
Collision of Trapped Topoisomerase 2 with Transcription and Replication: Generation and Repair of DNA Double-Strand Breaks with 5′ Adducts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. How Trapped Top2ccs Are Converted to DSBs: Collision with Transcription
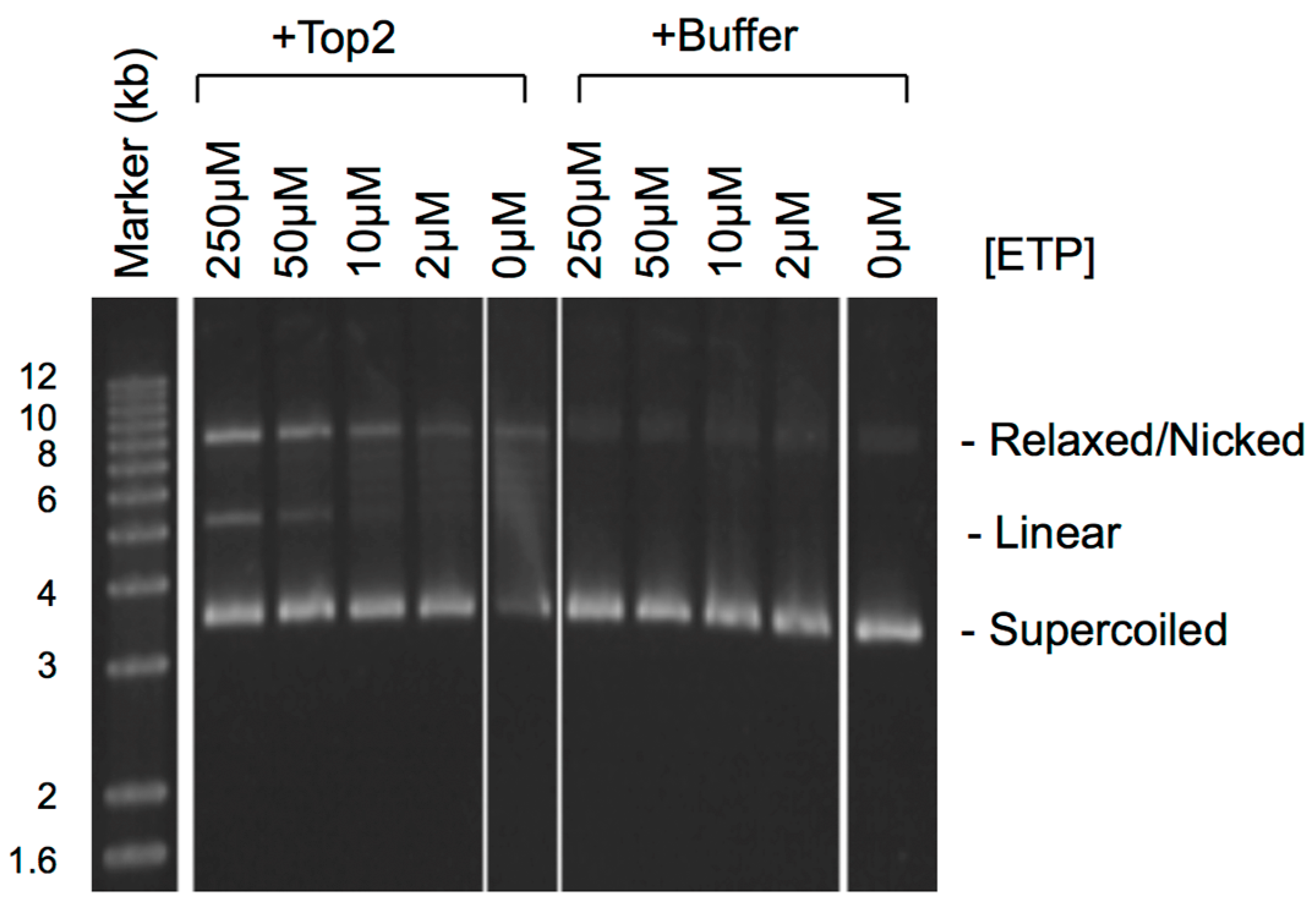
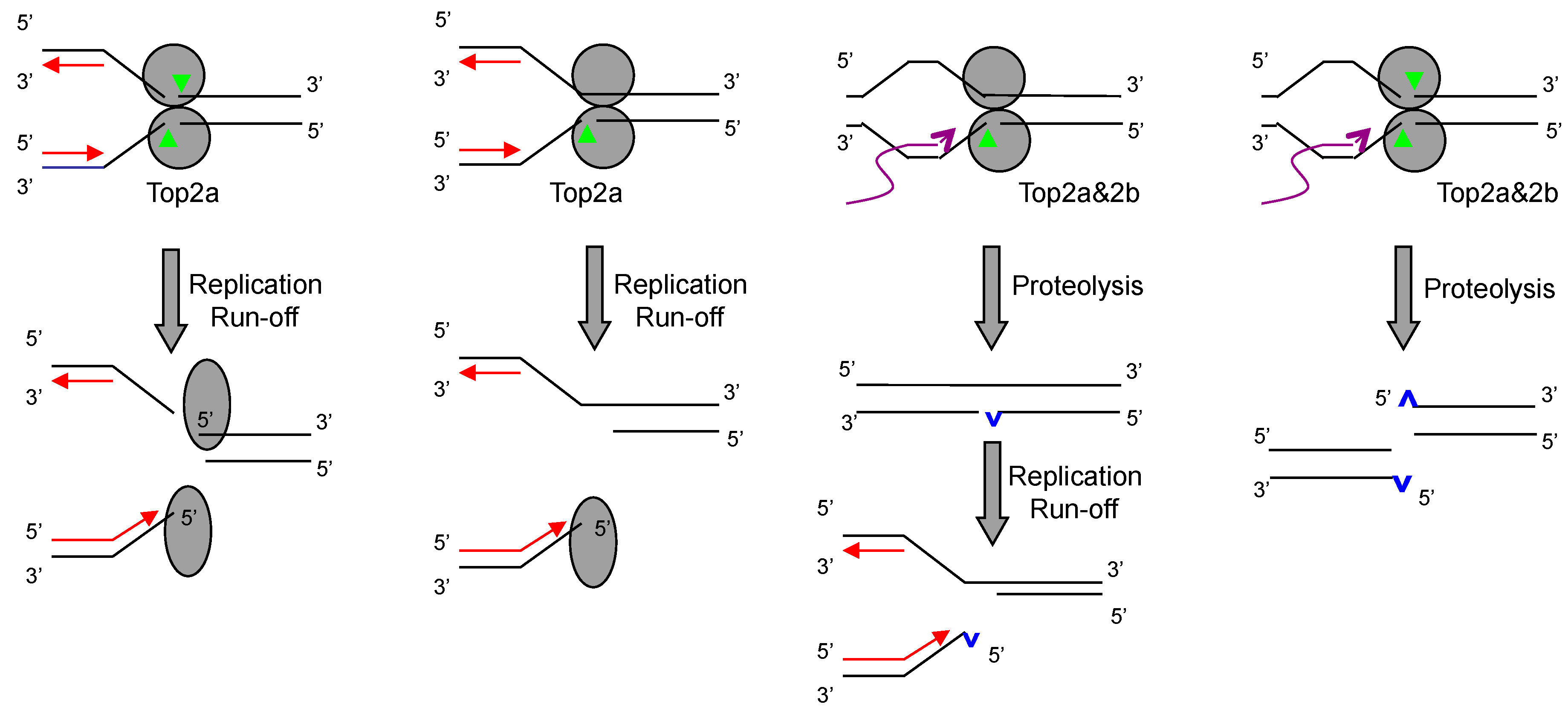
2. How Trapped Top2ccs Are Converted to DSBs: Collision with Replication
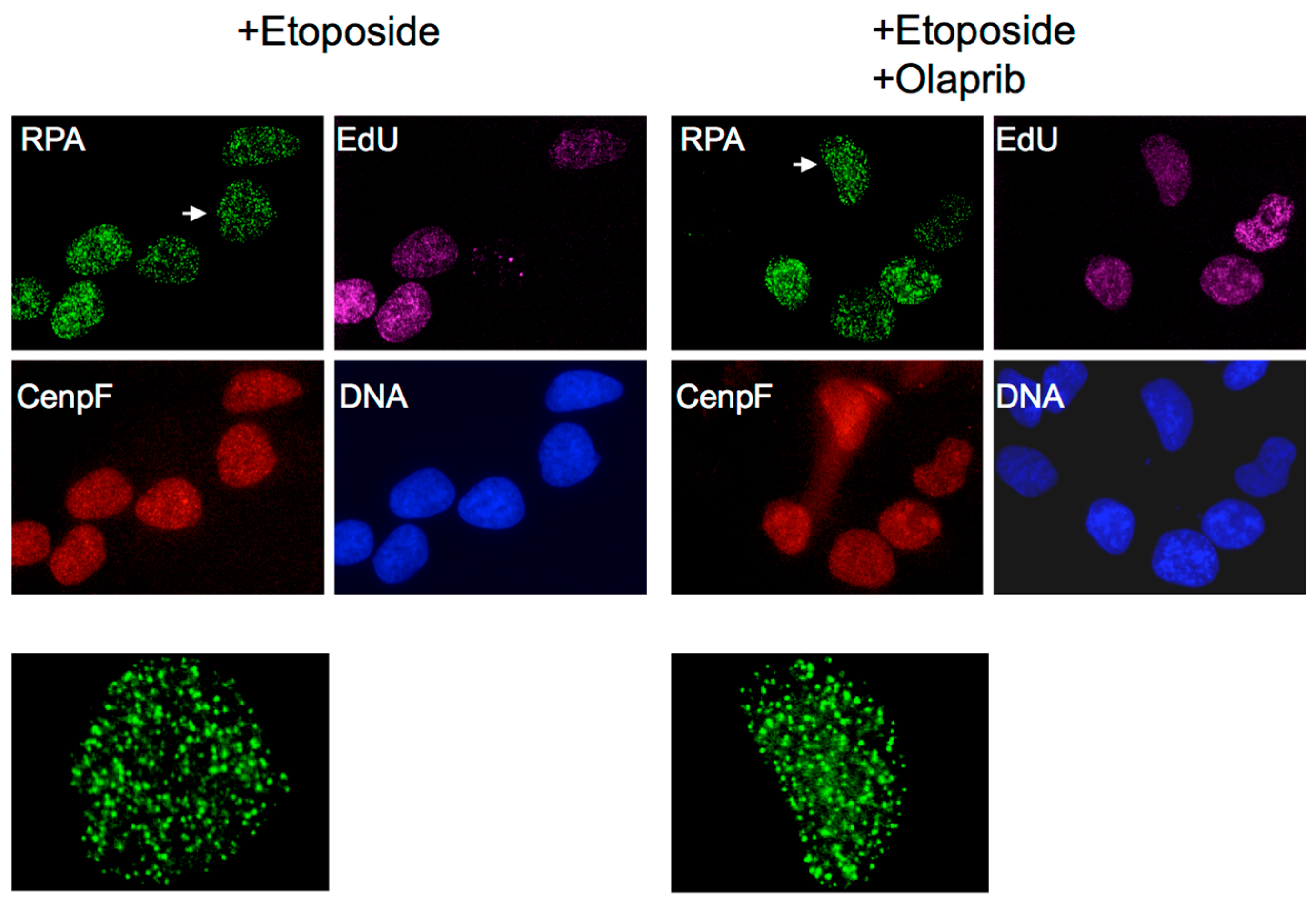
3. Do Top2ccs-Derived DSBs Carry 5′ Adducts?
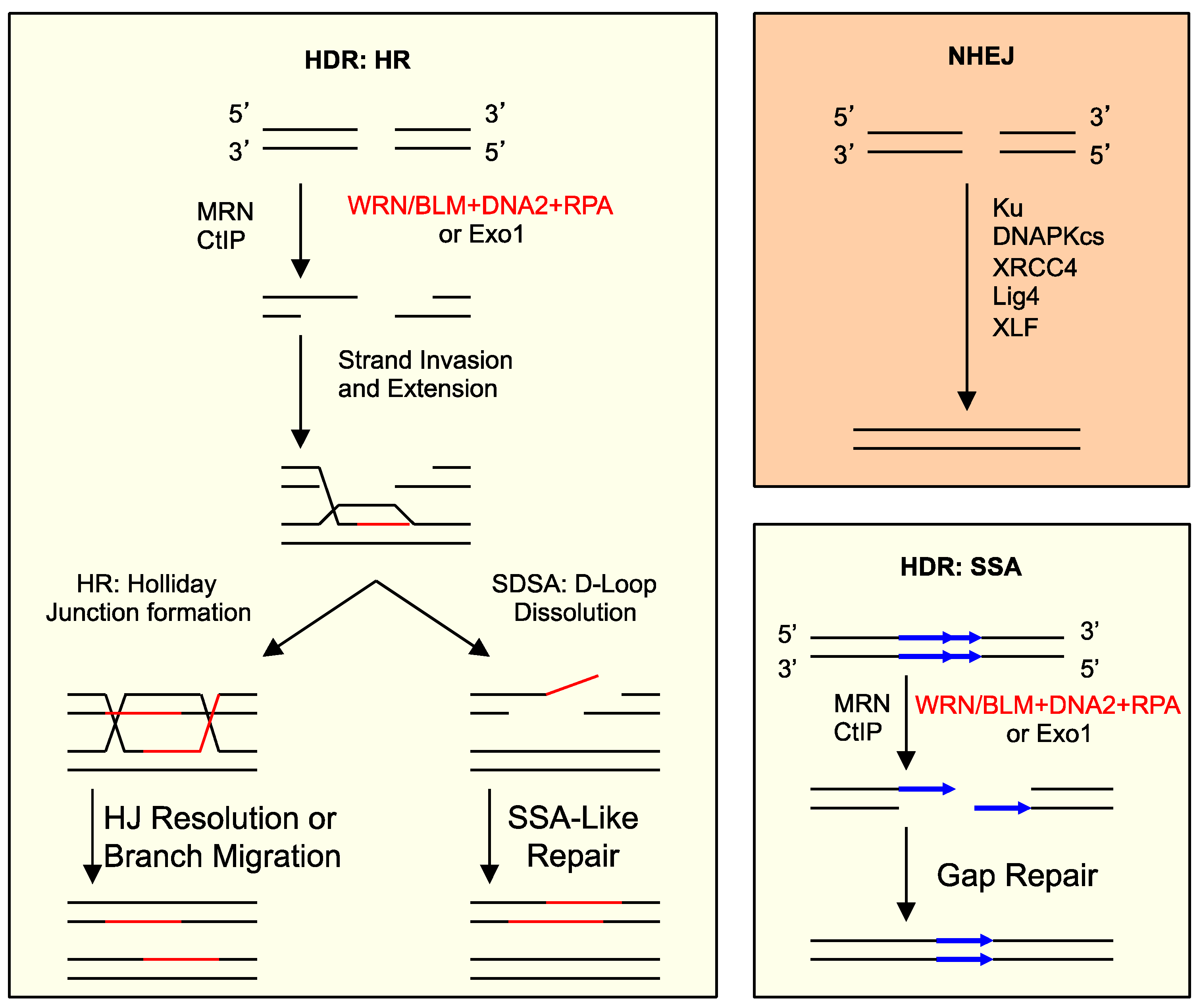
4. What Pathways Repair DSBs with 5′ Adducts?
5. Implications for Cancer Therapy
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, J.C. Untangling The Double Helix; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Deweese, J.E.; Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 2009, 37, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Coldwasser, F. Topoisomerase ii inhibitors: The epipodophyllotoxins. In Cancer Chemotherapy and Biotherapy; Chabner, B.A., Longo, D.L., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2011; pp. 392–410. [Google Scholar]
- Liu, L.F. Degradation of topoisomerase cleavable complexes. In DNA Topoisomerases in Cancer Therapy: Present and Future; Andoh, T., Ed.; Springer: New York, NY, USA, 2003; pp. 79–88. [Google Scholar]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Yang, L.; Rowe, T.C.; Halliqan, B.D.; Tewey, K.M.; Liu, L.F. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984, 259, 13560–13566. [Google Scholar] [PubMed]
- Bromberg, K.D.; Burgin, A.B.; Osheroff, N. A two-drug model for etoposide action against human topoisomerase IIalpha. J. Biol. Chem. 2003, 278, 7406–7412. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; LIU, Y.X.; Harbury, P.; Jannatipour, M.; Wasserman, R.; Wang, J.C. Amsacrine and etoposide hypersensitivity of yeast cells overexpressing DNA topoisomerase II. Cancer Res. 1992, 52, 4467–4472. [Google Scholar] [PubMed]
- Doroshow, J.H. Topoisomerase II inhibitors: Anthracyclines. In Cancer Chemotherapy and Biotherapy; Chabner, B.A., Longo, D.L., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2011; pp. 356–391. [Google Scholar]
