
Telomerase: The Devil Inside


1
Department of Urology, Ulm University, 89081 Ulm, Germany
2
Department of Internal Medicine I, Ulm University, 89081 Ulm, Germany
*
Author to whom correspondence should be addressed.
Genes 2016, 7(8), 43; https://0-doi-org.brum.beds.ac.uk/10.3390/genes7080043
Submission received: 25 May 2016
/
Revised: 23 July 2016
/
Accepted: 25 July 2016
/
Published: 29 July 2016
(This article belongs to the Special Issue Telomerase Activity in Human Cells)
{kind=link}
{kind=link}
Abstract
:High telomerase activity is detected in nearly all human cancers but most human cells are devoid of telomerase activity. There is well-documented evidence that reactivation of telomerase occurs during cellular transformation. In humans, tumors can rely in reactivation of telomerase or originate in a telomerase positive stem/progenitor cell, or rely in alternative lengthening of telomeres, a telomerase-independent telomere-length maintenance mechanism. In this review, we will focus on the telomerase positive tumors. In this context, the recent findings that telomerase reverse transcriptase (TERT) promoter mutations represent the most common non-coding mutations in human cancer have flared up the long-standing discussion whether cancer originates from telomerase positive stem cells or telomerase reactivation is a final step in cellular transformation. Here, we will discuss the pros and cons of both concepts in the context of telomere length-dependent and telomere length-independent functions of telomerase. Together, these observations may provoke a re-evaluation of telomere and telomerase based therapies, both in telomerase inhibition for cancer therapy and telomerase activation for tissue regeneration and anti-ageing strategies.
1. Telomerase Activity Is Required for Continuous Proliferation Capacity of Human Cells and Is Regulated through TERT Gene Expression
The activity of telomerase is necessary to overcome the end replication problem [1]. This enzyme extends the telomeric DNA at the very tips of linear chromosomes by the addition of a short GT-rich repetitive DNA sequence thus preventing loss of genetic material in proliferating cells [1,2,3]. Emerging evidence indicates additional roles of telomerase beyond this prime function (see below and [4] for a review on this aspect). In humans, telomerase activity is progressively downregulated during embryogenesis and only adult stem cell compartments still maintain low levels of telomerase activity [5,6,7,8]. Induction of telomerase was also observed in a subset of highly proliferating cells—such as proliferating B- and T-cells and the regenerating hepatocytes [9,10,11,12].
It is now well documented that telomerase activation is primarily regulated at the transcriptional level of its catalytic subunit, telomerase reverse transcriptase (TERT) [13,14,15]. In recent years, a number of additional factors, including dyskerin, TCAB1, and NOP10 have been identified to be constantly or transiently associated with the telomerase complex and have important functions in telomerase recruitment to telomeres or subcellular localization of the telomerase complex [16,17,18]. Although telomerase activity could be detected in the majority of human cancers [19], it is worth mentioning that about 10%–15% of human tumors were devoid of telomerase activity. Tumors that lack telomerase activity maintain their telomere length via a recombination-based mechanism (alternative lengthening of telomeres, ALT) [20]. Thus, telomerase or another mechanism for telomere maintenance is required for continuous tumor cell proliferation. Importantly, the prominent occurrence of telomerase in human cancers motivated the development of telomerase inhibitors to suppress tumor growth [21,22,23,24,25]. As a flaw, however, anti-telomerase therapy may provoke ALT mechanism in cancer cells [26].
2. Regulatory Mechanisms Involved in TERT Gene Regulation in Normal and Tumor Cells
In the light of these findings and considering the close correlation of TERT gene expression and telomerase activity with cancer, investigations focused on deciphering the underlying mechanisms that govern TERT promoter activity. A number of factors have been identified to be involved in the regulation of TERT expression. These factors include c-Myc and its antagonist Mad1, the receptors for the hormones estrogen and progesterone, AP-1 (Activator Protein 1), NF-κB (nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells), Rb/E2F factors (Retinoblastoma tumor suppressor protein/ E2 factor family of transcription factors), CEBP-α (CCAAT/Enhancer Binding Protein Alpha) and CEBP-β (CCAAT/Enhancer Binding Protein Beta) (see [27,28] for a comprehensive review on TERT promoter regulation). In the stem cell context, Wnt/β-catenin pathway and Kruppel-like factor 4 (KLF4) were described to regulate TERT gene expression and telomerase activity [29,30].
Although these factors were shown to regulate human telomerase reverse transcriptase (hTERT) promoter and telomerase activity in experimental systems, it remains to be shown whether these factors and mechanisms are also responsible for high telomerase activity in human cancer. Based on the current knowledge, it seems tempting to speculate that the same cis-regulatory elements can act as a site of positive and negative regulation of hTERT gene expression depending on the proliferation status, at least in some tissues; e.g., E-box binding by c-Myc activates the hTERT promoter whereas the binding of its counterpart Mad1 to the E-box sequences results in hTERT/telomerase downregulation [15,31,32]. Similarly, CEBP-α represses hTERT in normal breast cells, but CEBP-β activates it in breast cancer cells by binding to the same regulatory sites [33]; in resting hepatocytes, Rb factors repress hTERT expression whereas E2F2/E2F7 factors are involved in hTERT promoter activation in proliferating hepatocytes by binding to the same sequence [12]. Interestingly, ETS2 transcription factor was described both as a positive and negative regulator of wild-type hTERT promoter by regulating promoter activity through a native ETS binding site [32,34,35,36]. Recent data indicates that the ETS factor GA-binding protein alpha chain (GABPA) binds to the mutated hTERT promoter (generating a new potential ETS binding site), likely in combination with the juxtaposed native ETS binding site (Figure 1) leading to elevated hTERT expression and telomerase activation in cancer cells [37].
Beyond this assumption, several additional mechanisms were described for hTERT activation in human tumor cells:
- Alteration of regulatory factors: In line with the above regulatory factors, changes in the expression levels of the respective factors could account for the upregulation of hTERT gene in cancer cells. One example may be the increase of c-Myc due to gene amplification in breast cancer [38]. Another potential mechanism could be the loss of negative regulatory factors resulting in derepression of hTERT transcription, e.g., loss of the candidate tumor suppressor Mad-1 expression in many cancer types or loss of the Wilms’ tumor suppressor 1 (WT1) in clear cell renal cell carcinoma [15,39].
