitin ligase and the proteasome [43,44,46]. Importantly, degradation of Claspin also reduces Chk1 kinase signaling, thus inhibiting checkpoint signaling. This pathway is highly regulated, as a number of de-ubiquitinating enzymes have been identified that act to stabilize the Claspin protein, including USP7, USP28, USP29, and HERC2/USP20 [47,48,49,50,51]. Other types of replication stress also trigger Claspin degradation, but the ubiquitination pathway may differ, as the BRCA1 ubiquitin ligase can target Claspin degradation in response to topoisomerase inhibition [52].
In budding yeast, the Claspin ortholog Mrc1 is also targeted for degradation during recovery from the S-phase checkpoint (Figure 1C), indicating that removal of Mrc1 function during recovery is evolutionarily conserved. In this case, Mrc1 has been shown to be targeted for degradation via the SCFDia2 ubiquitin ligase during recovery from the DNA alkylating agent methyl methanesulfonate (MMS). Both checkpoint-phosphorylated Mrc1 and unmodified Mrc1 are degraded, and a Mrc1 mutant protein that cannot be phosphorylated by checkpoint kinases exhibits partial stabilization. Induced degradation of Mrc1 only during recovery rescues the checkpoint recovery defect in cells lacking SCFDia2 ubiquitin ligase activity, indicating that the predominant role of this complex during S-phase checkpoint recovery is degradation of Mrc1. However, induced degradation of a checkpoint-defective version of Mrc1 during the same time period cannot rescue the recovery defect in these cells, suggesting that removal of checkpoint-activated Mrc1 is key to the recovery process [53]. In addition, the Rtt101Mms22 ubiquitin ligase counteracts the replicative function of Mrc1 (although not via a degradation mechanism) to also facilitate replication fork restart or repair [54].
4. Resumption of DNA Replication
Completion of DNA replication after the activation of checkpoint signaling is critical to successful S-phase checkpoint recovery. During checkpoint activation, some proteins at the fork involved in checkpoint signaling, such as ATR/Mec1 and Claspin/Mrc1, promote replication fork stabilization [23,27,55,56,57], presumably to maintain forks so that they can be restarted after the replication stress is removed. Evidence suggests that in response to low deoxynucleotide triphosphates (dNTP) levels, Mec1 and Rad53 regulate replisome function rather than the integrity of the complex, as the replisome is stably associated with replication forks in the absence of Mec1 and Rad53 [58]. Additional proteins are recruited to help stabilize forks, many of which are also involved in fork restart mechanisms. Prolonged fork stalling may lead to the replisome moving away from the fork or replisome components dissociating from chromatin. Stalled forks may also undergo structural rearrangements such as fork reversal and rewinding of parental and newly-replicated DNA strands into “chicken foot” structures that are difficult to resolve.
A growing number of proteins have been identified to have roles in replication fork restart, and multiple fork restart mechanisms have been proposed. In general, these mechanisms can be divided into two groups: direct restart or a broad group of alternative restart mechanisms that require remodeling or recombination to restore DNA replication. The type of replication stress or DNA lesion that led to the fork stall may influence the type of restart mechanism. For example, recovery of a fork stalled by limiting replication components presents a different challenge than a leading strand lesion that has a led to uncoupling of the leading and lagging strand polymerases.
Direct restart of a stable, stalled fork is a straightforward approach to complete DNA synthesis during checkpoint recovery. However, we know very little about mechanistic steps required for initiation of direct restart of a stalled fork (re-priming) in eukaryotic cells. For example, it is not clear if re-priming is linked to inhibition of checkpoint signaling. In primates, the methyltransferase and nuclease protein METNASE (SETMAR) is required for restart of the majority of forks following a hydroxyurea-induced checkpoint [59,60]. Interestingly, METNASE is involved in a feedback mechanism with Chk1, in which Chk1-mediated phosphorylation of METNASE decreases its function in fork restart and increases Chk1 protein stability [61,62], thereby prolonging checkpoint activation and preventing premature fork restart. Thus, substantial coordination between checkpoint signaling inactivation and fork restart mechanisms may exist.
Alternative ways to restart DNA replication include a variety of mechanisms that may require remodeling by fork reversal, nucleolytic processing of nascent DNA strands, or recombination mechanisms (for comprehensive review, see [63,64]). Proteins capable of fork reversal include the Rad5 helicase in yeast [65] (and human ortholog HLTF [66]) and Fanconi Anemia protein FANCM [67] and its budding yeast homolog Mph1 [68], as well as its fission yeast homolog Fml1 [69]. In higher eukaryotes, fork reversal in vivo also depends on poly (ADP-ribose) polymerase (PARP1) [70]. In humans, the helicase SMARCAL1 can remodel forks to achieve branch migration and trigger fork restart [71,72,73,74,75]. The EEPD1 nuclease is recruited to stalled forks, and promotes DNA end resection and fork restart [76].
