
The MST/Hippo Pathway and Cell Death: A Non-Canonical Affair


{kind=link}
{kind=link}
Abstract
:1. Introduction
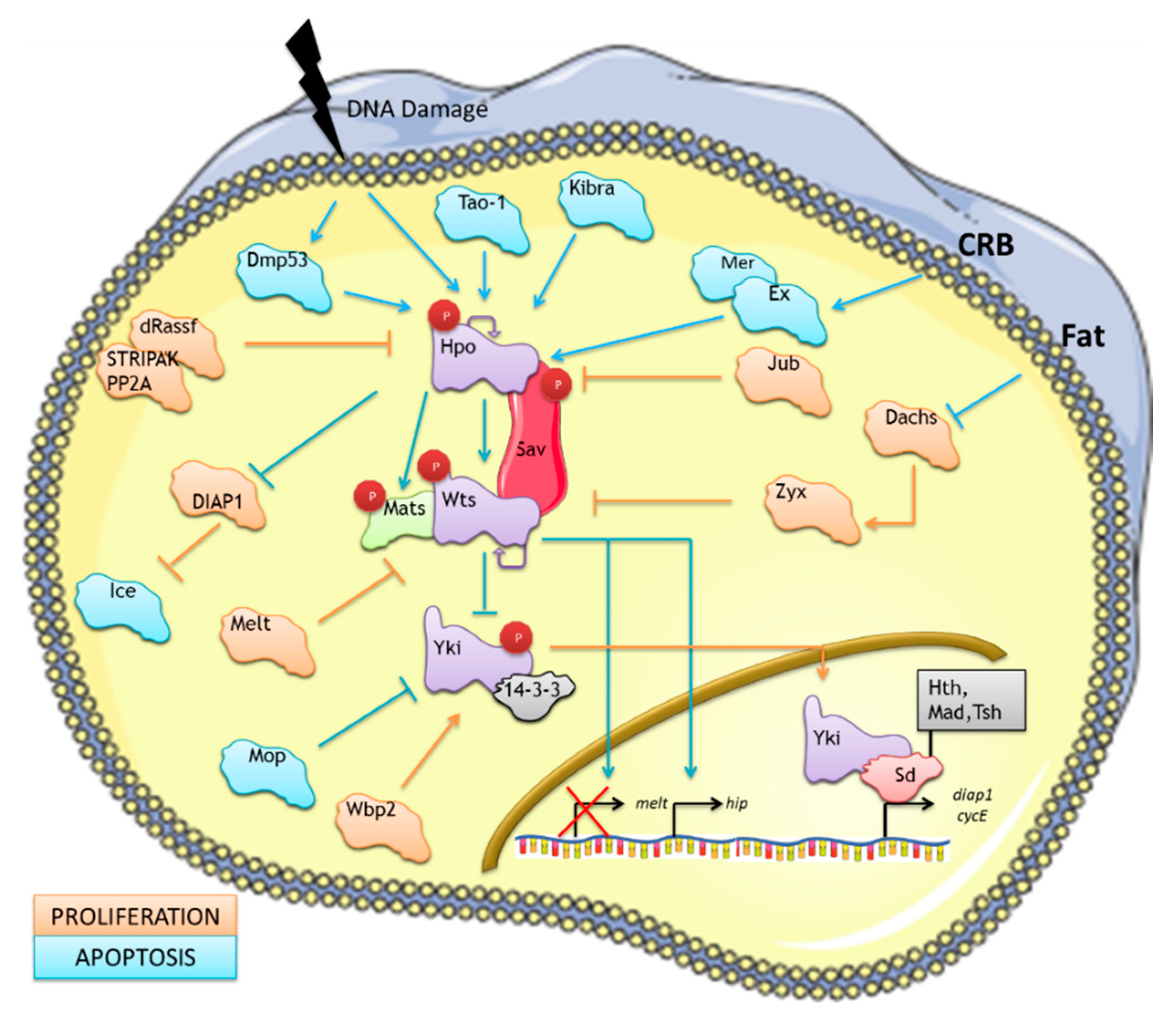
2. The Canonical Drosophila Hippo Pathway
2.1. The Hippo Pathway Regulation of Pro-apoptotic Signals
2.2. Non-canonical Regulation of the Hippo Pathway: Evidence for the Existence of a Hippo Signalling Network
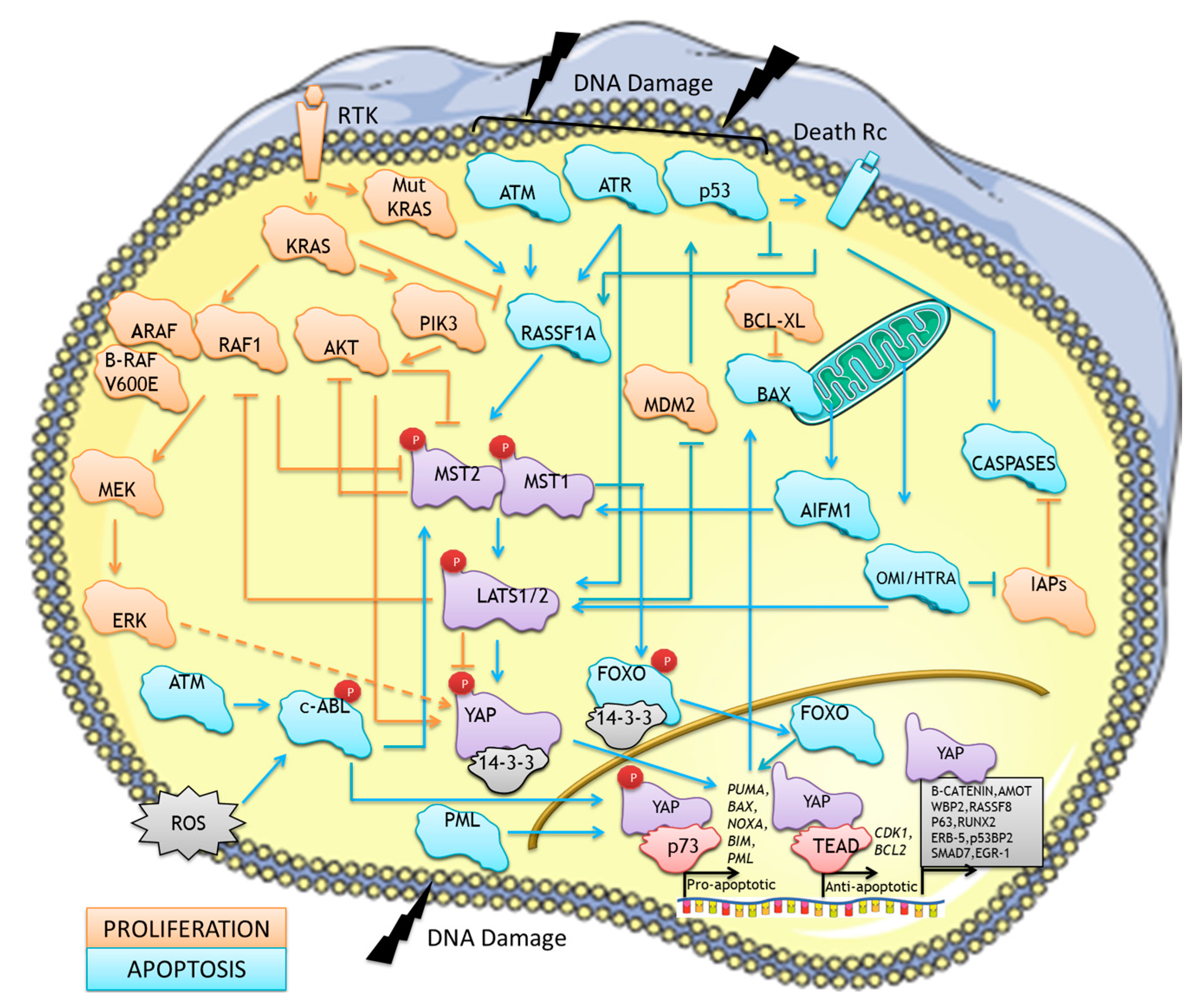
3. Mammalian MST/Hippo Pathway, a Regulator of Cell Death at Many Levels
3.1. The Canonical Mammalian Hippo Pathway
3.2. The Non-canonical MST/Hippo Pro-apoptotic Pathway
3.3. From Pathway to Network
3.3.1. Regulators
RASSF Family
Family of RAS GTPases
RAS Effector Signalling Network
ATM and ATR
3.3.2. Core Kinases, at the Crossroad of Many Pathways
MST1/2 Apoptotic Network
LATS1/2 Pro-apoptotic Network
3.3.3. YAP the Most Famous Effector of the MST/ Hippo Pathway
4. The MST/Hippo Pro-apoptotic Network in Human Disease
4.1. Cancer
4.2. Neurodegenerative Diseases
4.3. Diabetes
4.4. Cardiomyopathy
5. Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MAPK | Mitogen activated protein kinase |
| YAP | Yes-associated protein |
| LATS1/2 | large tumour suppressor 1/2 |
| MST1/2 | mammalian sterile 20-like kinase ½ |
| RASSF | Ras association domain family |
| SARAH | Salvador-RASSF-Hippo |
| TEAD | TEA domain family member |
| TAZ | Tafazzin |
| dSTRIPAK | Drosophila Striatin-interacting phosphatase and kinase WNT: wingless-type MMTV site family |
| SHH | sonic hedgehog |
| TGF | transforming growth factor |
| AKT | v-akt murine thymoma viral oncogene homolog |
| Ras | rat sarcoma virus oncogene |
| RAF | Rat fibrosarcoma |
| PI3K | phosphatidylinositol-4,5-bisphosphate 3-kinase |
References
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Hamilton, G.; Kolch, W.; O’Neill, E. A hippo in the ointment: Mst signalling beyond the fly. Cell Cycle 2008, 7, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. Rassf1a elicits apoptosis through an mst2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell. 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The hippo pathway effectors taz and yap in development, homeostasis and disease. Developmet 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The hippo-yap pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lei, Q.Y.; Guan, K.L. The hippo-yap pathway: New connections between regulation of organ size and cancer. Curr. Opin. Cell Biol. 2008, 20, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.E.; Matallanas, D.; Kolch, W. Mammalian sterile 20-like kinases in tumor suppression: An emerging pathway. Cancer Res. 2005, 65, 5485–5487. [Google Scholar] [CrossRef] [PubMed]
- Radu, M.; Chernoff, J. The demstification of mammalian ste20 kinases. Curr. Biol. 2009, 19, R421–R425. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Zhou, D.; Fitamant, J.; Bardeesy, N.; Mou, F.; Barrufet, L.R. Protein kinases of the hippo pathway: Regulation and substrates. Semin. Cell Dev. Biol. 2012, 23, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.; Yang, X. Lats tumor suppressor: A new governor of cellular homeostasis. Cell Cycle 2010, 9, 3892–3903. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Matallanas, D.; Frederick, D.T.; Flaherty, K.T.; Kolch, W. One hippo and many masters: Differential regulation of the hippo pathway in cancer. Biochem. Soc. Trans. 2014, 42, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate raf-1 and mst2/hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A molecular mechanism that links hippo signalling to the inhibition of wnt/beta-catenin signalling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.T.; Gough, N.R. Beyond canonical: The wnt and β-catenin story. Sci. Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Eisinger-Mathason, T.S. Targeting the hippo pathway: Clinical implications and therapeutics. Pharmacol. Res. 2016, 103, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of hippo: Targeting the hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Tomlinson, V.; Lara, R.; Holliday, D.; Chelala, C.; Harada, T.; Gangeswaran, R.; Manson-Bishop, C.; Smith, P.; Danovi, S.A.; et al. Yes-associated protein (yap) functions as a tumor suppressor in breast. Cell Death Differ. 2008, 15, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Du, Y.C.; Zhou, X.J.; Liu, H.; Tang, S.C. The dual functions of yap-1 to promote and inhibit cell growth in human malignancy. Cancer Metastasis Rev. 2014, 33, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, V.; Gudmundsdottir, K.; Luong, P.; Leung, K.Y.; Knebel, A.; Basu, S. Jnk phosphorylates yes-associated protein (yap) to regulate apoptosis. Cell Death Dis. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bertini, E.; Oka, T.; Sudol, M.; Strano, S.; Blandino, G. Yap: At the crossroad between transformation and tumor suppression. Cell Cycle 2009, 8, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Yu, A.; Tao, W. The non-canonical hippo/mst pathway in lymphocyte development and functions. Acta Biochim. Biophys. Sin. 2015, 47, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Scrace, S.F.; O’Neill, E. Rassf signalling and DNA damage: Monitoring the integrity of the genome? Mol. Biol. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. Yap mediates crosstalk between the hippo and pi(3)k-tor pathways by suppressing pten via mir-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Bondar, T.; Zhou, X.; Zhang, C.; He, H.; Medzhitov, R. Two-signal requirement for growth-promoting function of yap in hepatocytes. Elife 2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Ohyama, T.; Yajima, N.; Tsubuki, S.; Yonehara, S. Mst, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J. Biol. Chem. 2001, 276, 19276–19285. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant k-ras activation of the proapoptotic mst2 pathway is antagonized by wild-type k-ras. Mol. Cell 2011, 44, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Ofir-Rosenfeld, Y.; Yabuta, N.; Lapi, E.; Nojima, H.; Lu, X.; Oren, M. The lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of aspp1. Genes Dev. 2010, 24, 2420–2429. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Yabuta, N.; Besserglick, H.; Buganim, Y.; Rotter, V.; Nojima, H.; Oren, M. Silencing of the lats2 tumor suppressor overrides a p53-dependent oncogenic stress checkpoint and enables mutant h-ras-driven cell transformation. Oncogene 2009, 28, 4469–4479. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of hippo coactivator yap1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Staley, B.K.; Irvine, K.D. Hippo signaling in drosophila: Recent advances and insights. Dev. Dyn. 2012, 241, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating yorkie, the drosophila homolog of yap. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer Cell 2002, 1, 19–30. [Google Scholar] [CrossRef]
- Conlon, I.; Raff, M. Size control in animal development. Cell 1999, 96, 235–244. [Google Scholar] [CrossRef]
- Harvey, K.; Tapon, N. The salvador-warts-hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.D.; Harvey, K.F. Control of organ growth by patterning and hippo signaling in drosophila. Cold Spring Harb. Perspect Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.L.; Lin, J.I.; Zhang, X.; Harvey, K.F. The sterile 20-like kinase tao-1 controls tissue growth by regulating the salvador-warts-hippo pathway. Dev. Cell 2011, 21, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Polesello, C.; Huelsmann, S.; Brown, N.H.; Tapon, N. The drosophila rassf homolog antagonizes the hippo pathway. Curr. Biol. 2006, 16, 2459–2465. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.S.; Josue, F.; Wepf, A.; Wehr, M.C.; Rinner, O.; Kelly, G.; Tapon, N.; Gstaiger, M. Combined functional genomic and proteomic approaches identify a pp2a complex as a negative regulator of hippo signaling. Mol. Cell. 2010, 39, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Vos, M.D.; Clark, G.J. The rassf1a tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Rauskolb, C.; Pan, G.; Reddy, B.V.; Oh, H.; Irvine, K.D. Zyxin links fat signaling to the hippo pathway. PLoS Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.M.; Tipping, M.; Veraksa, A.; Moberg, K.H. A screen for conditional growth suppressor genes identifies the drosophila homolog of hd-ptp as a regulator of the oncoprotein yorkie. Dev. Cell 2011, 20, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Milton, C.C.; Poon, C.L.; Hong, W.; Harvey, K.F. Wbp2 cooperates with yorkie to drive tissue growth downstream of the salvador-warts-hippo pathway. Cell Death Differ. 2011, 18, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the salvador/warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The drosophila mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef]
- Pantalacci, S.; Tapon, N.; Leopold, P. The salvador partner hippo promotes apoptosis and cell-cycle exit in drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Colombani, J.; Polesello, C.; Josue, F.; Tapon, N. Dmp53 activates the hippo pathway to promote cell death in response to DNA damage. Curr. Biol. 2006, 16, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cohen, S.M. The hippo pathway acts via p53 and micrornas to control proliferation and proapoptotic gene expression during tissue growth. Biol. Open 2013, 2, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Vinayagam, A.; Sun, X.; Dephoure, N.; Gygi, S.P.; Hong, P.; Perrimon, N. The hippo signaling pathway interactome. Science 2013, 342, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Mikeladze-Dvali, T.; Wernet, M.F.; Pistillo, D.; Mazzoni, E.O.; Teleman, A.A.; Chen, Y.W.; Cohen, S.; Desplan, C. The growth regulators warts/lats and melted interact in a bistable loop to specify opposite fates in drosophila r8 photoreceptors. Cell 2005, 122, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Jukam, D.; Johnston, R.; Desplan, C. The neuronal transcription factor erect wing regulates specification and maintenance of drosophila r8 photoreceptor subtypes. Dev. Biol. 2013, 381, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Emoto, K.; Parrish, J.Z.; Jan, L.Y.; Jan, Y.N. The tumour suppressor hippo acts with the ndr kinases in dendritic tiling and maintenance. Nature 2006, 443, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Parrish, J.Z.; Emoto, K.; Jan, L.Y.; Jan, Y.N. Polycomb genes interact with the tumor suppressor genes hippo and warts in the maintenance of drosophila sensory neuron dendrites. Genes Dev. 2007, 21, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.W.; Slattery, M.; Mann, R.S. Transcription factor choice in the hippo signaling pathway: Homothorax and yorkie regulation of the microrna bantam in the progenitor domain of the drosophila eye imaginal disc. Genes Dev. 2009, 23, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, Q.; Pignoni, F. Yki/yap, sd/tead and hth/meis control tissue specification in the drosophila eye disc epithelium. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Robbins, R.M.; Gbur, S.C.; Beitel, G.J. Non-canonical roles for yorkie and drosophila inhibitor of apoptosis 1 in epithelial tube size control. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Irvine, K.D. In vivo regulation of yorkie phosphorylation and localization. Development 2008, 135, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Irvine, K.D. In vivo analysis of yorkie phosphorylation sites. Oncogene 2009, 28, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Blandino, G. Yap1 meets tumor suppression. Mol. Cell 2007, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. Yap1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, T.S.; Yang, T.H.; Koo, B.K.; Oh, S.P.; Lee, K.P.; Oh, H.J.; Lee, S.H.; Kong, Y.Y.; Kim, J.M.; et al. A crucial role of ww45 in developing epithelial tissues in the mouse. EMBO J. 2008, 27, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. Tead/tef transcription factors utilize the activation domain of yap65, a src/yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. Tead mediates yap-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Couzens, A.L.; Knight, J.D.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein interaction network of the mammalian hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, Q.; Gutkind, J.S. Oncotargeting G proteins: The hippo in the room. Oncotarget 2014, 5, 10997–10999. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Wepf, A.; van Drogen, A.; Varjosalo, M.; Tapon, N.; Aebersold, R.; Gstaiger, M. Interaction proteome of human hippo signaling: Modular control of the co-activator yap1. Mol. Syst. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.; Yang, X. Identification of lats transcriptional targets in hela cells using whole human genome oligonucleotide microarray. Gene 2010, 449, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.L. Hippo unleashed! Proteome-scale analysis reveals new views of hippo pathway biology. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kodaka, M.; Hata, Y. The mammalian hippo pathway: Regulation and function of yap1 and taz. Cell Mol. Life Sci. 2015, 72, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Hori, T.; Chonabayashi, K.; Oka, T.; Sudol, M.; Uchiyama, T. Kpm/lats2 is linked to chemosensitivity of leukemic cells through the stabilization of p73. Blood 2008, 112, 3856–3866. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Mazack, V.; Sudol, M. Mst2 and lats kinases regulate apoptotic function of yes kinase-associated protein (yap). J. Biol. Chem. 2008, 283, 27534–27546. [Google Scholar] [CrossRef] [PubMed]
- Preisinger, C.; von Kriegsheim, A.; Matallanas, D.; Kolch, W. Proteomics and phosphoproteomics for the mapping of cellular signalling networks. Proteomics 2008, 8, 4402–4415. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Rushworth, L.; Baccarini, M.; Kolch, W. Role of the kinase mst2 in suppression of apoptosis by the proto-oncogene product raf-1. Science 2004, 306, 2267–2270. [Google Scholar] [CrossRef] [PubMed]
- Creasy, C.L.; Ambrose, D.M.; Chernoff, J. The ste20-like protein kinase, mst1, dimerizes and contains an inhibitory domain. J. Biol. Chem. 1996, 271, 21049–21053. [Google Scholar] [PubMed]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the mst1 kinase by autophosphorylation, by the growth inhibitory proteins, rassf1 and nore1, and by ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Khokhlatchev, A.; Rabizadeh, S.; Xavier, R.; Nedwidek, M.; Chen, T.; Zhang, X.F.; Seed, B.; Avruch, J. Identification of a novel ras-regulated proapoptotic pathway. Curr. Biol. 2002, 12, 253–265. [Google Scholar] [CrossRef]
- Oh, H.J.; Lee, K.K.; Song, S.J.; Jin, M.S.; Song, M.S.; Lee, J.H.; Im, C.R.; Lee, J.O.; Yonehara, S.; Lim, D.S. Role of the tumor suppressor rassf1a in mst1-mediated apoptosis. Cancer Res. 2006, 66, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.K.; Dammann, R.; Pfeifer, G.P. Rassf1a is part of a complex similar to the drosophila hippo/salvador/lats tumor-suppressor network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the yes-associated protein, yap, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003, 11, 11–23. [Google Scholar] [CrossRef]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. Pml, yap, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Tufail, R.; Jorda, M.; Zhao, W.; Reis, I.; Nawaz, Z. Loss of yes-associated protein (yap) expression is associated with estrogen and progesterone receptors negativity in invasive breast carcinomas. Breast Cancer Res. Treat. 