- Epigenetic regulation of promoter activity: Another form of hTERT upregulation is through demethylation of histones proximal to the promoter region, imitating the low density of trimethylated histones seen in embryonic stem cells [40]. This permits the recruitment of histone acetyltransferases (HATs) allowing the transcription of the gene [41]. However, current data indicate a complex epigenetic regulation of hTERT promoter activity: it has been demonstrated that methylation status is crucial for the regulation of hTERT gene expression [42,43] and that the chromatin remodeling factor CTCF (CCCTC-binding factor) can bind to the hTERT promoter depending on the methylation status and suppress its expression [44]. On the other hand, demethylation of specific regions on hTERT promoter caused downregulation of hTERT gene expression, telomerase activity, and telomere shortening, indicating that a certain level of methylation is required for hTERT promoter activation [45]. In the same line, aberrant hTERT promoter hypermethylation correlates with elevated hTERT gene expression in the majority of non-infant Sonic-Hedgehog subgroup medulloblastoma tumors [46] whereas hTERT promoter methylation was shown to correlate with reduced hTERT gene expression in B-cell lymphocytic leukemia and childhood acute lymphoblastic leukemia (ALL) [47,48]. Alterations in the epigenetic pattern of hTERT promoter, e.g., by point mutations, may also lead to a deregulation of promoter activity [49,50]. In this line, recurrent genomic rearrangements in the chromosomal region at 5p15.33 proximal of the hTERT gene induced massive chromatin remodeling and DNA methylation leading to the transcriptional upregulation of hTERT expression and telomerase activity in high-risk neuroblastomas [51]. Future studies will clarify the exact mechanisms and the potential tissue-specific factors involved in the methylation-dependent hTERT promoter regulation.
- Amplification and genomic rearrangements: Structural and numerical changes in the genome organization can result in TERT promoter activation. Amplification of the hTERT gene locus was proposed as a mechanism for increased hTERT mRNA levels and telomerase activity [52,53,54]. It should be noted however, that the elevated hTERT mRNA levels may not necessarily be attributable to increased hTERT transcription.
- Promoter mutations: Over the last few years, mutations in the hTERT gene promoter, first described in melanoma [55,56], were found in several tumor types, including bladder cancer (29%–90%), hepatocellular carcinoma (HCC) (30%–63%), melanoma (29%–73%), thyroid cancer (10%), and tumors from the central nervous system (43%–51%), representing one of the most frequent non-coding mutations in human cancer (see [57,58,59,60,61,62] for a comprehensive overview on hTERT promoter mutations). On the other hand, other tumor entities have a low frequency or no hTERT promoter mutations which are described up to the recent date, including testicular germ cell tumors, colorectal adenocarcinoma, pancreatic cancer, papillary thyroid cancers, and most types of leukemia [63,64,65] despite high telomerase activity in these tumors [66,67,68]. These observations indicate that other mechanisms, e.g., loss of negative regulatory factors or chromatin remodeling may be responsible for telomerase reactivation in the tumor types (e.g., loss of suppression: see above). Alternatively, tumors may originate from telomerase positive stem or progenitor cells (see below).
3. Telomerase Reactivation versus Telomerase Positive Stem Cell Hypothesis
It is a long-standing debate whether telomerase is reactivated during tumor progression or if tumors initiate from telomerase-positive stem cells [69,70,71,72,73,74,75,76,77,78]. The telomerase reactivation concept is based on two early observations: (i) Telomerase activity is detectable in human tumors and transformed cells but not in most primary human cells, e.g., normal human fibroblasts or embryonic kidney cells [19,79,80]; (ii) in the absence of telomerase, telomere shortening coincides with the induction of the ‘cellular senescence’ phenotype in cells with functional checkpoints [81,82]. Consequently, cells devoid of functional p53 and Rb proteins overcome the senescence barrier but telomeres continue to shorten until a ‘crisis’ checkpoint. Cells that survive the crisis checkpoint possess telomerase activity [80]. In this concept, telomere shortening in the absence of telomerase acts as a tumor suppressor mechanism but dysfunctional telomeres in the absence of functional checkpoints can initiate cancer formation due to increased genome instability leading to oncogene activation/loss of tumor protective factors [83]. Studies in a variety of human and mouse cancer models, including breast cancer, HCC, and prostate cancer provided evidence for the telomere hypothesis [77,84,85,86,87,88,89]. In this model, the activation of telomerase is an essential instrument to stabilize telomere length and may function as an active driver of tumor progression [77]. This telomere hypothesis implies that the reactivation of telomerase is a late step during cellular transformation supported by low/undetectable telomerase activity in normal tissues and premalignant lesions [19] (Figure 2, reactivation concept 1).
This idea was questioned, arguing that telomerase activity may be present in a few cells in premalignant lesions, difficult to detect by the conventional methods, e.g., telomere repeat amplification protocol (TRAP). Instead, tumors may arise from telomerase positive stem cells [71,74]. There is emerging evidence that stem cells accumulate mutations during ageing and can be the cancer-initiating cell type [90,91]. Since stem and progenitor cells possess telomerase activity, these cells do not require a re-activation of telomerase during tumorigenesis. There is evidence from a number of tumor types (such as most leukemia or testicular tumors) supporting this hypothesis (Figure 2, stem/progenitor cell concept). In fact, tumors can originate from different sources within the same tissue, depending on the genetic event and on the activated pathways initiating tumorigenesis [91].
The new findings that promoter mutations can be found at early steps during carcinogenesis (Reactivation concept 2) open up new perspectives on this debate. The most noticeable conclusion from these findings is that, irrespective of the reactivation mechanism, the data support the idea that at least a subset of tumors originate from an initially telomerase negative cell. Remarkably, hTERT promoter mutations are frequently detected in cancers with low self-renewal rates [55,56,57,58,59,60,61,62] whereas they are extremely rare in tumors originating from highly proliferating tissues [63,64,65]. We suggest that hTERT promoter mutations may account for both reactivation concepts 1 and 2. For example, late telomerase reactivation can result from hTERT promoter mutations in thyroid cancer and gliomas (reactivation concept 1) but hTERT promoter mutations in bladder and some skin tumors can result in reactivation of telomerase at an early stage of tumorigenesis (reactivation concept 2). However, it should be noted that mutations in the hTERT promoter region are not found in all telomerase positive bladder cancer or HCC samples and are missing in some tumor types altogether (e.g., pancreatic cancer) despite high levels of telomerase activity in these tumors indicating that other telomerase activating mechanisms must account for increased hTERT expression and telomerase activity.