- Tennyson, R.B.; Lindsley, J.E. Type II DNA topoisomerase from Saccharomyces cerevisiae is a stable dimer. Biochemistry 1997, 36, 6107–6114. [Google Scholar] [CrossRef] [PubMed]
- Tsai-Pflugfelder, M.; Liu, L.F.; Liu, A.A.; Tewey, K.M.; Whang-Peng, J.; Knutsen, T.; Kubener, K.; Crece, C.M.; Wang, J.C. Cloning and sequencing of cDNA encoding human DNA topoisomerase II and localization of the gene to chromosome region 17q21–22. Proc. Natl. Acad. Sci. USA 1988, 85, 7177–7181. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.R.; Ayton, P.; Jones, T.; Davies, S.L.; Simmons, D.L.; Harris, A.L.; Sheer, D.; Hickson, I.D. Isolation of cDNA clones encoding the beta isozyme of human DNA topoisomerase II and localisation of the gene to chromosome 3p24. Nucleic Acids Res. 1992, 20, 5587–5592. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.B.; Dorman, T.E.; Falls, K.M.; Chung, T.D.; Mirabelli, C.K.; Crooke, S.T.; Mao, J. Topoisomerase II alpha and topoisomerase II beta genes: Characterization and mapping to human chromosomes 17 and 3, respectively. Cancer Res. 1992, 52, 231–234. [Google Scholar] [PubMed]
- Austin, C.A.; Sng, J.H.; Patel, S.; Fisher, L.M. Novel HeLa topoisomerase II is the II beta isoform: Complete coding sequence and homology with other type II topoisomerases. Biochim. Biophys. Acta 1993, 1172, 283–291. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell. Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Heck, M.M.; Earnshaw, W.C. Topoisomerase II: A specific marker for cell proliferation. J. Cell Biol. 1986, 103, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Wu, H.Y.; Liu, L.F. Proliferation-dependent regulation of DNA topoisomerase II in cultured human cells. Cancer Res. 1988, 48, 3230–3235. [Google Scholar] [PubMed]
- Goswami, P.C.; Roti Roti, J.L.; Hunt, C.R. The cell cycle-coupled expression of topoisomerase IIalpha during S phase is regulated by mRNA stability and is disrupted by heat shock or ionizing radiation. Mol. Cell Biol. 1996, 16, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Woessner, R.D.; Mattern, M.R.; Mirabelli, C.K.; Johnson, R.K.; Drake, F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2, 209–214. [Google Scholar] [PubMed]
