
Ups and Downs: Mechanisms of Repeat Instability in the Fragile X-Related Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
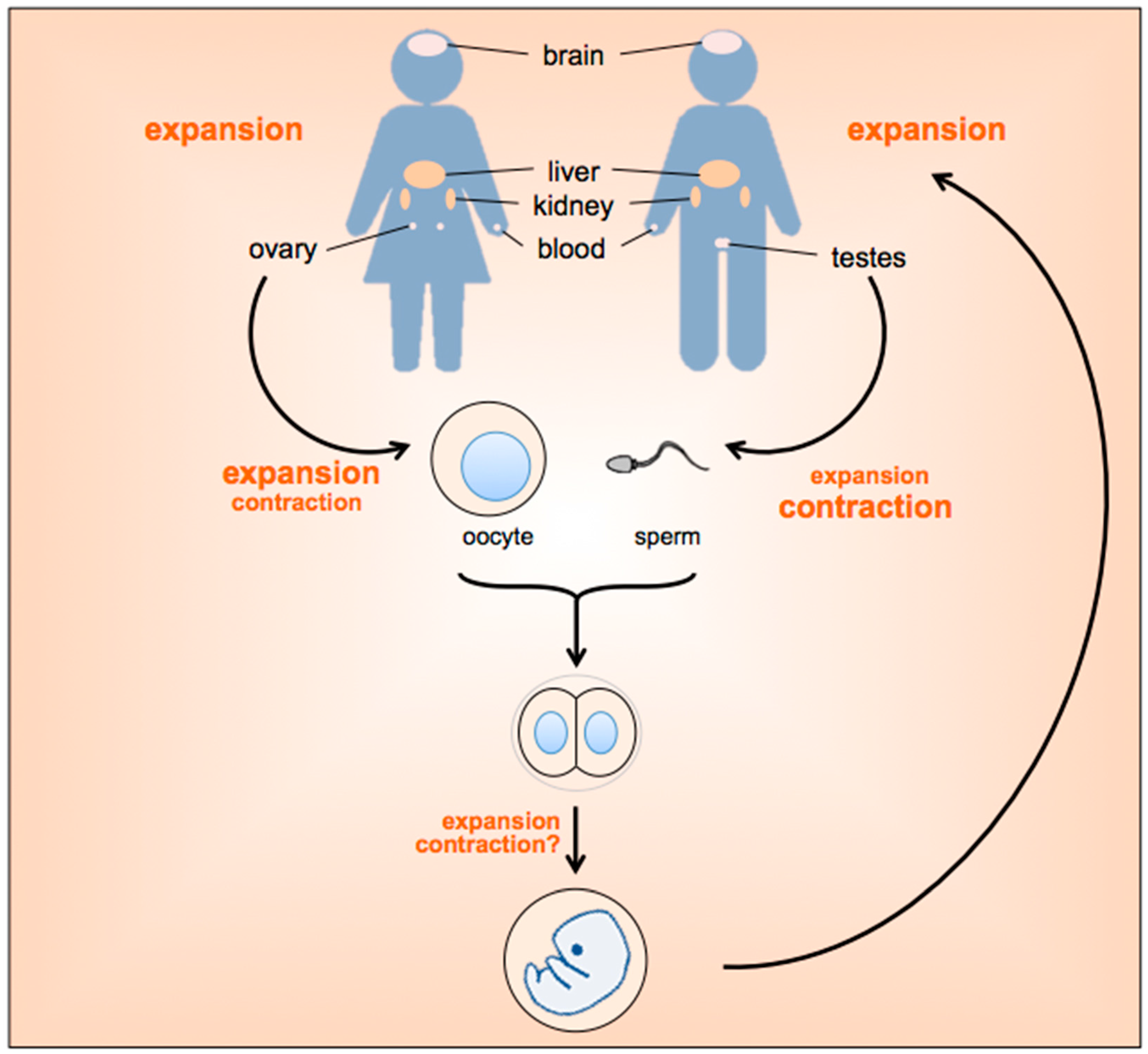
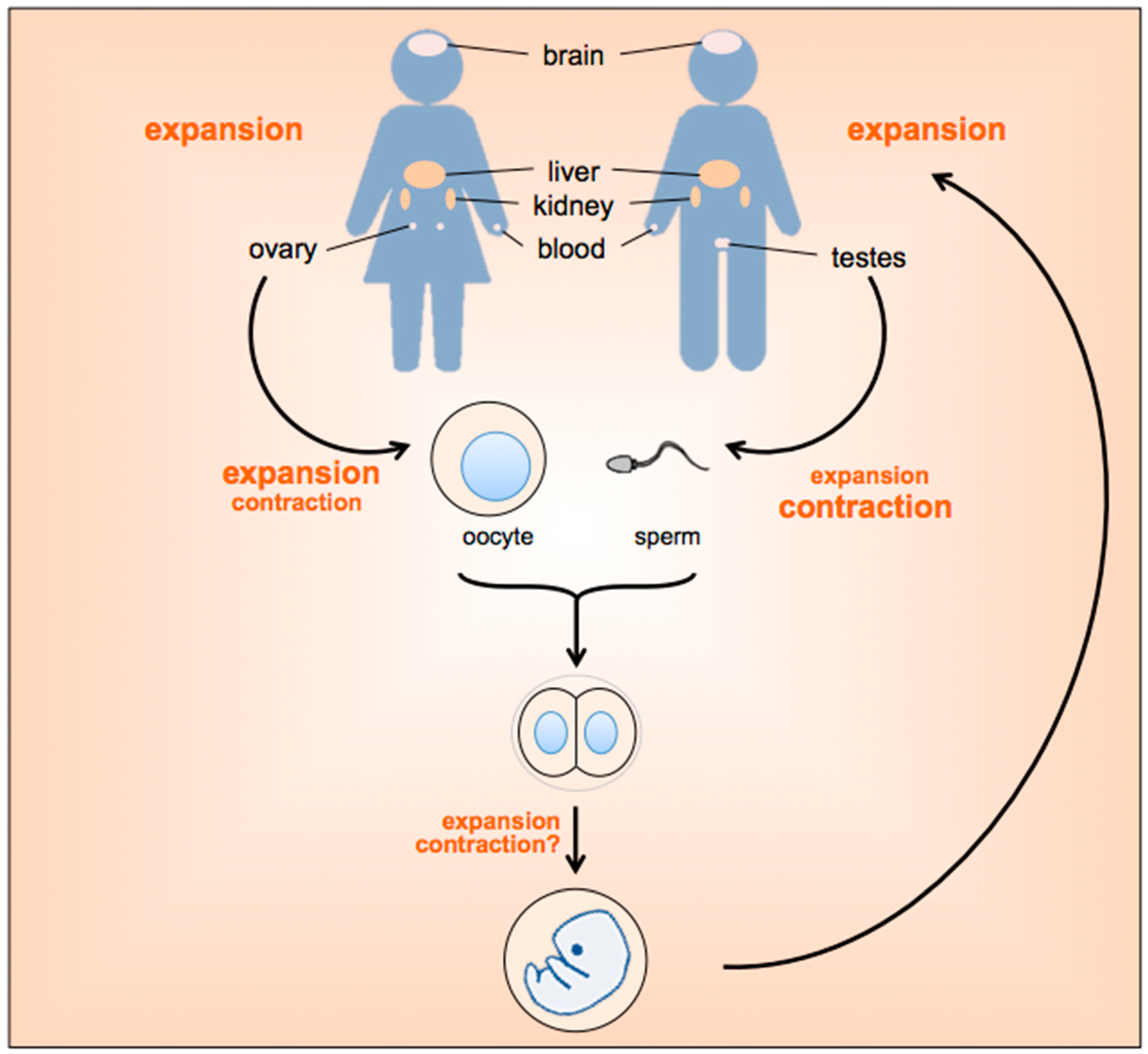
2. When and Where Does Repeat Instability Occur?
3. What Factors Affect Expansion Risk?