One interesting aspect remains as to whether reactivation of telomerase is an early or late event during tumorigenesis (Figure 2, reactivation concept 1 versus reactivation concept 2). As discussed above, there is well-documented evidence from human cancers that telomerase reactivation is rather a late event during tumor progression (telomere hypothesis) supported by cell culture experiments [80] although difficult to prove formally in progressing tumors. The new findings show that hTERT promoter mutations occur already at early stages and across all grades of bladder cancer [93,94,95,96]. Similarly, hTERT promoter mutations were frequent in HCC, detectable early in the process of tumor development and were already present in dysplastic macronodules but not in cirrhotic liver [58], indicating that reactivation of telomerase represents a very early event in the multistep progress of carcinogenesis and might be a key driver. Telomerase reactivation occurring early during tumorigenesis would generate a ‘stem-like’ phenotype preventing telomere shortening and the senescence pathway. Interestingly, human embryonic stem cells that were genetically modified to carry the common TERT promoter mutations failed to suppress telomerase activity during differentiation and maintained telomere length, providing evidence for this idea [76].
Taken together, the activity/reactivation of telomerase in cancer supports its classical role on telomere length maintenance. Telomerase has also been implicated in non-canonical functions which may contribute to cancer cell survival in a telomere length-independent fashion.
4. Providing Cellular Survival Advantage Telomere Length-Independent Mechanisms
There is well-documented evidence for telomere length-independent roles of telomerase, both under physiological conditions and in cancer cells. These non-canonical functions include providing growth advantage by activating cellular growth factor pathways, improved ribosomal biogenesis or improved stem cell functionality by modulating gene expression [97,98,99,100,101,102,103,104,105], suppressing apoptosis and/or telomere replication stress [106,107,108] and promoting cell adhesion and migration [109]. It was shown that a catalytically inactive hTERT can promote tumor formation, supporting a telomere length-independent functional role of telomerase in tumorigenesis [110]. Contrary to these findings, by analyzing mouse TERT (mTERT) or mouse telomerase RNA (mTR) knockout mice, Greider and colleagues did not find evidence for telomere length-independent roles of telomerase: (i) Telomerase knockout mTERT-G1 and mTR-G1 mice with long telomeres did not show significant differences in transcriptional profiling or in DNA damage responses compared to wild-type mice [111]; and (ii) phenotypes in mTERT+/− and mTERT−/− mice were similar to the phenotypes observed in mTR+/− and mTR−/− mice and were linked to progressive telomere shortening [112]. Greider and colleagues also questioned the previous observations that indicated a transcriptional regulation of the Wnt/β-catenin pathway by the telomerase protein component TERT [103,112]. They argued that the ectopic TERT that was used in some of the above mentioned studies may act non-specifically [112]. It remains to be clarified in future studies whether there are species-specific differences in the function of telomerase components. Interestingly, an increasing number of hTERT splice variants have been identified in human cells, some of them lacking the catalytic activity region. Alternative splicing could induce changes in expression intensity, but splice variants can also act as dominant negative proteins to counteract wild-type telomerase function, both canonical and non-canonical [107]. One prominent isoform is the β-deletion variant lacking the exons 7 and 8, creating an activity-deficient protein that was detected both in normal and cancer cells [113]. It is therefore conceivable that the canonical and non-canonical functions are exerted by different splice variants. This idea opens up the opportunity that modulating the levels of the splice variants could be a tool to target telomerase activity in cancer cells specifically.
5. Final Conclusions
High-throughput sequencing technologies have provided a new insight into the mechanisms of telomerase activation in cancer cells. Whole genome sequencing analyses revealed highly prevalent hTERT promoter mutations in human cancer. The two key hot-spot mutations (positions C228T and C250T in hTERT promoter region, corresponding to positions −124 and −146 upstream of the ATG transcription start site, respectively) represent the most common non-coding mutations found in human tumors [59,114,115,116]. These mutations result in the activation of hTERT gene expression, much in line with the well-documented evidence that reactivation of telomerase occurs during cellular transformation. The reactivation concept is closely linked to the classical view that the downregulation of telomerase is a cancer protective mechanism by leading to telomere shortening and, eventually, to cellular senescence in checkpoint proficient cells, whereas the activation of telomerase is required to stabilize telomere length after telomere dysfunction-driven genetic instability initiates tumorigenesis [80]. The findings support the concept that some tumors can arise from telomerase positive cells—such as stem cells—in humans. TERT promoter mutations at early steps during tumorigenesis (e.g., bladder cancer) may therefore generate a ‘stem-like’ cell that can accumulate further mutations without activating senescence checkpoints.
Emerging evidence over the last decade supports the idea that telomere length-independent functions of telomerase are also important for its function, both in normal and tumor cells. Interestingly, current research also revealed that telomeres may sense cellular stress (such as genotoxic stress, oncogenic or aneuploidy-inducing mutations) that result from harmful mutations that lead to genome instability and induce senescence in cells with intact checkpoints [108]. Although the mechanistic details of the ‘sensing’ process are yet to be revealed, this new function of telomeres, thought to be a result of accumulating replication stress at the telomeres, seems to be independent of telomere length. In this context, telomerase relieves this cellular protective mechanism by mitigating telomere replication stress and this function of telomerase apparently is separate from its telomere elongation activity [75,78,117]. In light of the recent discoveries hinting at novel, telomere length-independent roles of telomeres and telomerase, attempts at modulating telomerase activity to improve organ function and longevity must be seriously reconsidered. In this line, interfering with telomerase activity and its extracurricular functions for cancer therapy seems to be an attractive strategy again but new concepts need to be taken into account.