2012, 131, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Mauviel, A.; Nallet-Staub, F.; Varelas, X. Integrating developmental signals: A hippo in the (path)way. Oncogene 2012, 31, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The hippo pathway regulates wnt/beta-catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Matallanas, D.; Weitsman, G.; Preisinger, C.; Ng, T.; Kolch, W. Proapoptotic kinase mst2 coordinates signaling crosstalk between rassf1a, raf-1, and akt. Cancer Res. 2010, 70, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Gordon, M.; Salla, M.; Ghazaleh, H.A.; Baksh, S. Rassf tumor suppressor gene family: Biological functions and regulation. FEBS Lett. 2014, 588, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.S.; Carneiro, F.; Oliveira, C.; Seruca, R. Colorectal cancer and rassf family—A special emphasis on rassf1a. Int. J. Cancer 2013, 132, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The rassf proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Van der Weyden, L.; Adams, D.J. The ras-association domain family (rassf) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, X.; Huang, J.; Feng, L.; Dolinta, K.G.; Chen, J. Defining the protein-protein interaction network of the human hippo pathway. Mol. Cell Proteomics 2014, 13, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Hauri, S.; Wepf, A.; van Drogen, A.; Varjosalo, M.; Tapon, N.; Aebersold, R.; Gstaiger, M. Interaction proteome of human hippo signaling: Modular control of the co-activator yap1. Mol. Syst. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.N.; Hesson, L.B.; Matallanas, D.; Dallol, A.; von Kriegsheim, A.; Ward, R.; Kolch, W.; Latif, F. Rassf2 associates with and stabilizes the proapoptotic kinase mst2. Oncogene 2009, 28, 2988–2998. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Cooper, W.N.; Latif, F. Role of the ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005, 65, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Grawenda, A.M.; O’Neill, E. Clinical utility of rassf1a methylation in human malignancies. Br. J. Cancer 2015, 113, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.E.; Firek, M.; Chen, S.T.; Amaar, Y. The rassf1 gene and the opposing effects of the rassf1a and rassf1c isoforms on cell proliferation and apoptosis. Mol. Biol. Int. 2013. [Google Scholar] [CrossRef] [PubMed]
- Malpeli, G.; Amato, E.; Dandrea, M.; Fumagalli, C.; Debattisti, V.; Boninsegna, L.; Pelosi, G.; Falconi, M.; Scarpa, A. Methylation-associated down-regulation of rassf1a and up-regulation of rassf1c in pancreatic endocrine tumors. BMC Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Coordinating erk/mapk signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, Y.; Avruch, J.; Zhang, X.F. Nore1 inhibits tumor cell growth independent of ras or the mst1/2 kinases. Oncogene 2004, 23, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Praskova, M.; Ortiz-Vega, S.; Liu, M.; Zhang, X.F. Nore1 and rassf1 regulation of cell proliferation and of the mst1/2 kinases. Meth. Enzymol. 2006, 407, 290–310. [Google Scholar] [PubMed]
- Zhou, X.H.; Yang, C.Q.; Zhang, C.L.; Gao, Y.; Yuan, H.B.; Wang, C. Rassf5 inhibits growth and invasion and induces apoptosis in osteosarcoma cells through activation of mst1/lats1 signaling. Oncol. Rep. 2014, 32, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Keller, M.; Calvayrac, O.; Soncin, F.; Hoa, L.; Hergovich, A.; Parrini, M.C.; Mazieres, J.; Vaisse-Lesteven, M.; Camonis, J.; et al. Rassf1a suppresses the invasion and metastatic potential of human non-small cell lung cancer cells by inhibiting yap activation through the gef-h1/rhob pathway. Cancer Res. 2016, 76, 1627–1640. [Google Scholar] [CrossRef] [PubMed]
- Kohli, P.; Bartram, M.P.; Habbig, S.; Pahmeyer, C.; Lamkemeyer, T.; Benzing, T.; Schermer, B.; Rinschen, M.M. Label-free quantitative proteomic analysis of the yap/taz interactome. Am. J. Physiol. Cell Physiol. 2014, 306, C805–C818. [Google Scholar] [CrossRef] [PubMed]
- Feig, L.A.; Buchsbaum, R.J. Cell signaling: Life or death decisions of ras proteins. Curr. Biol. 2002, 12, R259–R261. [Google Scholar] [CrossRef]
- Ortiz-Vega, S.; Khokhlatchev, A.; Nedwidek, M.; Zhang, X.F.; Dammann, R.; Pfeifer, G.P.; Avruch, J. The putative tumor suppressor rassf1a homodimerizes and heterodimerizes with the ras-gtp binding protein nore1. Oncogene 2002, 21, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Maccario, H.; Doherty, C.; Quinn, N.P.; Kolch, W.; Matallanas, D. The differential effects of wild-type and mutated k-ras on mst2 signaling are determined by k-ras activation kinetics. Mol. Cell. Biol. 2013, 33, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the ras and hippo pathways converge on regulation of yap protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rendueles, M.E.; Ricarte-Filho, J.C.; Untch, B.R.; Landa, I.; Knauf, J.A.; Voza, F.; Smith, V.E.; Ganly, I.; Taylor, B.S.; Persaud, Y.; et al. Nf2 loss promotes oncogenic ras-induced thyroid cancers via yap-dependent transactivation of ras proteins and sensitizes them to mek inhibition. Cancer Discov. 2015, 5, 1178–1193. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Li, C.; Luo, S.; Liu-Smith, F.; Yang, J.; Wang, X.; Wang, N.; Lai, B.; Lei, T.; Wang, Q.; et al. Yes-associated protein contributes to the development of human cutaneous squamous cell carcinoma via activation of ras. J. Investig. Dermatol. 