Recombination-based restart mechanisms are probably most relevant to collapsed forks where the replication machinery has been lost, thus facilitating Holliday junction formation. The recombination factor Rad51 (which catalyzes Holliday junctions) can be recruited to stalled forks [77,78,79,80]. Helicases that function in Holliday junction resolution during recombination, including the RecQ helicase family members Bloom Syndrome protein BLM and the Werner Syndrome protein WRN have demonstrated roles in fork restart [81,82,83]. This activity is conserved, as the related protein in budding yeast, Sgs1, is important for recombination-mediated fork restart [84,85]. Replication fork restart is also linked to Fanconi Anemia (for review, see [86]). Although members of this group are best known for their roles in interstrand crosslink repair, the FANCD1, FANCD2, and FANCJ proteins have distinct roles in replication fork restart [87]. In particular, FANCD2 is required to stabilize and recruit BLM to stalled forks [88]. In addition to recombination factors, conserved scaffold proteins such as Slx4 and Rtt107 that interact with structure-specific nucleases or fork repair proteins are also important for fork restart [89,90,91,92,93,94,95]. Finally, forks that cannot be recovered may be bypassed by the firing of nearby “back-up” origins, ensuring that chromosome duplication is completed [96,97].
5. Future Perspectives
We are just beginning to identify and investigate mechanisms of checkpoint signal inactivation. As these mechanisms become better understood, it will be interesting to determine whether they are coordinated into an overall cellular program that facilitates recovery from the S-phase checkpoint. One intriguing question is whether checkpoint activation in response to distinct damage or replication stress triggers specific signaling inactivation mechanisms. Moreover, are checkpoint recovery mechanisms themselves downstream targets of the initial checkpoint activation? It is tempting to imagine that mechanisms that turn off the signaling pathway are folded into the initial activation as a means of limiting prolonged activation.
Many of the proteins required for the DNA replication checkpoint and fork restart during recovery are compromised in human diseases. It is easy to imagine that defects in checkpoint recovery might lead to genome instability, and therefore also contribute to human disease. As checkpoint recovery mechanisms become better understood, we look forward to new information about the role of these pathways in protecting human health.
Acknowledgments
D.M.K. and I.C. are supported by the Department of Genetics, Cell Biology, and Development at the University of Minnesota.
Author Contributions
D.M.K. organized and wrote the paper. I.C. contributed to writing the paper and designed the figures.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsor had no role in the writing of the review manuscript.
Abbreviations and Protein Name Derivations
| Rad | Radiation sensitive |
| Mec | Mitosis entry checkpoint |
| Ddc1 | DNA damage checkpoint 1 |
| ATR | ataxia telangiectasia mutated- and Rad3-related |
| Chk | Checkpoint kinase |
| Mrc1 | Mediator of replication checkpoint protein 1 |
| Csm3 | Chromosome segregation in meiosis protein 3 |
| Tof1 | Topoisomerase 1-associated factor 1 |
| Sgs1 | slow growth suppressor 1 |
| RecQ | recombination Q family |
| BLM | Bloom Syndrome protein |
| PP2A | protein phosphatase 2A |
| Pph3 | protein phosphatase 3 |
| Psy2 | platinum sensitivity 2 |
| PP2C | protein phosphatase 2C |
| Slx4 | Structure-Specific Endonuclease Subunit |
| Rtt | Regulator of Ty1 transposition protein |
| Ino80 | inositol requiring 80 |
| Isw2 | imitation switch 2 |
| Plk1 | Polo-like kinase 1 |
| SCF | Skp, Cullin, F-box protein containing complex |
| βTrCP | beta-transducin repeat protein |
| USP | ubiquitin-specific protease |
| HERC2 | HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase 2 |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| Dia2 | Digs into agar 2 |
| Mms22 | Methyl Methanesulfonate sensitivity 22 |
| METNASE | methyl transferase and nuclease |
| SETMAR | SET domain and mariner transposase fusion |
| HLTF | Helicase-like transcription factor |
| FANC | Fanconi Anemia group protein |
| Mph1 | mutator phenotype 1 |
| Fml1 | FANCM ortholog |
| SMARCAL | SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A-Like 1 |
| EEPD1 | Endonuclease/exonuclease/phosphatase family domain-containing protein 1 |
| WRN | Werner Syndrome protein |
References
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.B.; Zhou, Z.; Siede, W.; Friedberg, E.C.; Elledge, S.J. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994, 8, 2401–2415. [Google Scholar] [CrossRef] [PubMed]