- Grue, P.; Grasser, A.; Sehested, M.; Jensen, P.B.; Uhse, A.; Straub, T.; Ness, W.; Boeqe, F. Essential mitotic functions of DNA topoisomerase IIalpha are not adopted by topoisomerase IIbeta in human H69 cells. J. Biol. Chem. 1998, 273, 33660–33666. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIbeta and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Lunyak, W.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Cornarotti, M.; Tinelli, S.; Willmore, E.; Zunino, F.; Fisher, L.M.; Austin, C.A.; Capranico, G. Drug sensitivity and sequence specificity of human recombinant DNA topoisomerases IIalpha (p170) and IIbeta (p180). Mol. Pharmacol. 1996, 50, 1463–1471. [Google Scholar] [PubMed]
- Willmore, E.; Frank, A.J.; Padqet, K.; Tilby, M.J.; Austin, C.A. Etoposide targets topoisomerase IIalpha and IIbeta in leukemic cells: Isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol. Pharmacol. 1998, 54, 78–85. [Google Scholar] [PubMed]
- Mao, Y.; Desai, S.D.; Ting, C.Y.; Hwang, J.; Liu, L.F. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 2001, 276, 40652–40658. [Google Scholar] [CrossRef] [PubMed]
- Sunter, N.J.; Cowell, I.G.; Willmore, E.; Watters, G.P.; Austin, C.A. Role of Topoisomerase IIbeta in DNA Damage Response following IR and Etoposide. J. Nucleic Acids 2010. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Bramlev, R.L.; Cowell, I.G.; Jackson, G.H.; Austin, C.A. Proteasomal inhibition potentiates drugs targeting DNA topoisomerase II. Biochem. Pharmacol. 2016, 103, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Mao, Y.; Desai, S.D.; Zhou, N.; Ting, C.Y.; Hwang, J.; Liu, L.Y. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Lyu, Y.L.; Lin, C.P.; Zhou, N.; Azarova, A.M.; Wood, L.M.; Liu, L.F. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J. Biol. Chem. 2006, 281, 35997–36003. [Google Scholar] [CrossRef] [PubMed]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.-P.; Tsai, Y.