2016, 136, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The hippo effector yap promotes resistance to raf- and mek-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kilili, G.K.; Kyriakis, J.M. Mammalian ste20-like kinase (mst2) indirectly supports raf-1/erk pathway activity via maintenance of protein phosphatase-2a catalytic subunit levels and consequent suppression of inhibitory raf-1 phosphorylation. J. Biol. Chem. 2010, 285, 15076–15087. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; O’Neill, E.; Mack, B.; Matthias, C.; Munz, M.; Kolch, W.; Gires, O. Heterogeneous nuclear ribonucleoprotein h blocks mst2-mediated apoptosis in cancer cells by regulating a-raf transcription. Cancer Res. 2010, 70, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, M.H.; Kim, D.W.; Lee, S.; Huang, S.; Ryu, M.J.; Kim, Y.K.; Kim, S.J.; Kim, S.J.; Hwang, J.H.; et al. Cross-regulation between oncogenic braf(v600e) kinase and the mst1 pathway in papillary thyroid carcinoma. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Meikle, S.; Yazici, Z.; Eulitz, M.; Kolch, W. Regulation of raf-1 activation and signalling by dephosphorylation. EMBO J. 2002, 21, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant kras, the transcription regulator yap is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/raf/mek/erk and pi3k/pten/akt/mtor inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shu, S.; Coppola, M.D.; Kaneko, S.; Yuan, Z.Q.; Cheng, J.Q. Regulation of proapoptotic mammalian ste20-like kinase mst2 by the igf1-akt pathway. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Cinar, B.; Fang, P.K.; Lutchman, M.; di Vizio, D.; Adam, R.M.; Pavlova, N.; Rubin, M.A.; Yelick, P.C.; Freeman, M.R. The pro-apoptotic kinase mst1 and its caspase cleavage products are direct inhibitors of akt1. EMBO J. 2007, 26, 4523–4534. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The pten/pi3k/akt signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Paluch, B.E.; Wang, X.; Jiang, X. Pten at a glance. J. Cell Sci. 2012, 125, 4687–4692. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.E.; O’Neill, E. Hippo pathway and protection of genome stability in response to DNA damage. FEBS J. 2016, 283, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. Atm regulates a rassf1a-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef] [PubMed]
- Pefani, D.E.; Latusek, R.; Pires, I.; Grawenda, A.M.; Yee, K.S.; Hamilton, G.; van der Weyden, L.; Esashi, F.; Hammond, E.M.; O’Neill, E. Rassf1a-lats1 signalling stabilizes replication forks by restricting cdk2-mediated phosphorylation of brca2. Nat. Cell Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Keshet, R.; Adler, J.; Ricardo Lax, I.; Shanzer, M.; Porat, Z.; Reuven, N.; Shaul, Y. C-abl antagonizes the yap oncogenic function. Cell Death Differ. 2015, 22, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A. Regulation and functions of mammalian lats/ndr kinases: Looking beyond canonical hippo signalling. Cell Biosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A. The roles of ndr protein kinases in hippo signalling. Genes 2016. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A. Mammalian hippo signalling: A kinase network regulated by protein-protein interactions. Biochem. Soc. Trans. 2012, 40, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Creasy, C.L.; Chernoff, J. Cloning and characterization of a member of the mst subfamily of ste20-like kinases. Gene 1995, 167, 303–306. [Google Scholar] [CrossRef]
- Creasy, C.L.; Chernoff, J. Cloning and characterization of a human protein kinase with homology to ste20. J. Biol. Chem. 1995, 270, 21695–21700. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Sanz, G.; Matallanas, D.; Nguyen, L.K.; Kholodenko, B.N.; Rosta, E.; Kolch, W.; Buchete, N.V. Mst2-rassf protein-protein interactions through sarah domains. Brief. Bioinform. 2015. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Gotoh, Y.; Draves, K.E.; Ambrose, D.; Han, D.K.; Wright, M.; Chernoff, J.; Clark, E.A.; Krebs, E.G. Caspase-mediated activation and induction of apoptosis by the mammalian ste20-like kinase mst1. EMBO J. 1998, 17, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Glantschnig, H.; Rodan, G.A.; Reszka, A.A. Mapping of mst1 kinase sites of phosphorylation. Activation and autophosphorylation. J. Biol. Chem. 2002, 277, 42987–42996. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Murakami, Y.; Kataoka, K.; Lin, H.; Connor, K.M.; Miller, J.W.; Zhou, D.; Avruch, J.; Vavvas, D.G. Mammalian ste20-like kinase 2, not kinase 1, mediates photoreceptor cell death during retinal detachment. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ura, S.; Masuyama, N.; Graves, J.D.; Gotoh, Y. Mst1-jnk promotes apoptosis via caspase-dependent and independent pathways. Genes Cells 2001, 6, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Ajiro, K.; Samejima, K.; Kloc, M.; Cheung, P.; Mizzen, C.A.; Beeser, A.; Etkin, L.D.; Chernoff, J.; Earnshaw, W.C.; et al. Apoptotic phosphorylation of histone h2b is mediated by mammalian sterile twenty kinase. Cell 2003, 113, 507–517. [Google Scholar] [CrossRef]
