
The Complete Genome of Brucella Suis 019 Provides Insights on Cross-Species Infection


Abstract
:1. Introduction
2. Results and Discussion
2.1. Complete Genome Sequencing, Assembly and Annotation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Chr1_ID | Length | Gen# | Chr2_ID | Length | Gen# | CG% |
|---|---|---|---|---|---|---|---|
| * B. abortus A13334 | NC_016795.1 | 2,123,773 | 2086 | NC_016777.1 | 1,162,259 | 1102 | 57.40 |
| B. abortus bv.1 str 9-941 | NC_006932.1 | 2,124,241 | 2082 | NC_006933.1 | 1,162,204 | 1103 | 57.22 |
| B. abortus S19 | NC_010742.1 | 2,122,487 | 2089 | NC_010740.1 | 1,161,449 | 1106 | 57.22 |
| B. canis ATCC 23365 | NC_010103.1 | 2,105,969 | 2022 | NC_010104.1 | 1,206,800 | 1131 | 57.24 |
| B. canis HSKA 52141 | NC_016778.1 | 2,107,023 | 2019 | NC_016796.1 | 1,170,489 | 1098 | 57.24 |
| B. melitensis 16M | NC_003317.1 | 2,117,144 | 2040 | NC_003318.1 | 1,177,787 | 1107 | 57.22 |
| B. melitensis ATCC 23457 | NC_012441.1 | 2,125,701 | 2059 | NC_012442.1 | 1,185,518 | 1117 | 57.22 |
| B. melitensis biovar Abortus 2308 | NC_007618.1 | 2,121,359 | 2086 | NC_007624.1 | 1,156,948 | 1104 | 57.22 |
| B. melitensis M28 | NC_017244.1 | 2,126,133 | 2058 | NC_017245.1 | 1,185,615 | 1118 | 57.22 |
| B. melitensis M5-90 | NC_017246.1 | 2,126,451 | 2062 | NC_017247.1 | 1,185,778 | 1118 | 57.22 |
| B. melitensis NI | NC_017248.1 | 2,117,717 | 2051 | NC_017283.1 | 1,176,758 | 1112 | 57.23 |
| B. microti CCM 4915 | NC_013119.1 | 2,117,050 | 2024 | NC_013118.1 | 1,220,319 | 1135 | 57.25 |
| B. ovis ATCC 25840 | NC_009505.1 | 2,111,370 | 2068 | NC_009504.1 | 1,164,220 | 1122 | 57.19 |
| B. pinnipedialis B2/94 | NC_015857.1 | 2,138,342 | 2081 | NC_015858.1 | 1,260,926 | 1188 | 57.20 |
| B. suis 1330 | NC_017251.1 | 2,107,783 | 2014 | NC_017250.1 | 1,207,380 | 1130 | 57.25 |
| * B. suis ATCC 23445 | NC_010169.1 | 1,923,763 | 1848 | NC_010167.1 | 1,400,844 | 1327 | 57.21 |
| B. suis VBI22 | NC_016797.1 | 2,108,637 | 2015 | NC_016775.1 | 1,207,451 | 1132 | 57.25 |
| B. suis 019 | CP013963.1 | 2,098,391 | 1972 | CP013964.1 | 1,204,433 | 1119 | 57.27 |
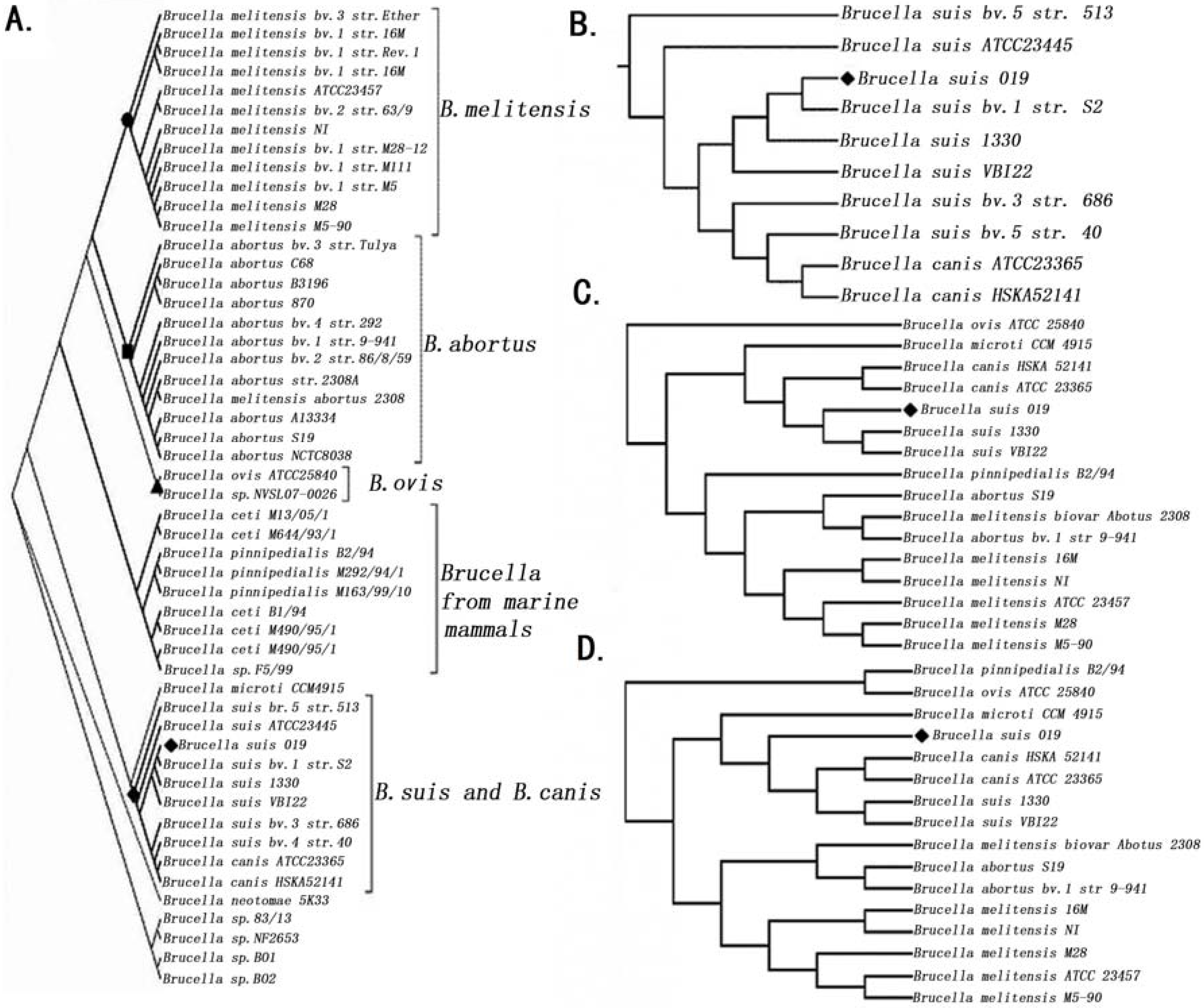
2.2. Phylogenetic Analysis

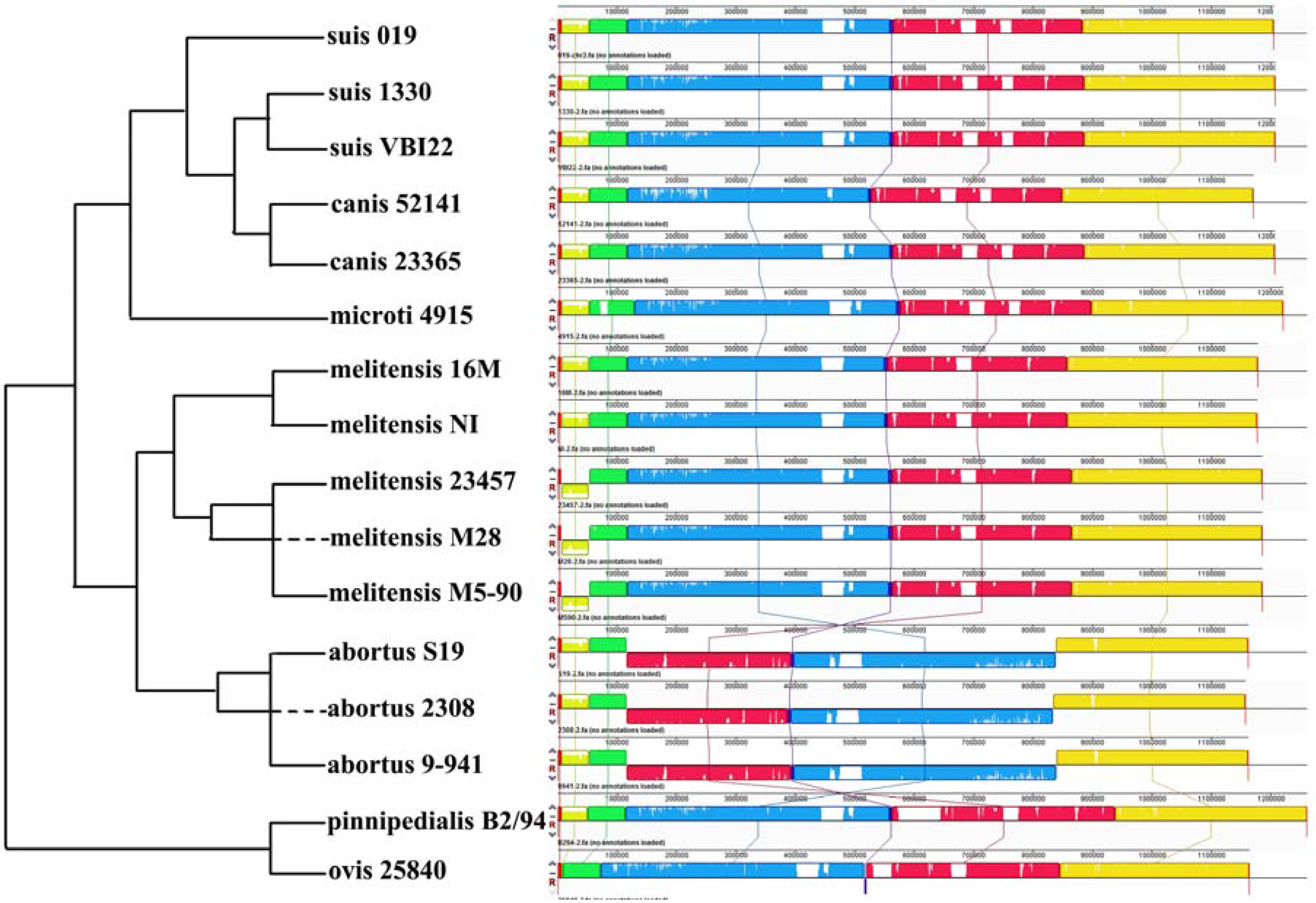
2.3. Comparative Genome Analysis Using 16 Complete Genomes