-C.; Lau, J.Y.-N.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.L.; Kerriqan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Ho, C.-W.; Lin, R.-K.; Lyu, Y.L.; Liu, L.F. Activation of a novel ubiquitin-independent proteasome pathway when RNA polymerase II encounters a protein roadblock. Mol. Cell Biol. 2013, 33, 4008–4016. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J.; Doles, J.; Zender, L.; Xue, W.; Ma, B.; Mccombie, W.R.; Hannon, G.J.; Lowe, S.W.; Hemann, M.T. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 9053–9058. [Google Scholar] [CrossRef] [PubMed]
- De Campos-Nebel, M.; Larripa, I.; Gonzalez-Cid, M. Topoisomerase II-mediated DNA damage is differently repaired during the cell cycle by non-homologous end joining and homologous recombination. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.; Covey, J.M.; Kerriqan, D.; Pommier, Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989, 49, 6365–6368. [Google Scholar] [PubMed]
- D’Arpa, P.; Beardmore, C.; Liu, L.F. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990, 50, 6919–6924. [Google Scholar] [PubMed]
- Fan, J.R.; Peng, A.L.; Chen, H.C.; Lo, S.C.; Huang, T.H.; Li, T.K. Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair (Amst) 2008, 7, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.J.; Fangman, W.L. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 1987, 51, 463–471. [Google Scholar] [CrossRef]
- Olive, P.L.; Banath, J.P. Detection of DNA double-strand breaks through the cell cycle after exposure to X-rays, bleomycin, etoposide and 125IdUrd. Int. J. Radiat. Biol. 1993, 64, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Rossi, R.; Ferrari, G.; Scovassi, A.I.; Prosperi, E.; Biamonti, G. Etoposide induces the dispersal of DNA ligase I from replication factories. Mol. Biol. Cell. 2001, 12, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Robison, J.G.; Dixon, K.; Bissler, J.J. Cell cycle-and proteasome-dependent formation of etoposide-induced replication protein A (RPA) or Mre11/Rad50/Nbs1 (MRN) complex repair foci. Cell Cycle 2007, 6, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, M.; Barr, P.; Ricci, B.; Yan, H. Replication-dependent and transcription-dependent mechanisms of DNA double-strand break induction by the topoisomerase 2-targeting drug Etoposide. PLoS ONE 2013, 8, e79202. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, M.; Liao, S.; Beeharrry, N.; Yan, H. DNA double-strand breaks with 5′ adducts are efficiently channeled to the DNA2-mediated resection pathway. Nucleic Acids Res. 2016, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Berti, M.; Levikova, M.; Pinto, C.; Gomathinayagam, S.; Vujanovic, M.; Zellweger, R.; Moore, H.; Lee, E.H.; Hendrickson, E.A.; et al. DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 2015, 208, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell. Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Long, B.H.; Musial, S.T.; Brattain, M.G. Single- and double-strand DNA breakage and repair in human lung adenocarcinoma cells exposed to etoposide and teniposide. Cancer Res. 1985, 45, 3106–3112. [Google Scholar] [PubMed]
- Muslimovic, A.; Nystrom, S.; Gao, Y.; Hammarsten, O. Numerical analysis of etoposide induced DNA breaks. PLoS ONE 2009, 4, e5859. [Google Scholar] [CrossRef]
- Strumberg, D.; Pilon, A.A.; Smith, M.; Hickey, R.; Malkas, L.; Pommier, Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell Biol. 2000, 20, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Tounekti, O.; Kenani, A.; Foray, N.; Orlowski, S.; Mir, L.M. The ratio of single- to double-strand DNA breaks and their absolute values determine cell death pathway. Br. J. Cancer 2001, 84, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef]
- Neale, M.J.; Pan, J.; Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 2005, 436, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Furbee, C.S.; Muller, M.T. ICE bioassay. Isolatingin vivo complexes of enzyme to DNA. Methods Mol. Biol. 2001, 95, 137–147. [Google Scholar] [PubMed]