Author Contributions
M.K., A.L. and C.G. wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Greider, C.W.; Henderson, E.; Lee, M.; Shampay, J.; Shippen-Lentz, D. Recognition and elongation of telomeres by telomerase. Genome 1989, 31, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhou, J.; Wang, M.; Cong, Y.S. Implications of telomere-independent activities of telomerase reverse transcriptase in human cancer. FEBS J. 2013, 280, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.D.; Baird, D.M. Telomere length maintenance in stem cell populations. Biochem. Biophys. Acta. 2009, 1792, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Weise, J.M.; Güneş, Ç. Differential regulation of human and mouse telomerase reverse transcriptase (TERT) promoter activity during testis development. Mol. Reprod. Dev. 2009, 76, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Broccoli, D.; Young, J.W.; de Lange, T. Telomerase activity in normal and malignant hematopoietic cells. Proc. Natl. Acad. Sci. USA 1995, 92, 9082–9086. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.T.; Lee, S.C.; Marin, E.; Ryan, D.H.; Insel, R.A. Telomerase is upregulated in human germinal center B cells in vivo and can be re-expressed in memory B cells activated in vitro. J. Immunol. 1997, 159, 1068–1071. [Google Scholar] [PubMed]
- Weng, N.P.; Levine, B.L.; June, C.H.; Hodes, R.J. Regulation of telomerase RNA template expression in human T lymphocyte development and activation. J. Exp. Med. 1996, 183, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Sirma, H.; Kumar, M.; Meena, J.K.; Witt, B.; Weise, J.M.; Lechel, A.; Ande, S.; Sakk, V.; Guguen-Guillouzo, C.; Zender, L.; et al. The promoter of human telomerase reverse transcriptase is activated during liver regeneration and hepatocyte proliferation. Gastroenterology 2011, 141, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, S.L.; Pruzan, R.; Ma, L.; Ouellette, M.; Tesmer, V.M.; Holt, S.E.; Bodnar, A.G.; Lichtsteiner, S.; Kim, N.W.; Trager, J.B.; et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 1997, 17, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Saito, M.; Nakamura, H.; Matsuura, A.; Ishikawa, F. TLP1: A gene encoding a protein component of mammalian telomerase is a novel member of WD repeats family. Cell 1997, 88, 875–884. [Google Scholar] [CrossRef]
- Güneş, Ç.; Lichtsteiner, S.; Vasserot, A.P.; Englert, C. Expression of the hTERT gene is regulated at the level of transcriptional initiation and repressed by Mad1. Cancer Res. 2000, 60, 2116–2121. [Google Scholar] [PubMed]
- Cohen, S.B.; Graham, M.E.; Lovrecz, G.O.; Bache, N.; Robinson, P.J.; Reddel, R.R. Protein composition of catalytically active human telomerase from immortal cells. Science 2007, 315, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Pogacić, V.; Dragon, F.; Filipowicz, W. Human H/ACA small nucleolar RNPs and telomerase share evolutionarily conserved proteins NHP2 and NOP10. Mol. Cell. Biol. 2000, 20, 9028–9040. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.C.; Piatyszek, M.A.; Wright, W.E.; Shay, J.W.; Corey, D.R. Inhibition of human telomerase activity by peptide nucleic acids. Nat. Biotechnol. 1996, 14, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B.; et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.S.; Pongracz, K.; Shay, J.W.; Gryaznov, S.M. Oligonucleotide N3′-->P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene 2002, 21, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Dikmen, Z.G.; Gellert, G.C.; Jackson, S.; Gryaznov, S.; Tressler, R.; Dogan, P.; Wright, W.E.; Shay, J.W. In vivo inhibition of lung cancer by GRN163L: A novel human telomerase inhibitor. Cancer Res. 2005, 65, 7866–7873. [Google Scholar] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.; Peek, G.W.; Tollefsbol, T.O. Regulation of the human catalytic subunit of telomerase (hTERT). Gene 2012, 498, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Hou, P.S.; Tseng, S.F.; Chien, C.L.; Wu, K.J.; Chen, H.F.; Ho, H.N.; Kyo, S.; Teng, S.C. Krüppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 2010, 28, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Björkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Phogat, S.K.; Sidorov, I.A.; Yang, J.; Horikawa, I.; Prieto, D.; Adelesberger, J.; Lempicki, R.; Barrett, J.C.; Dimitrov, D.S. Identification and characterization of rapidly dividing U937 clones with differential telomerase activity and gene expression profiles: Role of c-Myc/Mad1 and Id/Ets proteins. Leukemia 2002, 16, 1877–1880. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Witt, B.; Knippschild, U.; Koch, S.; Meena, J.K.; Heinlein, C.; Weise, J.M.; Krepulat, F.; Kuchenbauer, F.; Iben, S.; et al. CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int. J. Cancer 2013, 132, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Kyo, S.; Kanaya, T.; Wang, Z.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Ohmichi, M.; Gotoh, N.; Murakami, S.; et al. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene 2002, 21, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, D.; Li, J.; Berndt, M.C.; Liu, J.P. Transforming growth factor beta suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J. Biol. Chem. 2006, 281, 25588–25600. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Escot, C.; Theillet, C.; Lidereau, R.; Spyratos, F.; Champeme, M.H.; Gest, J.; Callahan, R. Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc. Natl. Acad. Sci. USA 1986, 83, 4834–4838. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, R.T.; Degerman, S.; Ljungberg, B.; Andersson, E.; Oji, Y.; Sugiyama, H.; Roos, G.; Li, A. Wilms’ tumour 1 can suppress hTERT gene expression and telomerase activity in clear cell renal cell carcinoma via multiple pathways. Br. J. Cancer 2010, 103, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008, 99, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Dessain, S.K.; Yu, H.; Reddel, R.R.; Beijersbergen, R.L.; Weinberg, R.A. Methylation of the human telomerase gene CpG island. Cancer Res. 2000, 60, 537–541. [Google Scholar] [PubMed]