| Gene-ID | Chr | Length | Copy | Product |
|---|---|---|---|---|
| BSS2_I0512 | 1 | 795 | 1 | integrase catalytic subunit |
| BSS2_I0517 | 1 | 1119 | >1 | mannosyltransferase |
| BSS2_I0518 | 1 | 369 | 1 | transposase |
| BSS2_I0519 | 1 | 342 | 1 | IS5 family transposase orfB |
| BSS2_I0898 | 1 | 165 | >1 | hypothetical protein |
| BSS2_I1794 | 1 | 1929 | 1 | hypothetical protein |
| BSS2_I1795 | 1 | 675 | 1 | hypothetical protein |
| BSS2_II0527 | 2 | 1965 | >1 | cell wall surface protein |
| chr1_239 | 1 | 2232 | 1 | gtp pyrophosphokinase rsh |
| chr1_277 | 1 | 1140 | 1 | cytochrome c-type biogenesis protein |
| chr1_995 | 1 | 4782 | 1 | outer membrane autotransporter barrel domain-containing protein |
| chr1_1847 | 1 | 777 | 1 | 3-mercaptopyruvate sulfurtransferase |
2.4. Beta-Ketoadipate Pathway and Lipopolysaccharide
| 1330-ID | Chr | Start | End | 019-ID | Start | End | Product |
|---|---|---|---|---|---|---|---|
| BRA0636 | 2 | 617674 | 618876 | chr2_454 | 490638 | 491840 | beta-ketoadipyl CoA thiolase |
| BRA0637 | 2 | 618885 | 619574 | chr2_453 | 489940 | 490629 | 3-oxoadipate CoA-transferase, beta subunit |
| BRA0638 | 2 | 619571 | 620278 | chr2_452 | 489236 | 489943 | 3-oxoadipate CoA-transferase, alpha subunit |
| BRA0639 | 2 | 620462 | 621232 | chr2_451 | 488282 | 489052 | transcriptional regulator PcaR, putative |
| BRA0640 | 2 | 621239 | 622156 | chr2_450 | 487358 | 488275 | pobR protein |
| BRA0641 | 2 | 622269 | 623438 | chr2_449 | 486076 | 487245 | p-hydroxybenzoate hydroxylase |
| BRA0642 | 2 | 623848 | 624756 | chr2_448 | 484758 | 485666 | transcriptional regulator PcaQ |
| BRA0643 | 2 | 624850 | 625653 | chr2_447 | 483861 | 484664 | 3-oxoadipate enol-lactone hydrolase |
| BRA0644 | 2 | 625657 | 626073 | chr2_446 | 483441 | 483860 | 4-carboxymuconolactone decarboxylase |
| BRA0645 | 2 | 626070 | 626810 | chr2_445 | 482704 | 483444 | protocatechuate 3,4-dioxygenase, beta subunit |
| BRA0646 | 2 | 626812 | 627429 | chr2_444 | 482085 | 482702 | protocatechuate 3,4-dioxygenase, alpha subunit |
| BRA0647 | 2 | 627433 | 628497 | chr2_443 | 481017 | 482081 | 3-carboxy-cis,cis-muconate cycloisomerase, putative |
| BRA1155 | 2 | 1155255 | 1156661 | chr2_1084 | 1157262 | 1158668 | aldehyde dehydrogenase family protein |