- Cortes Ledesma, F.; Ei Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Schellenberg, M.J.; Huang, S.Y.; Abdelmalak, M.; Marchand, C.; Nitiss, K.C.; Nitiss, J.L.; Williams, R.S.; Pommier, Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 2014, 289, 17960–17969. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Cortes-Ledesma, F.; Ei khamisy, S.F.; Caldecott, K.W. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2010, 286, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Alvarez-Quilon, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef] [PubMed]
- Maede, Y.; Shimizu, H.; Fukushima, T.; Kogame, T.; Nakamura, T.; Miki, T.; Takeda, S.; Pommier, Y. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2014, 13, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Hartsuiker, E.; Neale, M.J.; Carr, A.M. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 2009, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Padget, K.; Curitis, H.; Cowell, I.G.; Moiani, D.; Sondka, Z.; Morris, N.J.; Jackson, G.H.; Cockell, S.J.; Tainer, J.A.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell. Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.; Graham, C.W.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenburger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Toczylowski, T.; Yan, H. Mechanistic analysis of a DNA end processing pathway mediated by the Xenopus Werner syndrome protein. J. Biol. Chem. 2006, 281, 33198–33205. [Google Scholar] [CrossRef] [PubMed]
- Gravel, S.; Chapman, J.R.; Maqill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Toczylowski, T.; Yan, H. Identification of the Xenopus DNA2 protein as a major nuclease for the 5′- > 3′ strand-specific processing of DNA ends. Nucleic Acids Res. 2008, 36, 6091–6100. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases DNA2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Toceylowski, T.; Mccane, J.; Chen, C.; Liao, S. Replication protein A promotes 5′- > 3′ end processing during homology-dependent DNA double-strand break repair. J. Cell. Biol. 2011, 192, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Inoue, S.; Weaver, D.T. Differential etoposide sensitivity of cells deficient in the Ku and DNA-PKcs components of the DNA-dependent protein kinase. Carcinogenesis 1998, 19, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Suzuki, H.; Liizumi, S.; Koyama, H. Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: Implications for the repair of topoisomerase II-mediated DNA damage. J. Biol. Chem. 2003, 278, 35897–35902. [Google Scholar] [CrossRef] [PubMed]