- Alcasabas, A.A.; Osborn, A.J.; Bachant, J.; Hu, F.; Werler, P.J.; Bousset, K.; Furuya, K.; Diffley, J.F.; Carr, A.M.; Elledge, S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Osborn, A.J.; Elledge, S.J. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003, 17, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Diffley, J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998, 395, 615–618. [Google Scholar] [PubMed]
- Shirahige, K.; Hori, Y.; Shiraishi, K.; Yamashita, M.; Takahashi, K.; Obuse, C.; Tsurimoto, T.; Yoshikawa, H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998, 395, 618–621. [Google Scholar] [PubMed]
- Paulovich, A.G.; Hartwell, L.H. A checkpoint regulates the rate of progression through S-phase in S. Cerevisiae in response to DNA damage. Cell 1995, 82, 841–847. [Google Scholar] [CrossRef]
- Boddy, M.N.; Russell, P. DNA replication checkpoint. Curr. Biol. 2001, 11, R953–R956. [Google Scholar] [CrossRef]
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 2002, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.C.; Haber, J.E. Surviving the breakup: The DNA damage checkpoint. Annu. Rev. Genet. 2006, 40, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Majka, J.; Burgers, P.M. Yeast Rad17/Mec3/Ddc1: A sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2003, 100, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Venclovas, C.; Thelen, M.P. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000, 28, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of Rad53 by the ATM-like kinases Mec1 and TEL1 in yeast cell cycle checkpoint pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Fay, D.S.; Marini, F.; Foiani, M.; Stern, D.F. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 1996, 10, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Hekmat-Nejad, M.; You, Z.; Yee, M.C.; Newport, J.W.; Cimprich, K.A. Xenopus Atr is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 2000, 10, 1565–1573. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. Atr-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Foss, E.J. Tof1p regulates DNA damage responses during s phase in Saccharomyces cerevisiae. Genetics 2001, 157, 567–577. [Google Scholar] [PubMed]
- Tanaka, K.; Russell, P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001, 3, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Bando, M.; Katou, Y.; Komata, M.; Tanaka, H.; Itoh, T.; Sutani, T.; Shirahige, K. Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J. Biol. Chem. 2009, 284, 34355–34365. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.A.; Bjergbaek, L.; Shimada, K.; Frei, C.; Gasser, S.M. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. Embo J. 2003, 22, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Bjergbaek, L.; Cobb, J.A.; Tsai-Pflugfelder, M.; Gasser, S.M. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. Embo J. 2005, 24, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hegnauer, A.M.; Hustedt, N.; Shimada, K.; Pike, B.L.; Vogel, M.; Amsler, P.; Rubin, S.M.; van Leeuwen, F.; Guenole, A.; van Attikum, H.; et al. An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. Embo J. 2012, 31, 3768–3783. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Viggiani, C.J.; Aparicio, O.M. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell 2005, 19, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Versini, G.; Cordon-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Clemenson, C.; Marsolier-Kergoat, M.C. DNA damage checkpoint inactivation: Adaptation and recovery. DNA Repair 2009, 8, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbein, F.; Guerois, R.; Haber, J.E.; Marsolier-Kergoat, M.C. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 2003, 11, 827–835, Erratum 2003, 11, 1119–1119. [Google Scholar]
- Guillemain, G.; Ma, E.; Mauger, S.; Miron, S.; Thai, R.; Guerois, R.; Ochsenbein, F.; Marsolier-Kergoat, M.C. Mechanisms of checkpoint kinase Rad53 inactivation after a double-strand break in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.M.; Szyjka, S.J.; Lis, E.T.; Bailey, A.O.; Yates, J.R.; Aparicio, O.M.; Romesberg, F.E. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Travesa, A.; Duch, A.; Quintana, D.G. Distinct phosphatases mediate the deactivation of the DNA damage Checkpoint kinase Rad53. J. Biol. Chem. 2008, 283, 17123–17130. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Aparicio, J.G.; Viggiani, C.J.; Knott, S.; Xu, W.; Tavare, S.; Aparicio, O.M. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008, 22, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Liu, Y.; Ma, C.J.; Smolka, M.B. DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 2013, 493, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Balint, A.; Kim, T.; Gallo, D.; Cussiol, J.R.; Bastos de Oliveira, F.M.; Yimit, A.; Ou, J.; Nakato, R.; Gurevich, A.; Shirahige, K.; et al. Assembly of Slx4 signaling complexes behind DNA replication forks. Embo J. 2015, 34, 2182–2197. [Google Scholar] [CrossRef] [PubMed]