| BRA1156 | 2 | 1156818 | 1157627 | chr2_1082 | 1156296 | 1157105 | 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase |
| BRA1157 | 2 | 1157694 | 1157804 | chr2_1081x | 1156119 | 1156229 | hypothetical protein |
| BRA1158 | 2 | 1157845 | 1158648 | chr2_1081 | 1155275 | 1156078 | 2-keto-4-pentenoate hydratase |
| BRA1159 | 2 | 1158652 | 1159518 | chr2_1080 | 1154405 | 1155271 | fumarylacetoacetate hydrolase family protein |
| BRA1160 | 2 | 1159587 | 1160567 | chr2_1078 | 1153356 | 1154336 | catechol 2,3-dioxygenase (pseudo) |
| BRA1161 | 2 | 1160622 | 1162136 | chr2_1077 | 1151787 | 1153301 | 5-carboxy-2-hydroxymuconate semialdehyde dehydrogenase |
| BRA1162 | 2 | 1162206 | 1162598 | chr2_1076 | 1151325 | 1151717 | 5-carboxymethyl-2-hydroxymuconate delta isomerase |
| BR0058 | 1 | 64824 | 66455 | chr1_179 | 820216 | 821847 | phosphoglucomutase |
| BR0510 | 1 | 509856 | 511724 | chr1_359 | 374961 | 376829 | epimerase/dehydratase, putative |
| BR0511 | 1 | 511711 | 512718 | chr1_358 | 373967 | 374974 | glycosyl transferase, group 4 family protein |
| BR0517 * | 1 | 516587 | 517366 | chr1_353 | 369319 | 370098 | formyltransferase, putative |
| BR0519 * | 1 | 518244 | 519002 | chr1_351 | 367683 | 368441 | O-antigen export system ATP-binding protein RfbE |
| BR0520 | 1 | 518999 | 519781 | chr1_350 | 366904 | 367686 | O-antigen export system permease protein RfbD |
| BR0521 | 1 | 519796 | 520899 | chr1_349 | 365786 | 366889 | perosamine synthase, putative |
| BR0522 | 1 | 520907 | 521995 | chr1_348 | 364690 | 365778 | GDP-mannose 4,6-dehydratase |
| BR0529 | 1 | 525257 | 526375 | NA | 498887 | 498927 | mannosyltransferase, putative |
| BR0537 | 1 | 529702 | 531039 | chr1_345 | 361179 | 362516 | phosphomannomutase, putative |
| BR0538 * | 1 | 531064 | 532488 | chr1_344 | 359730 | 361154 | mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase |
| BR0539 * | 1 | 532521 | 533693 | chr1_343 | 358525 | 359697 | mannose-6-phosphate isomerase |
| BR0540 | 1 | 533776 | 534885 | chr1_342 | 357333 | 358442 | glycosyl transferase, group 1 family protein |
| BR0615 | 1 | 606884 | 608995 | chr1_269 | 283223 | 285334 | membrane protein, putative |
| BR0981 | 1 | 948161 | 949393 | chr1_1915 | 2041237 | 2042469 | glycosyl transferase WboA |