- Martensson, S.; Nygren, J.; Osheroff, N.; Hammarsten, O. Activation of the DNA-dependent protein kinase by drug-induced and radiation-induced DNA strand breaks. Radiat. Res. 2003, 160, 291–301. [Google Scholar] [CrossRef]
- Adachi, N.; Liizumi, S.; So, S.; Koyama, H. Genetic evidence for involvement of two distinct nonhomologous end-joining pathways in repair of topoisomerase II-mediated DNA damage. Biochem. Biophys. Res. Commun. 2004, 318, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, A.; Koyama, H.; Takayama, S.; Miki, K.; Ayusawa, D.; Fujii, M.; Liizumi, S.; Adachi, N. The requirement of Artemis in double-strand break repair depends on the type of DNA damage. DNA Cell Biol. 2008, 27, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L.; Soans, E.; Berk, J.; Seth, A.; Mishina, M.; Nitiss, K.C. Repair of Topoisomerase II-Mediated DNA. In DNA Topoisomerases and Cancer; Pommier, Y., Ed.; Springer-Verlag: New York, NY, USA, 2011. [Google Scholar]
- Xiao, H.; Goodrich, D.W. The retinoblastoma tumor suppressor protein is required for efficient processing and repair of trapped topoisomerase II-DNA-cleavable complexes. Oncogene 2005, 24, 8105–8113. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Tammaro, M.; Yan, H. The structure of ends determines the pathway choice and Mre11 nuclease dependency of DNA double-strand break repair. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Felix, C.A. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta 1998, 1400, 233–255. [Google Scholar] [CrossRef]
- Smith, M.A.; Rubinstein, L.; Anderson, J.R.; Arthur, D.; Catalano, P.J.; Freidlin, B.; Heyn, R.; Khayat, A.; Krailo, M.; Land, V.J.; et al. Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins. J. Clin. Oncol. 1999, 17, 569–577. [Google Scholar] [PubMed]