- Renaud, S.; Loukinov, D.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. CTCF binds the proximal exonic region of hTERT and inhibits its transcription. Nucleic Acids Res. 2005, 33, 6850–6860. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.; Loukinov, D.; Abdullaev, Z.; Guilleret, I.; Bosman, F.T.; Lobanenkov, V.; Benhattar, J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007, 35, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Guilleret, I.; Benhattar, J. Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp. Cell Res. 2003, 289, 326–334. [Google Scholar] [CrossRef]
- Lindsey, J.C.; Schwalbe, E.C.; Potluri, S.; Bailey, S.; Williamson, D.; Clifford, S.C. TERT promoter mutation and aberrant hypermethylation are associated with elevated expression in medulloblastoma and characterise the majority of non-infant SHH subgroup tumours. Acta Neuropathol. 2014, 127, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Bechter, O.E.; Eisterer, W.; Dlaska, M.; Kühr, T.; Thaler, J. CpG island methylation of the hTERT promoter is associated with lower telomerase activity in B-cell lymphocytic leukemia. Exp. Hematol. 2002, 30, 26–33. [Google Scholar] [CrossRef]
- Borssén, M.; Cullman, I.; Norén-Nyström, U.; Sundström, C.; Porwit, A.; Forestier, E.; Roos, G. hTERT promoter methylation and telomere length in childhood acute lymphoblastic leukemia: Associations with immunophenotype and cytogenetic subgroup. Exp. Hematol. 2011, 39, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Azouz, A.; Wu, Y.L.; Hillion, J.; Tarkanyi, I.; Karniguian, A.; Aradi, J.; Lanotte, M.; Chen, G.Q.; Chehna, M.; Ségal-Bendirdjian, E. Epigenetic plasticity of hTERT gene promoter determines retinoid capacity to repress telomerase in maturation-resistant acute promyelocytic leukemia cells. Leukemia 2010, 24, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 2015, 29, 2219–2224. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zheng, C.; Lindvall, C.; Hou, M.; Ekedahl, J.; Lewensohn, R.; Yan, Z.; Yang, X.; Henriksson, M.; Blennow, E.; et al. Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res. 2000, 60, 6230–6235. [Google Scholar] [PubMed]
- Fan, X.; Wang, Y.; Kratz, J.; Brat, D.J.; Robitaille, Y.; Moghrabi, A.; Perlman, E.J.; Dang, C.V.; Burger, P.C.; Eberhart, C.G. hTERT gene amplification and increased mRNA expression in central nervous system embryonal tumors. Am. J. Pathol. 2003, 162, 1763–1769. [Google Scholar] [CrossRef]
- Palmqvist, R.; Zhang, A.; Xu, D.; Golovleva, I.; Norrback, K.F.; Gruber, A.; Oberg, A.; Stenling, R.; Roos, G. hTERT gene copy number is not associated with hTERT RNA expression or telomerase activity in colorectal cancer. Int. J. Cancer 2005, 116, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bishop, J.; Shan, Y.; Pai, S.; Liu, D.; Murugan, A.K.; Sun, H.; El-Naggar, A.K.; Xing, M. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr. Relat. Cancer 2013, 20, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Gallia, G.L.; Giovanella, B.C.; Grollman, A.P.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Pinto, V.; Celestino, R.; Reis, M.; Pópulo, H.; Boaventura, P.; Melo, M.; Catarino, T.; Lima, J.; Lopes, J.M.; et al. Telomerase promoter mutations in cancer: An emerging molecular biomarker? Virchows Arch. 2014, 465, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol. Cancer Res. 2016, 14, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Cárcano, F.M.; Vidal, D.O.; van Helvoort Lengert, A.; Neto, C.S.; Queiroz, L.; Marques, H.; Baltazar, F.; da Silva Martinelli, C.M.; Soares, P.; da Silva, E.C.; et al. Hotspot TERT promoter mutations are rare events in testicular germ cell tumors. Tumour Biol. 2016, 37, 4901–4907. [Google Scholar] [CrossRef] [PubMed]
- Campanella, N.C.; Celestino, R.; Pestana, A.; Scapulatempo-Neto, C.; de Oliveira, A.T.; Brito, M.J.; Gouveia, A.; Lopes, J.M.; Guimarães, D.P.; Soares, P.; et al. Low frequency of TERT promoter mutations in gastrointestinal stromal tumors (GISTs). Eur. J. Hum. Genet. 2015, 23, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, M.J.; Kim, W.G.; Sim, S.; Lim, S.; Kwon, H.; Kim, T.Y.; Shong, Y.K.; Kim, W.B. Low Prevalence of Somatic TERT Promoter Mutations in Classic Papillary Thyroid Carcinoma. Endocrinol. Metab. (Seoul) 2016, 31, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Suehara, N.; Mizumoto, K.; Muta, T.; Tominaga, Y.; Shimura, H.; Kitajima, S.; Hamasaki, N.; Tsuneyoshi, M.; Tanaka, M. Telomerase elevation in pancreatic ductal carcinoma compared to nonmalignant pathological states. Clin. Cancer Res. 1997, 3, 993–998. [Google Scholar] [PubMed]