| BR0982 | 1 | 949390 | 950949 | chr1_1914 | 2039681 | 2041240 | glycosyl transferase, group 1 family protein |
| BR1503 * | 1 | 1457802 | 1458866 | chr1_1441 | 1531664 | 1532728 | lipopolysaccharide core biosynthesis mannosyltransferase LpcC |
| BRA0347 * | 2 | 326808 | 328223 | chr2_723 | 778336 | 779751 | mannose-1-phosphate guanylyltransferase/mannose-6-phosphate isomerase |
| BRA0348 | 2 | 328220 | 329653 | chr2_722 | 776906 | 778339 | phosphoglucomutase, putative |
3. Materials and Methods
3.1. Sample Preparation, DNA-seq Library Construction
3.2. Draft Genome Sequencing, Assembly and Annotation
3.3. Complete Genome Sequencing, Assembly and Annotation
3.4. Phylogenetic Analysis Using Homologous Genes
3.5. Comparative Genome Analysis Using 16 Complete Genomes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NGS | Next-generation sequencing |
| LCBs | Locally Collinear Blocks |
| LPS | Lipopolysaccharide |
| O-PS | O-polysaccharide |
| GO | Gene Ontology |
| UPGMA | unweighted pair-group method with arithmetic means |
References
- Carvalho, N.A.; Mol, J.P.; Xavier, M.N.; Paixao, T.A.; Lage, A.P.; Santos, R.L. Pathogenesis of bovine brucellosis. Vet. J. 2010, 184, 146–155. [Google Scholar]
- Roth, F.; Zinsstag, J.; Orkhon, D.; Chimed-Ochir, G.; Hutton, G.; Cosivi, O.; Carrin, G.; Otte, J. Human health benefits from livestock vaccination for brucellosis: Case study. Bull. World Health Organ. 2003, 81, 867–876. [Google Scholar] [PubMed]
- O'Callaghan, D.; Whatmore, A.M. Brucella genomics as we enter the multi-genome era. Brief. Funct. Genomics 2011, 10, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Foster, J.T.; Mane, S.P.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.M.; Dickerman, A.W.; Keim, P.; Pearson, T.; Shukla, M.; Ward, D.V. Comparative phylogenomics and evolution of the brucellae reveal a path to virulence. J. Bacteriol. 2014, 196, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Miao, L. Brucella ovis was first isolated and identified in xinjiang province. Shihezi Sci.Technol. 1992, S1–S4. [Google Scholar]
- Liu, Z.; Ren, J. A study on brucella ovis in xinjiang. Endemic Dis. Bull. 1993, 8, 52–60. [Google Scholar]
- Zhang, G.; Zhang, Y.; Liu, Z.; Cheng, F.; Miao, L.; Guo, X.; Wang, C.; Bai, C.; Cai, Q. Artifical infection or rheus monkey with brucella ovis and pathological observation and isolation of pathogen. Chin. J. Zoonoses 1999, 15, 78–80. [Google Scholar]