- Cussiol, J.R.; Jablonowski, C.M.; Yimit, A.; Brown, G.W.; Smolka, M.B. Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. Embo J. 2015, 34, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Dibitetto, D.; Ferrari, M.; Rawal, C.C.; Balint, A.; Kim, T.; Zhang, Z.; Smolka, M.B.; Brown, G.W.; Marini, F.; Pellicioli, A. Slx4 and Rtt107 control checkpoint signalling and DNA resection at double-strand breaks. Nucleic Acids Res. 2016, 44, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Jablonowski, C.M.; Cussiol, J.R.; Oberly, S.; Yimit, A.; Balint, A.; Kim, T.; Zhang, Z.; Brown, G.W.; Smolka, M.B. Termination of replication stress signaling via concerted action of the Slx4 scaffold and the PP4 phosphatase. Genetics 2015, 201, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Rodriguez, J.; Tsukiyama, T. Chromatin remodeling factors Isw2 and Ino80 regulate checkpoint activity and chromatin structure in S phase. Genetics 2015, 199, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCF beta TrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of claspin by SCF beta TrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.N.; Clarke, P.R. Regulation of claspin degradation by the ubiquitin-proteosome pathway during the cell cycle and in response to Atr-dependent checkpoint activation. FEBS Lett. 2006, 580, 4176–4181. [Google Scholar] [CrossRef] [PubMed]
- Mamely, I.; van Vugt, M.A.T.M.; Smits, V.A.J.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Freire, R.; Medema, R.H. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Faustrup, H.; Bekker-Jensen, S.; Bartek, J.; Lukas, J.; Mailand, N. USP7 counteracts SCFβTrCP- but not APCCdh1-mediated proteolysis of claspin. J. Cell. Biol. 2009, 184, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.; Cabrera, E.; Amoedo, H.; Hernandez-Perez, S.; Dominguez-Kelly, R.; Freire, R. USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination. Oncogene 2015, 34, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhao, H.; Liao, J.; Xu, X. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Res. 2014, 42, 13074–13081. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Deng, M.; Li, Y.; Yin, P.; Gao, B.; Fang, Y.; Wu, P.; Liu, T.; Lou, Z. HERC2-USP20 axis regulates DNA damage checkpoint through CLASPIN. Nucleic Acids Res. 2014, 42, 13110–13121. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sundaramoorthy, E.; Rajendra, E.; Hattori, H.; Jeyasekharan, A.D.; Ayoub, N.; Schiess, R.; Aebersold, R.; Nishikawa, H.; Sedukhina, A.S.; et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr. Biol. 2012, 22, 1659–1666. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.M.; Arumugam, A.; Koepp, D.M. The Saccharomyces cerevisiae f-box protein Dia2 is a mediator of S-phase checkpoint recovery from DNA damage. Genetics 2013, 193, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Buser, R.; Kellner, V.; Melnik, A.; Wilson-Zbinden, C.; Schellhaas, R.; Kastner, L.; Piwko, W.; Dees, M.; Picotti, P.; Maric, M.; et al. The replisome-coupled E3 ubiquitin ligase Rtt101Mms22 counteracts Mrc1 function to tolerate genotoxic stress. PLoS Genet. 2016, 12, e1005843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Scorah, J.; McGowan, C.H. Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle 2009, 8, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- De Piccoli, G.; Katou, Y.; Itoh, T.; Nakato, R.; Shirahige, K.; Labib, K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol. Cell 2012, 45, 696–704. [Google Scholar] [CrossRef] [PubMed]
- De Haro, L.P.; Wray, J.; Williamson, E.A.; Durant, S.T.; Corwin, L.; Gentry, A.C.; Osheroff, N.; Lee, S.H.; Hromas, R.; Nickoloff, J.A. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010, 38, 5681–5691. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Williamson, E.; Nickoloff, J.; Lee, S.H.; Hromas, R. Metnase/setmar: A domesticated primate transposase that enhances DNA repair, replication, and decatenation. Genetica 2010, 138, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Williamson, E.A.; Fnu, S.; Lee, Y.J.; Park, S.J.; Beck, B.D.; You, J.S.; Leitao, A.; Nickoloff, J.A.; Lee, S.H. Chk1 phosphorylation of metnase enhances DNA repair but inhibits replication fork restart. Oncogene 2012, 31, 4245–4254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, E.A.; Wu, Y.; Singh, S.; Byrne, M.; Wray, J.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. The DNA repair component metnase regulates Chk1 stability. Cell Div. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.; Poli, J.; Marians, K.J.; Pasero, P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013, 5, a012815. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Blastyak, A.; Pinter, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Haracska, L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. USA 2011, 108, 14073–14078. [Google Scholar] [CrossRef] [PubMed]