- Mishra, G.; Zhao, Y.; Sweeney, J.; Pineau, B.C.; Case, D.; Ho, C.; Blackstock, A.W.; Geisinger, K.; Howerton, R.; Levine, E.; et al. Determination of qualitative telomerase activity as an adjunct to the diagnosis of pancreatic adenocarcinoma by EUS-guided fine-needle aspiration. Gastrointest. Endosc. 2006, 63, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Kammori, M.; Nakamura, K.; Kawahara, M.; Mimura, Y.; Kaminishi, M.; Takubo, K. Telomere shortening with aging in human thyroid and parathyroid tissue. Exp. Gerontol. 2002, 37, 513–521. [Google Scholar] [CrossRef]
- Vicente-Dueñas, C.; Romero-Camarero, I.; Sánchez-García, I. Understanding telomerase in cancer stem cell biology. Cell Cycle 2012, 11, 1479–1480. [Google Scholar] [PubMed]
- Greaves, M. Is telomerase activity in cancer due to selection of stem cells and differentiation arrest? Trends Genet. 1996, 12, 127–128. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. The reactivation of telomerase activity in cancer progression. Trends Genet. 1996, 12, 129–131. [Google Scholar] [CrossRef]
- Weise, J.M.; Güneş, Ç. Telomeres and telomerase. A survey about methods and recent advances in cancer diagnostic and therapy. Histol. Histopathol. 2006, 21, 1249–1261. [Google Scholar] [PubMed]
- Stindl, R. Defining the steps that lead to cancer: Replicative telomere erosion, aneuploidy and an epigenetic maturation arrest of tissue stem cells. Med. Hypotheses 2008, 71, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.; Greider, C.W. Telomerase and cancer stem cells. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Meena, J.K.; Cerutti, A.; Beichler, C.; Morita, Y.; Bruhn, C.; Kumar, M.; Kraus, J.M.; Speicher, M.R.; Wang, Z.Q.; Kestler, H.A.; et al. Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J. 2015, 34, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Johnson, J.Z.; Vogan, J.M.; Wagner, T.; Boyle, J.M.; Hockemeyer, D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife 2015, 4, e07918. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, C.J.; Jaskelioff, M.; Ivanova, E.; Kost-Alimova, M.; Protopopov, A.; Chu, G.C.; Wang, G.; Lu, X.; Labrot, E.S.; et al. Telomerase Reactivation following Telomere Dysfunction Yields Murine Prostate Tumors with Bone Metastases. Cell 2012, 148, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Güneş, Ç.; Rudolph, K.L. The role of telomeres in stem cells and cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.B. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Pereira-Smith, O.M.; Wright, W.E. A role for both RB and p53 in the regulation of human cellular senescence. Exp. Cell Res. 1991, 196, 33–39. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; de Solorzano, C.O.; Knowles, D.; Jones, A.; Chou, W.; Rodriguez, E.G.; Kuo, W.L.; Ljung, B.M.; Chew, K.; Myambo, K.; et al. In situ analyses of genome instability in breast cancer. Nat. Genet. 2004, 36, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Shpitz, B.; Zimlichman, S.; Zemer, R.; Bomstein, Y.; Zehavi, T.; Liverant, S.; Bernehim, J.; Kaufman, Z.; Klein, E.; Shapira, Y.; et al. Telomerase activity in ductal carcinoma in situ of the breast. Breast Cancer Res. Treat. 1999, 58, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Chadeneau, C.; Hay, K.; Hirte, H.W.; Gallinger, S.; Bacchetti, S. Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer Res. 1995, 55, 2533–2536. [Google Scholar] [PubMed]
- O’Sullivan, J.N.; Bronner, M.P.; Brentnall, T.A.; Finley, J.C.; Shen, W.T.; Emerson, S.; Emond, M.J.; Gollahon, K.A.; Moskovitz, A.H.; Crispin, D.A.; et al. Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat. Genet. 2002, 32, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. Telomeres and telomerase in cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C. Tracing the cellular origin of cancer. Nat. Cell Biol. 2013, 15, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Bruedigam, C.; Bagger, F.O.; Heidel, F.H.; Paine Kuhn, C.; Guignes, S.; Song, A.; Austin, R.; Vu, T.; Lee, E.; Riyat, S.; et al. Telomerase inhibition effectively targets mouse and human AML stem cells and delays relapse following chemotherapy. Cell Stem Cell 2014, 15, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Munari, E.; Faraj, S.F.; Hruban, R.H.; Schoenberg, M.; Bivalacqua, T.; Allaf, M.; Springer, S.; Wang, Y.; Diaz, L.A., Jr.; et al. TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res. 2013, 73, 7162–7167. [Google Scholar] [CrossRef] [PubMed]
- Hurst, C.D.; Platt, F.M.; Knowles, M.A. Comprehensive mutation analysis of the TERT promoter in bladder cancer and detection of mutations in voided urine. Eur. Urol. 2014, 65, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Allory, Y.; Beukers, W.; Sagrera, A.; Flandez, M.; Marques, M.; Marquez, M.; van der Keur, K.A.; Dyrskjot, L.; Lurkin, I.; Vermeij, M.; et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: High frequency across stages, detection in urine, and lack of association with outcome. Eur. Urol. 2014, 65, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Coller, H.A.; Roberts, J.M. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat. Cell Biol. 2003, 5, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Iben, S.; Güneş, Ç. Telomerase exerts physiological and tumor promoting functions by stimulating ribosomal biogenesis. Telomere Telomerase 2014, 1, e433. [Google Scholar]