- Vichalkovski, A.; Gresko, E.; Cornils, H.; Hergovich, A.; Schmitz, D.; Hemmings, B.A. Ndr kinase is activated by rassf1a/mst1 in response to fas receptor stimulation and promotes apoptosis. Curr. Biol. 2008, 18, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Lee, Y.J. Differential cleavage of mst1 by caspase-7/-3 is responsible for trail-induced activation of the mapk superfamily. Cell. Signal. 2008, 20, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.S. The tumour suppressor rassf1a promotes mdm2 self-ubiquitination by disrupting the mdm2-daxx-hausp complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Yoon, J.H.; Lee, J.K.; Noh, K.T.; Yoon, K.W.; Oh, S.P.; Oh, H.J.; Chae, J.S.; Hwang, S.G.; Kim, E.H.; et al. Daxx mediates activation-induced cell death in microglia by triggering mst1 signalling. EMBO J. 2011, 30, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Navas, T.A.; Baldwin, D.T.; Stewart, T.A. Rip2 is a raf1-activated mitogen-activated protein kinase kinase. J. Biol. Chem. 1999, 274, 33684–33690. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wu, H.; Nie, H.; Yue, L.; Jiang, H.; Xiao, S.; Li, Y. Aif downregulation and its interaction with stk3 in renal cell carcinoma. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Maedler, K. Mst1: A promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia 2016. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Yang, L.; Wang, J.; Wang, Y.; Wang, J.; Zhou, X.; Yin, X.; Zhang, Z.; Zhao, D. C-abl tyrosine kinase mediates neurotoxic prion peptide-induced neuronal apoptosis via regulating mitochondrial homeostasis. Mol. Neurobiol. 2014, 49, 1102–1116. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.P.; Sadoshima, J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of bcl-xl. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Xie, Q.; Wu, J.; Bai, Y.; Mao, B.; Dong, Y.; Bi, W.; Ji, G.; Tao, W.; Wang, Y.; et al. Mst1 promotes apoptosis through regulating sirt1-dependent p53 deacetylation. J. Biol. Chem. 2011, 286, 6940–6945. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved mst-foxo signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Lehtinen, M.K.; Merlo, P.; Villen, J.; Gygi, S.; Bonni, A. Regulation of neuronal cell death by mst1-foxo1 signaling. J. Biol. Chem. 2009, 284, 11285–11292. [Google Scholar] [CrossRef] [PubMed]
- Sanphui, P.; Biswas, S.C. Foxo3a is activated and executes neuron death via bim in response to beta-amyloid. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Valis, K.; Prochazka, L.; Boura, E.; Chladova, J.; Obsil, T.; Rohlena, J.; Truksa, J.; Dong, L.F.; Ralph, S.J.; Neuzil, J. Hippo/mst1 stimulates transcription of the proapoptotic mediator noxa in a foxo1-dependent manner. Cancer Res. 2011, 71, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Yang, S.J.; Srinivasan, S.; Ye, K. Akt phosphorylates msti and prevents its proteolytic activation, blocking foxo3 phosphorylation and nuclear translocation. J. Biol. Chem. 2007, 282, 30836–30844. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, J.H.; Hwang, S.G.; Gwag, B.J.; McKee, A.C.; Lee, J.; Kowall, N.W.; Ryu, H.; Lim, D.S.; Choi, E.J. Mst1 functions as a key modulator of neurodegeneration in a mouse model of als. Proc. Natl. Acad. Sci. USA 2013, 110, 12066–12071. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, D.; Hu, P.; Wu, J.; Liu, W.; Zhao, Y.; Cao, M.; Fang, Y.; Bi, W.; Zheng, Z.; et al. The c-abl-mst1 signaling pathway mediates oxidative stress-induced neuronal cell death. J. Neurosci. 2011, 31, 9611–9619. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, J.; Xiao, L.; Bai, Y.; Qu, A.; Zheng, Z.; Yuan, Z. Regulation of neuronal cell death by c-abl-hippo/mst2 signaling pathway. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.S.; Jariwala, J.S.; Anderson, E.; Mitra, K.; Meisenhelder, J.; Chang, J.T.; Ideker, T.; Hunter, T.; Nizet, V.; Dillin, A.; et al. Phosphorylation of lc3 by the hippo kinases stk3/stk4 is essential for autophagy. Mol. Cell. 2015, 57, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.J.; Yan, L.; Vatner, D.E.; Vatner, S.F. Mst1 inhibition rescues beta1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis. Basic. Res. Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yabuta, N.; Fujii, T.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Nishiguchi, H.; Endo, Y.; Toji, S.; Tanaka, H.; Nishimune, Y.; et al. Structure, expression, and chromosome mapping of lats2, a mammalian homologue of the drosophila tumor suppressor gene lats/warts. Genomics 2000, 63, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, Y.; Hirota, T.; Morisaki, T.; Hara, T.; Marumoto, T.; Iida, S.; Makino, K.; Yamamoto, H.; Hiraoka, T.; Kitamura, N.; et al. A human homolog of drosophila warts tumor suppressor, h-warts, localized to mitotic apparatus and specifically phosphorylated during mitosis. FEBS Lett. 1999, 459, 159–165. [Google Scholar] [CrossRef]