- Schonn, I.; Hennesen, J.; Dartsch, D.C. Cellular responses to etoposide: Cell death despite cell cycle arrest and repair of DNA damage. Apoptosis 2010, 15, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruqe, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIbeta-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. USA 2012, 109, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Raoof, A.; Depledge, P.; Niall, M.; Hamilton, N.M.; Hamilton, N.S.; Hitchin, J.R.; Hopkins, G.V.; Jordan, A.M.; Maguire, L.A.; Mcgonagie, A.E.; et al. Toxoflavins and deazaflavins as the first reported selective small molecule inhibitors of tyrosyl-DNA phosphodiesterase II. J. Med. Chem. 2013, 56, 6352–6370. [Google Scholar] [CrossRef] [PubMed]
- Kankanala, J.; Marchand, C.; Abdelmalak, M.; Aihara, H.; Pommier, Y.; Wang, Z. Isoquinoline-1,3-diones as Selective Inhibitors of Tyrosyl DNA Phosphodiesterase II (TDP2). J. Med. Chem. 2016, 59, 2734–2746. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, ML.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van rossum-fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, M.; Li, Z.; Li, H.; Polaczek, P.; Dai, H.; Wu, Q.; Liu, C.; Kranjia, K.K.; Popuri, V.; et al. A Selective Small Molecule DNA2 Inhibitor for Sensitization of Human Cancer Cells to Chemotherapy. EBioMedicine 2016, 6, 73–86. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Tammaro, M.; Liao, S. Collision of Trapped Topoisomerase 2 with Transcription and Replication: Generation and Repair of DNA Double-Strand Breaks with 5′ Adducts. Genes 2016, 7, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7070032
Yan H, Tammaro M, Liao S. Collision of Trapped Topoisomerase 2 with Transcription and Replication: Generation and Repair of DNA Double-Strand Breaks with 5′ Adducts. Genes. 2016; 7(7):32. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7070032
Chicago/Turabian StyleYan, Hong, Margaret Tammaro, and Shuren Liao. 2016. "Collision of Trapped Topoisomerase 2 with Transcription and Replication: Generation and Repair of DNA Double-Strand Breaks with 5′ Adducts" Genes 7, no. 7: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7070032