- Gari, K.; Decaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Prakash, R.; Saro, D.; Longerich, S.; Niu, H.; Sung, P. Processing of DNA structures via DNA unwinding and branch migration by the S. cerevisiae mph1 protein. DNA Repair (Amst) 2011, 10, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Nandi, S.; Osman, F.; Ahn, J.S.; Jakovleska, J.; Lorenz, A.; Whitby, M.C. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol. Cell 2008, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Bredemeyer, A.L.; Sowa, M.E.; Terret, M.E.; Jallepalli, P.V.; Harper, J.W.; Elledge, S.J. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009, 23, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Bansbach, C.E.; Betous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Yusufzai, T.; Kong, X.; Yokomori, K.; Kadonaga, J.T. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009, 23, 2400–2404. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ghosal, G.; Chen, J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009, 23, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different Rad51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Franchitto, A.; Pirzio, L.M.; Prosperi, E.; Sapora, O.; Bignami, M.; Pichierri, P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008, 183, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.M.; Li, N.; Folch, A.; Monnat, R.J., Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 2008, 7, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.B.; Hickson, I.D. RecQ helicases: Conserved guardians of genomic integrity. Adv. Exp. Med. Biol. 2013, 767, 161–184. [Google Scholar] [PubMed]
- Ashton, T.M.; Hickson, I.D. Yeast as a model system to study RecQ helicase function. DNA Repair (Amst) 2010, 9, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. To the rescue: The Fanconi anemia genome stability pathway salvages replication forks. Cancer Cell 2012, 22, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Raghunandan, M.; Chaudhury, I.; Kelich, S.L.; Hanenberg, H.; Sobeck, A. FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi anemia core complex. Cell Cycle 2015, 14, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Sareen, A.; Raghunandan, M.; Sobeck, A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013, 41, 6444–6459. [Google Scholar] [CrossRef] [PubMed]
- Flott, S.; Alabert, C.; Toh, G.W.; Toth, R.; Sugawara, N.; Campbell, D.G.; Haber, J.E.; Pasero, P.; Rouse, J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol. Cell. Biol. 2007, 27, 6433–6445. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Bergstralh, D.T.; Kohl, K.P.; LaRocque, J.R.; Moore, C.B.; Sekelsky, J. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell 2009, 35, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R., 3rd; Russell, P.; et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Munoz, I.M.; Hain, K.; Declais, A.C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.M.; Zaidi, I.W.; Vaisica, J.A.; Peter, M.; Brown, G.W. Regulation of Rtt107 recruitment to stalled DNA replication forks by the cullin Rtt101 and the Rtt109 acetyltransferase. Mol. Biol. Cell 2008, 19, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ohouo, P.Y.; Bastos de Oliveira, F.M.; Almeida, B.S.; Smolka, M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell 2010, 39, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.M.; Gohler, T.; Luciani, M.G.; Oehlmann, M.; Ge, X.; Gartner, A.; Jackson, D.A.; Blow, J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006, 173, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of DNA replication checkpoint signaling inactivation mechanisms. (A) During recovery, phosphatases from the PP2A and PP2C families de-phosphorylate Rad53, thus abrogating checkpoint signaling; (B) Competition for binding Rad9 (a Rad53 adaptor) by the Rtt107/Slx4 complex can dampen checkpoint signaling; (C) Ubiquitin-mediated degradation of the Rad53 kinase adaptor Mrc1, facilitated by the SCFDia2 ubiquitin ligase, promotes checkpoint recovery.
Figure 1.
Diagram of DNA replication checkpoint signaling inactivation mechanisms. (A) During recovery, phosphatases from the PP2A and PP2C families de-phosphorylate Rad53, thus abrogating checkpoint signaling; (B) Competition for binding Rad9 (a Rad53 adaptor) by the Rtt107/Slx4 complex can dampen checkpoint signaling; (C) Ubiquitin-mediated degradation of the Rad53 kinase adaptor Mrc1, facilitated by the SCFDia2 ubiquitin ligase, promotes checkpoint recovery.

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaudhury, I.; Koepp, D.M. Recovery from the DNA Replication Checkpoint. Genes 2016, 7, 94. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110094
AMA Style
Chaudhury I, Koepp DM. Recovery from the DNA Replication Checkpoint. Genes. 2016; 7(11):94. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110094
Chicago/Turabian StyleChaudhury, Indrajit, and Deanna M. Koepp. 2016. "Recovery from the DNA Replication Checkpoint" Genes 7, no. 11: 94. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7110094
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.