- Sarin, K.Y.; Cheung, P.; Gilison, D.; Lee, E.; Tennen, R.I.; Wang, E.; Artandi, M.K.; Oro, A.E.; Artandi, S.E. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005, 436, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Saretzki, G.; Peters, H.; Wappler, I.; Evans, J.; Hole, N.; von Zglinicki, T.; Lako, M. Overexpression of telomerase confers growth advantage, stress resistance, and enhanced differentiation of ESCs toward the hematopoietic lineage. Stem Cells 2005, 23, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sung, Y.H.; Cheong, C.; Choi, Y.S.; Jeon, H.K.; Sun, W.; Hahn, W.C.; Ishikawa, F.; Lee, H.W. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene 2008, 27, 3754–3760. [Google Scholar] [CrossRef] [PubMed]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial Telomerase Protects Cancer Cells from Nuclear DNA Damage and Apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.G.; Assfalg, R.; Koch, S.; Schelling, A.; Meena, J.K.; Kraus, J.; Lechel, A.; Katz, S.F.; Benes, V.; Scharffetter-Kochanek, K.; et al. Telomerase stimulates ribosomal DNA transcription under hyperproliferative conditions. Nat. Commun. 2014, 13, 4599. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Beck, S.; Sohn, Y.W.; Kim, J.K.; Kim, S.H.; Yin, J.; Pian, X.; Kim, S.C.; Choi, Y.J.; Kim, H. Human telomerase catalytic subunit (hTERT) suppresses p53-mediated anti-apoptotic response via induction of basic fibroblast growth factor. Exp. Mol. Med. 2010, 42, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase-incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- Meena, J.; Rudolph, K.L.; Güneş, Ç. Telomere Dysfunction, Chromosomal Instability and Cancer. Recent Results Cancer Res. 2015, 200, 61–79. [Google Scholar] [PubMed]
- Liu, H.; Liu, Q.; Ge, Y.; Zhao, Q.; Zheng, X.; Zhao, Y. hTERT promotes cell adhesion and migration independent of telomerase activity. Sci Rep. 2016, 14, 22886. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Hahn, W.C.; O’Connor, B.F.; Banner, E.N.; Lundberg, A.S.; Modha, P.; Mizuno, H.; Brooks, M.W.; Fleming, M.; Zimonjic, D.B.; et al. Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 12606–12611. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Cardenas, S.L.; Greider, C.W. Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: A critical examination of telomere length maintenance-independent roles of telomerase. Nucleic Acids Res. 2010, 38, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.A.; Vidal-Cardenas, S.L.; Karim, B.; Yu, H.; Guo, N.; Greider, C.W. Phenotypes in mTERT+/− and mTERT−/− mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol. Cell Biol. 2011, 31, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Oruganti, H.; Giudice, L.C.; Hoffman, A.R. Regulation of telomerase by alternate splicing of human telomerase reverse transcriptase (hTERT) in normal and neoplastic ovary, endometrium and myometrium. Int. J. Cancer 2000, 85, 330–335. [Google Scholar] [CrossRef]
- Melton, C.; Reuter, J.A.; Spacek, D.V.; Snyder, M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat. Genet. 2015, 47, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
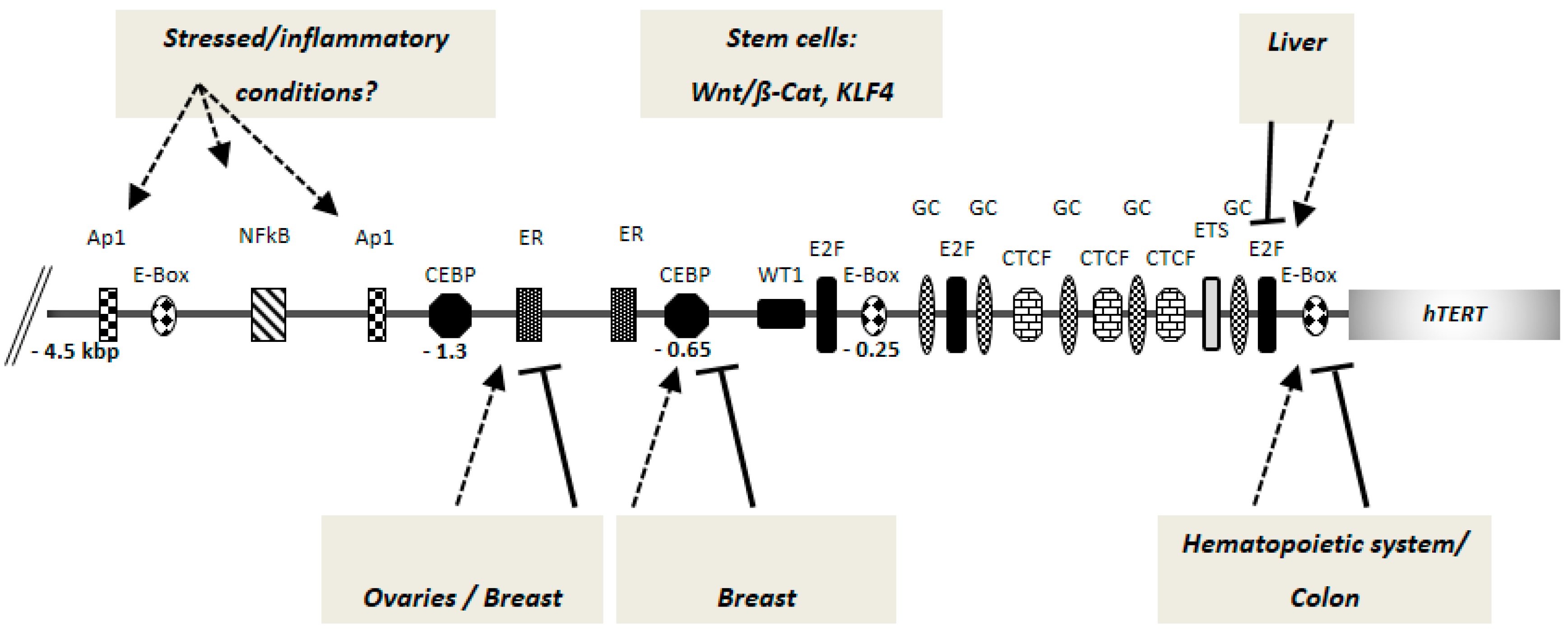
Figure 1.
Putative and bona fide cis-regulatory elements of human telomerase reverse transcriptase (hTERT) gene promoter. Current research indicates that different factors can act in a tissue specific manner for hTERT/telomerase regulation. A 4.5 kbp hTERT promoter fragment upstream of the AUG start codon is shown (not drawn to scale). The numbers denote the distance from the AUG start site. The arrows indicate activation and the lines indicate repression of hTERT promoter by the same binding site, depending on the occupation by the respective tissue-specific regulatory factors (see text for details). The mode of action by the stem cell specific factors is not fully clear. E-box: c-Myc/Mad-family factor binding sites; E2F: E2 factor family transcription factor binding sites; GC: GC-box binding sites; ETS: ETS-domain binding sites; CTCF: CTCF factor binding sites; CEBP: CEBP family factor binding sites; ER: estrogen receptor binding sites; Ap1: Ap1 factor binding sites; NFκB: NFκB factor binding site; WT1: binding site for the Wilms’ tumor 1 transcription factor.
Figure 1.
Putative and bona fide cis-regulatory elements of human telomerase reverse transcriptase (hTERT) gene promoter. Current research indicates that different factors can act in a tissue specific manner for hTERT/telomerase regulation. A 4.5 kbp hTERT promoter fragment upstream of the AUG start codon is shown (not drawn to scale). The numbers denote the distance from the AUG start site. The arrows indicate activation and the lines indicate repression of hTERT promoter by the same binding site, depending on the occupation by the respective tissue-specific regulatory factors (see text for details). The mode of action by the stem cell specific factors is not fully clear. E-box: c-Myc/Mad-family factor binding sites; E2F: E2 factor family transcription factor binding sites; GC: GC-box binding sites; ETS: ETS-domain binding sites; CTCF: CTCF factor binding sites; CEBP: CEBP family factor binding sites; ER: estrogen receptor binding sites; Ap1: Ap1 factor binding sites; NFκB: NFκB factor binding site; WT1: binding site for the Wilms’ tumor 1 transcription factor.