- Hori, T.; Takaori-Kondo, A.; Kamikubo, Y.; Uchiyama, T. Molecular cloning of a novel human protein kinase, kpm, that is homologous to warts/lats, a drosophila tumor suppressor. Oncogene 2000, 19, 3101–3109. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Qi, H.; Li, Y.; Pei, J.; Barton, J.; Blackstad, M.; Xu, T.; Tao, W. Lats1 tumor suppressor regulates g2/m transition and apoptosis. Oncogene 2002, 21, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Kuninaka, S.; Nomura, M.; Hirota, T.; Iida, S.; Hara, T.; Honda, S.; Kunitoku, N.; Sasayama, T.; Arima, Y.; Marumoto, T.; et al. The tumor suppressor warts activates the omi / htra2-dependent pathway of cell death. Oncogene 2005, 24, 5287–5298. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Pei, J.; Ni, Z.; Xia, H.; Qi, H.; Woods, T.; Kelekar, A.; Tao, W. Putative tumor suppressor lats2 induces apoptosis through downregulation of bcl-2 and bcl-x(l). Exp. Cell Res. 2004, 298, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Hirota, T.; Morisaki, T.; Marumoto, T.; Hara, T.; Kuninaka, S.; Honda, S.; Kosai, K.; Kawasuji, M.; Pallas, D.C.; et al. Tumor suppressor warts ensures genomic integrity by regulating both mitotic progression and g1 tetraploidy checkpoint function. Oncogene 2004, 23, 5266–5274. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the mammalian yap (yes-associated protein) gene and its role in defining a novel protein module, the ww domain. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor lats1 is a negative regulator of oncogene yap. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of yap/taz: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, N.; Xie, R.; Wang, W.; Cai, J.; Choi, K.S.; David, K.K.; Huang, B.; Yabuta, N.; Nojima, H.; et al. Homeostatic control of hippo signaling activity revealed by an endogenous activating mutation in yap. Genes Dev. 2015, 29, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Fausti, F.; Di Agostino, S.; Cioce, M.; Bielli, P.; Sette, C.; Pandolfi, P.P.; Oren, M.; Sudol, M.; Strano, S.; Blandino, G. Atm kinase enables the functional axis of yap, pml and p53 to ameliorate loss of werner protein-mediated oncogenic senescence. Cell Death Differ. 2013, 20, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Zagurovskaya, M.; Shareef, M.M.; Das, A.; Reeves, A.; Gupta, S.; Sudol, M.; Bedford, M.T.; Prichard, J.; Mohiuddin, M.; Ahmed, M.M. Egr-1 forms a complex with yap-1 and upregulates bax expression in irradiated prostate carcinoma cells. Oncogene 2009, 28, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Yonehara, S. Identification of mechanism that couples multisite phosphorylation of yes-associated protein (yap) with transcriptional coactivation and regulation of apoptosis. J. Biol. Chem. 2012, 287, 9568–9578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by lats and ck1 regulates yap stability through scf(beta-trcp). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; and Cinar, B. Yap1 and ar interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-catenin-driven cancers require a yap1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers yap and taz. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Landin Malt, A.; Cagliero, J.; Legent, K.; Silber, J.; Zider, A.; Flagiello, D. Alteration of tead1 expression levels confers apoptotic resistance through the transcriptional up-regulation of livin. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Salah, Z.; Itzhaki, E.; Aqeilan, R.I. The ubiquitin e3 ligase itch enhances breast tumor progression by inhibiting the hippo tumor suppressor pathway. Oncotarget 2014, 5, 10886–10900. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I.; Donati, V.; Palamarchuk, A.; Trapasso, F.; Kaou, M.; Pekarsky, Y.; Sudol, M.; Croce, C.M. Ww domain-containing proteins, wwox and yap, compete for interaction with erbb-4 and modulate its transcriptional function. Cancer Res. 2005, 65, 6764–6772. [Google Scholar] [CrossRef] [PubMed]
- Salah, Z.; Cohen, S.; Itzhaki, E.; Aqeilan, R.I. Nedd4 e3 ligase inhibits the activity of the hippo pathway by targeting lats1 for degradation. Cell Cycle 2013, 12, 3817–3823. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Aqeilan, R.I.; Neale, M.; Candi, E.; Salomoni, P.; Knight, R.A.; Croce, C.M.; Melino, G. The e3 ubiquitin ligase itch controls the protein stability of p63. Proc. Natl. Acad. Sci. USA 2006, 103, 12753–12758. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.; Kelly, C.; Rauch, J.; Kida, K.; Garcia-Munoz, A.; Monsefi, N.; Turriziani, B.; Doherty, C.; Mehta, J.P.; Matallanas, D.; et al. Hgf induces epithelial-to-mesenchymal transition by modulating the mammalian hippo/mst2 and isg15 pathways. J. Proteome Res. 2014, 13, 2874–2886. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Kreda, S.M.; Boucher, R.C.; Sudol, M.; Stutts, M.J.; Milgram, S.L. Yes-associated protein 65 localizes p62(c-yes) to the apical compartment of airway epithelia by association with ebp50. J. Cell Biol. 1999, 147, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by yap. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Bui, D.A.; Lee, W.; White, A.E.; Harper, J.W.; Schackmann, R.C.; Overholtzer, M.; Selfors, L.M.; Brugge, J.S. Cytokinesis involves a nontranscriptional function of the hippo pathway effector yap. Sci. Signal. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M. Yap1 oncogene and its eight isoforms. Oncogene 2013. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bexiga, M.; Castillo, F.; Cobos, E.S.; Oka, T.; Sudol, M.; Luque, I. Ww domains of the yes-kinase-associated-protein (yap) transcriptional regulator behave as independent units with different binding preferences for ppxy motif-containing ligands. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, N.; Nagai, M.; Miyazaki, K.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; Okazawa, H.; Abe, K. Progressive decrease in the level of yapdeltacs, prosurvival isoforms of yap, in the spinal cord of transgenic mouse carrying a mutant sod1 gene. J. Neurosci. Res. 2009, 87, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, M.; Qi, M.L.; Yoshimura, N.; Miyashita, T.; Tagawa, K.; Wada, Y.; Enokido, Y.; Marubuchi, S.; Harjes, P.; Arai, N.; et al. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of yap isoforms and p73. J. Cell Biol. 2006, 172, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; del Sal, G.; Blandino, G. Yap enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, K.; Al-Moujahed, A.; Marmalidou, A.; Meyer Zu Horste, M.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. The clinically used photosensitizer verteporfin (vp) inhibits yap-tead and human retinoblastoma cell growth in vitro without light activation. Exp. Eye. Res. 2014, 124, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the tead-yap complex suppresses the oncogenic activity of yap. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Crespo, P. New druggable targets in the ras pathway? Curr. Opin. Mol. Ther. 2010, 12, 674–683. [Google Scholar] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Kempf, C.R.; Chappell, W.H.; Abrams, S.L.; Stivala, F.; Malaponte, G.; Nicoletti, F.; Libra, M.; Basecke, J.; et al. Therapeutic resistance resulting from mutations in raf/mek/erk and pi3k/pten/akt/mtor signaling pathways. J. Cell Physiol. 2011, 226, 2762–2781. [Google Scholar] [CrossRef] [PubMed]
- Shtivelman, E.; Davies, M.Q.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.J.; Pinnamaneni, R. Targeted therapies in melanoma. Surg. Oncol. Clin. N. Am. 2015, 24, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [PubMed]
- Zhang, X.; Guo, C.; Wu, X.; Li, A.X.; Liu, L.; Tsark, W.; Dammann, R.; Shen, H.; Vonderfecht, S.L.; Pfeifer, G.P. Analysis of liver tumor-prone mouse models of the hippo kinase scaffold proteins rassf1a and sav1. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in hbv- and hcv-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Nagasaka, T.; Nishimura, T.; Ikai, I.; Boland, C.R.; Goel, A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology 2008, 47, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ma, Z.; Wang, D.; Zhao, W.; Chen, L.; Wang, G. Microrna-602 regulating tumor suppressive gene rassf1a is overexpressed in hepatitis b virus-infected liver and hepatocellular carcinoma. Cancer Biol. Ther. 2010, 9, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Zekri, A.R.; Bahnasy, A.A.; Shoeab, F.E.; Mohamed, W.S.; El-Dahshan, D.H.; Ali, F.T.; Sabry, G.M.; Dasgupta, N.; Daoud, S.S. Methylation of multiple genes in hepatitis c virus associated hepatocellular carcinoma. J. Adv. Res. 2014, 5, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Petrucelli, L. “What kills neurons in neurodegenerative diseases?”, a review series in an open access journal. Mol. Neurodegener 2009. [Google Scholar] [CrossRef] [PubMed]
- Emoto, K. The growing role of the hippo—NDR kinase signalling in neuronal development and disease. J. Biochem. 2011, 150, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Steel, A.J.; de la Monte, S.M.; Sutherland, G.T. The rise and fall of insulin signaling in alzheimer’s disease. Metab. Brain Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of yap/taz activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Odashima, M.; Usui, S.; Takagi, H.; Hong, C.; Liu, J.; Yokota, M.; Sadoshima, J. Inhibition of endogenous mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ. Res. 2007, 100, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Leach, J.; Wang, J.; Martin, J.F. Hippo/yap signaling in cardiac development and regeneration. Curr. Treat. Options Cardiovasc. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.W.; Meng, Z.; Guan, K.L. The hippo pathway in intestinal regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Gershoni, A.; Rotkopf, R.; Biton, I.E.; Porat, Z.; Koh, A.P.; Sun, X.; Lee, Y.; Fiel, M.I.; Hoshida, Y.; et al. The lats2 tumor suppressor inhibits srebp and suppresses hepatic cholesterol accumulation. Genes Dev. 2016, 30, 786–797. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fallahi, E.; O’Driscoll, N.A.; Matallanas, D. The MST/Hippo Pathway and Cell Death: A Non-Canonical Affair. Genes 2016, 7, 28. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7060028
Fallahi E, O’Driscoll NA, Matallanas D. The MST/Hippo Pathway and Cell Death: A Non-Canonical Affair. Genes. 2016; 7(6):28. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7060028
Chicago/Turabian StyleFallahi, Emma, Niamh A. O’Driscoll, and David Matallanas. 2016. "The MST/Hippo Pathway and Cell Death: A Non-Canonical Affair" Genes 7, no. 6: 28. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7060028