- Liu, J.; Chen, C.; Tian, J. Cloning and sequence analysis of the omp25 gene of brucella ovis in xinjiang sheep. Chin. J. Vet. Med. 2006, 41, 6–8. [Google Scholar]
- Wang, Y.; Chen, C.; Cui, B.; Cao, X.; Zhang, H. Comparative study on omp gene sequences of brucella ovis 019 strain. Dis. Surveill. 2010, 25, 737–740. [Google Scholar]
- Darling, A.E.; Miklos, I.; Ragan, M.A. Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 2008, 4, e1000128. [Google Scholar] [CrossRef] [PubMed]
- Xin, X. Orally administrable brucellosis vaccine: Brucella suis strain 2 vaccine. Vaccine 1986, 4, 212–216. [Google Scholar] [CrossRef]
- Foster, J.T.; Beckstrom-Sternberg, S.M.; Pearson, T.; Beckstrom-Sternberg, J.S.; Chain, P.S.; Roberto, F.F.; Hnath, J.; Brettin, T.; Keim, P. Whole-genome-based phylogeny and divergence of the genus brucella. J. Bacteriol. 2009, 191, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Dikow, R.B. Genome-level homology and phylogeny of shewanella (gammaproteobacteria: Lteromonadales: Shewanellaceae). BMC Genomics 2011, 12, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Dozot, M.; Boigegrain, R.A.; Delrue, R.M.; Hallez, R.G.; Ouahrani-Bettache, S.; Danese, I.; Letesson, J.J.; De Bolle, X.; K hler, S. The stringent response mediator rsh is required for brucella melitensis and brucella suis virulence, and for expression of the type iv secretion system virb. Cell. Microbiol. 2006, 8, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Ouahrani-Bettache, S.; Drake, K.L.; Adams, L.G.; Köhler, S.; Occhialini, A. Global rsh-dependent transcription profile of brucella suis during stringent response unravels adaptation to nutrient starvation and cross-talk with other stress responses. BMC Genomics 2013, 14, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T.; Seshadri, R.; Nelson, K.E.; Eisen, J.A.; Heidelberg, J.F.; Read, T.D.; Dodson, R.J.; Umayam, L.; Brinkac, L.M.; Beanan, M.J. The brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. 2002, 99, 13148–13153. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Williams, K.P.; Snyder, E.E.; Almeida, N.F.; Shukla, M.; Dickerman, A.; Crasta, O.; Kenyon, R.; Lu, J.; Shallom, J. Analysis of ten brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J. Bacteriol. 2009, 191, 3569–3579. [Google Scholar] [CrossRef] [PubMed]