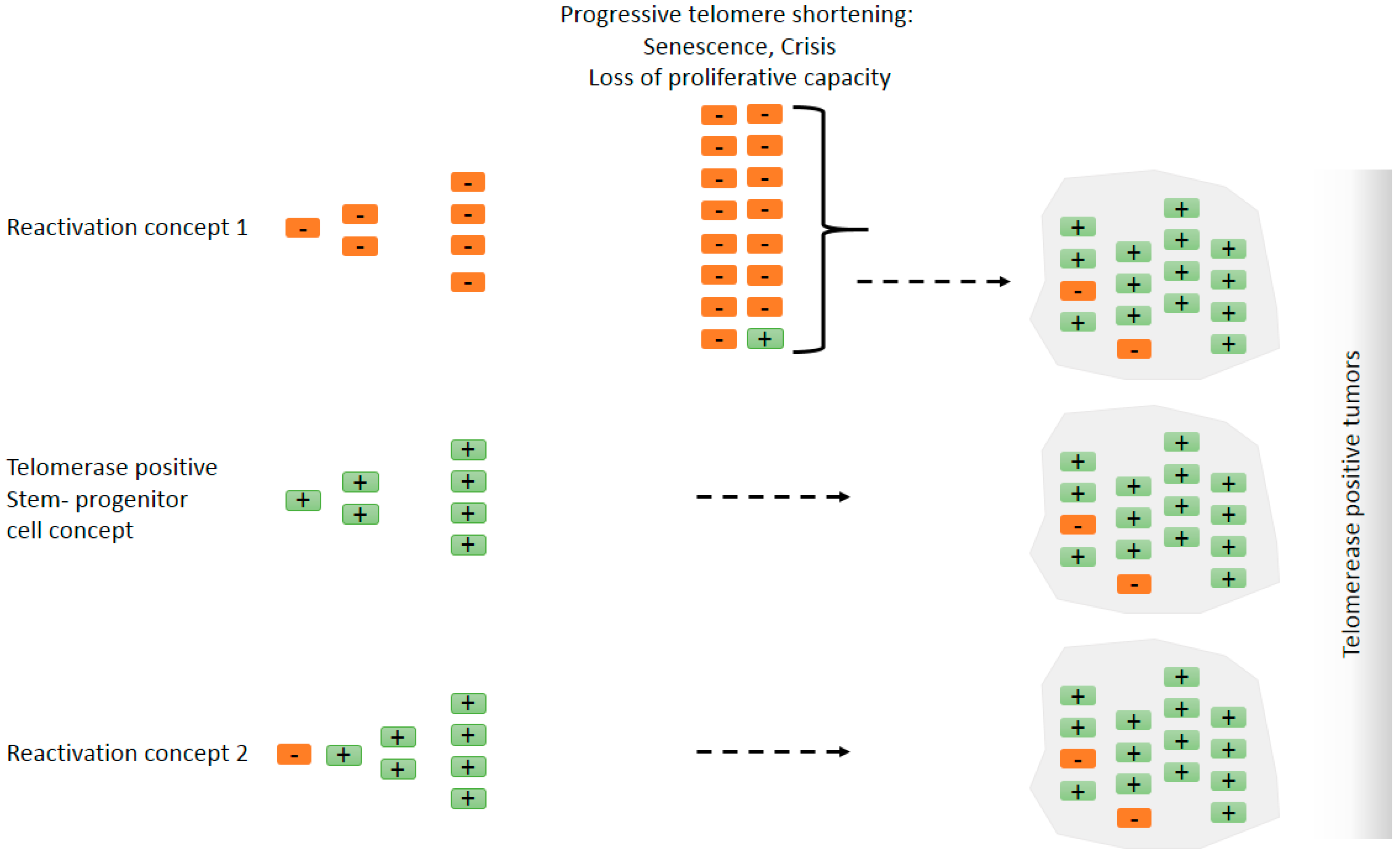
Figure 2.
Does telomerase reactivation occur in tumors, or do tumors originate from telomerase positive cells such as stem cells? Simplified models explaining ‘detectable’ telomerase activity in cancer. To what extent the final tumor mass contains telomerase negative cells is unclear. There is evidence, however, that there is a subset of cells that are telomerase-positive cancer stem cells [92]. Orange rectangles with a negative mark represent telomerase negative cells and green rectangles with a positive mark represent telomerase positive cells. Reactivation concept 1: Telomerase negative cells with a tumorigenic mutation can proliferate but eventually enter senescence and crisis due to progressive telomere shortening. Cells that evade hTERT repression can continue to proliferate and constitute the major tumor mass. In this scenario, telomerase reactivation occurs late during tumorigenesis or it is not detectable because only a minority of patient-derived early lesion cells are telomerase positive. Stem/progenitor cell concept: The tumorigenic mutation, e.g., aneuploidy-inducing mutation [75], occurs in a telomerase positive stem/progenitor cell. In the presence of telomerase, cells continue to proliferate where telomerase activity may suppress telomere shortening during extended proliferation and cellular differentiation [76] and/or confer telomere-length-independent survival advantage. Reactivation concept 2: Telomerase reactivation occurs early in tumorigenesis, e.g., by hTERT promoter mutations or by loss of negative regulatory mechanisms, and telomerase activity is high at all stages. Telomerase reactivation may precede other tumorigenic events. It is important to note that the alternative models proposed here are non-exclusive. The three mechanisms are very likely to take place under different scenarios and different cancer types.
Figure 2.
Does telomerase reactivation occur in tumors, or do tumors originate from telomerase positive cells such as stem cells? Simplified models explaining ‘detectable’ telomerase activity in cancer. To what extent the final tumor mass contains telomerase negative cells is unclear. There is evidence, however, that there is a subset of cells that are telomerase-positive cancer stem cells [92]. Orange rectangles with a negative mark represent telomerase negative cells and green rectangles with a positive mark represent telomerase positive cells. Reactivation concept 1: Telomerase negative cells with a tumorigenic mutation can proliferate but eventually enter senescence and crisis due to progressive telomere shortening. Cells that evade hTERT repression can continue to proliferate and constitute the major tumor mass. In this scenario, telomerase reactivation occurs late during tumorigenesis or it is not detectable because only a minority of patient-derived early lesion cells are telomerase positive. Stem/progenitor cell concept: The tumorigenic mutation, e.g., aneuploidy-inducing mutation [75], occurs in a telomerase positive stem/progenitor cell. In the presence of telomerase, cells continue to proliferate where telomerase activity may suppress telomere shortening during extended proliferation and cellular differentiation [76] and/or confer telomere-length-independent survival advantage. Reactivation concept 2: Telomerase reactivation occurs early in tumorigenesis, e.g., by hTERT promoter mutations or by loss of negative regulatory mechanisms, and telomerase activity is high at all stages. Telomerase reactivation may precede other tumorigenic events. It is important to note that the alternative models proposed here are non-exclusive. The three mechanisms are very likely to take place under different scenarios and different cancer types.

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kumar, M.; Lechel, A.; Güneş, Ç. Telomerase: The Devil Inside. Genes 2016, 7, 43. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7080043
AMA Style
Kumar M, Lechel A, Güneş Ç. Telomerase: The Devil Inside. Genes. 2016; 7(8):43. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7080043
Chicago/Turabian StyleKumar, Mukesh, Andre Lechel, and Çagatay Güneş. 2016. "Telomerase: The Devil Inside" Genes 7, no. 8: 43. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7080043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.