- Godfroid, F.; Cloeckaert, A.; Taminiau, B.; Danese, I.; Tibor, A.; de Bolle, X.; Mertens, P.; Letesson, J.-J. Genetic organisation of the lipopolysaccharide o-antigen biosynthesis region of brucella melitensis 16m (wbk). Res. Microbiol. 2000, 151, 655–668. [Google Scholar] [CrossRef]
- Mancilla, M.; Marín, C.M.; Blasco, J.M.; Zárraga, A.M.; López-Goñi, I.; Moriyón, I. Spontaneous excision of the o-polysaccharide wbka glycosyltranferase gene is a cause of dissociation of smooth to rough brucella colonies. J. Bacteriol. 2012, 194, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. Soapdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with glimmer. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Fei, Z.; Zhan, F.; Gong, X.; Gao, S. Fastq_clean: An optimized pipeline to clean the illumina sequencing data with quality control. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, November 2014; pp. 44–48.
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A. A whole-genome assembly of drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J.Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Nadalin, F.; Vezzi, F.; Policriti, A. Gapfiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13, S8–S23. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evolut. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, Z.; Chen, X.; Zhang, H.; Guo, F.; Zhang, K.; Feng, H.; Gu, W.; Wu, C.; Ma, L.; et al. The Complete Genome of Brucella Suis 019 Provides Insights on Cross-Species Infection. Genes 2016, 7, 7. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7020007
Wang Y, Wang Z, Chen X, Zhang H, Guo F, Zhang K, Feng H, Gu W, Wu C, Ma L, et al. The Complete Genome of Brucella Suis 019 Provides Insights on Cross-Species Infection. Genes. 2016; 7(2):7. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7020007
Chicago/Turabian StyleWang, Yuanzhi, Zhen Wang, Xin Chen, Hui Zhang, Fei Guo, Ke Zhang, Hanping Feng, Wenyi Gu, Changxin Wu, Lei Ma, and et al. 2016. "The Complete Genome of Brucella Suis 019 Provides Insights on Cross-Species Infection" Genes 7, no. 2: 7. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7020007