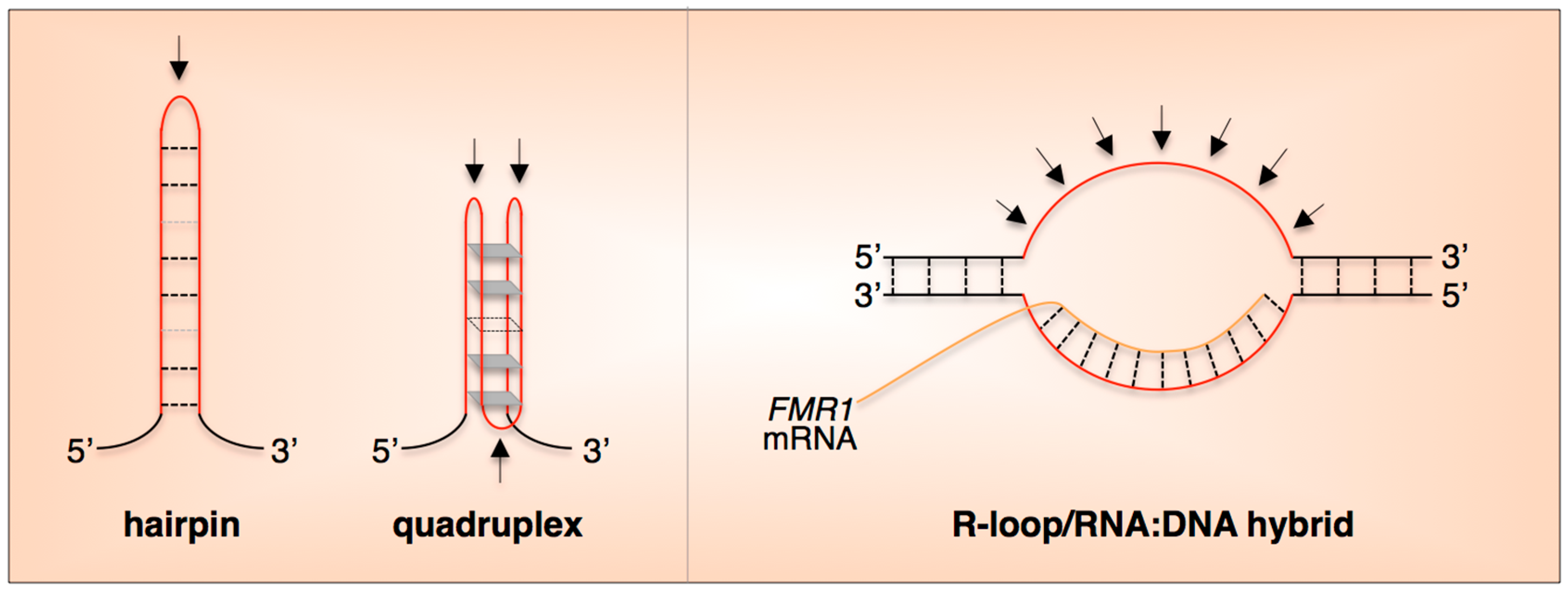
4. What Is It about the Repeat that Makes It Unstable?
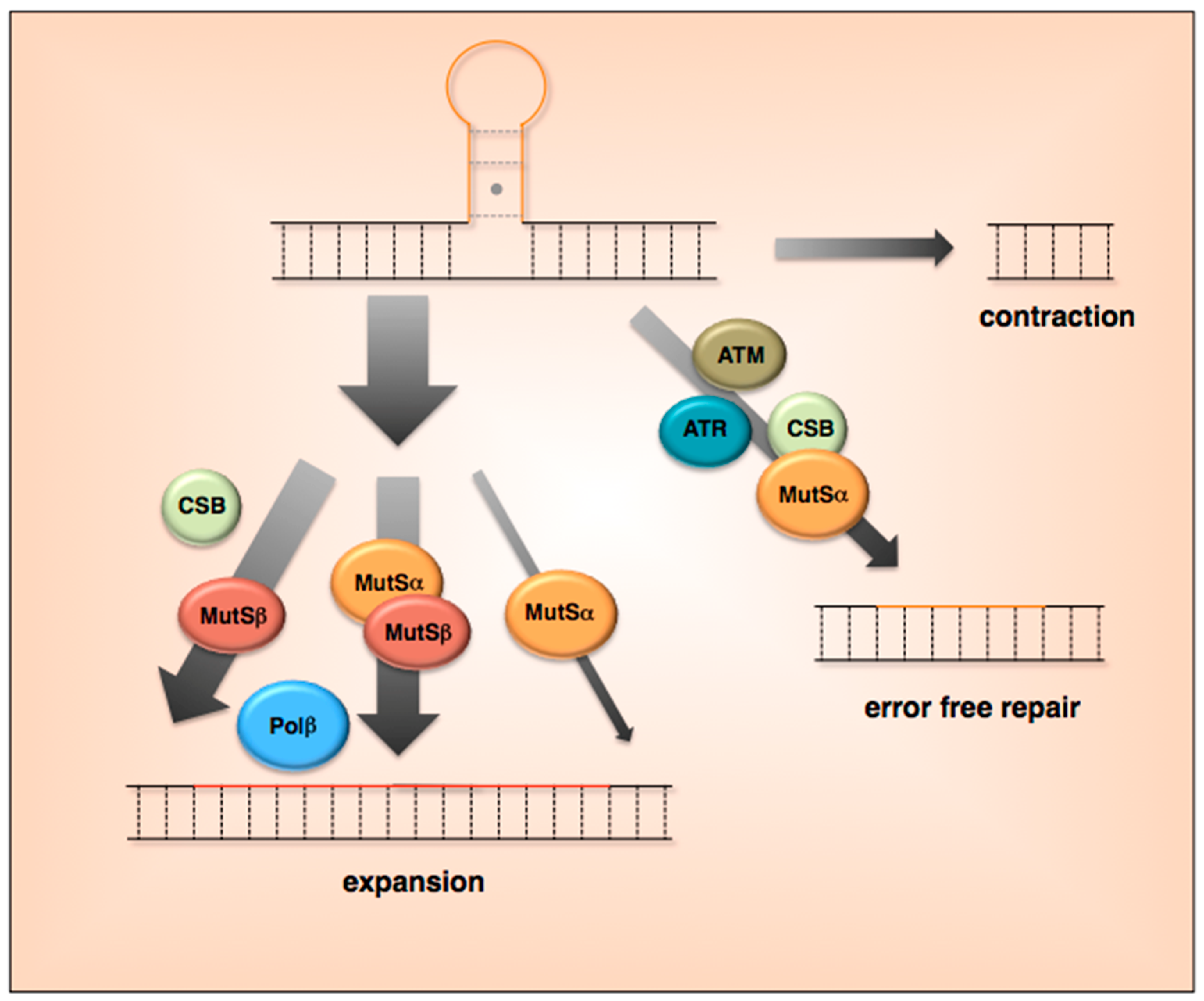
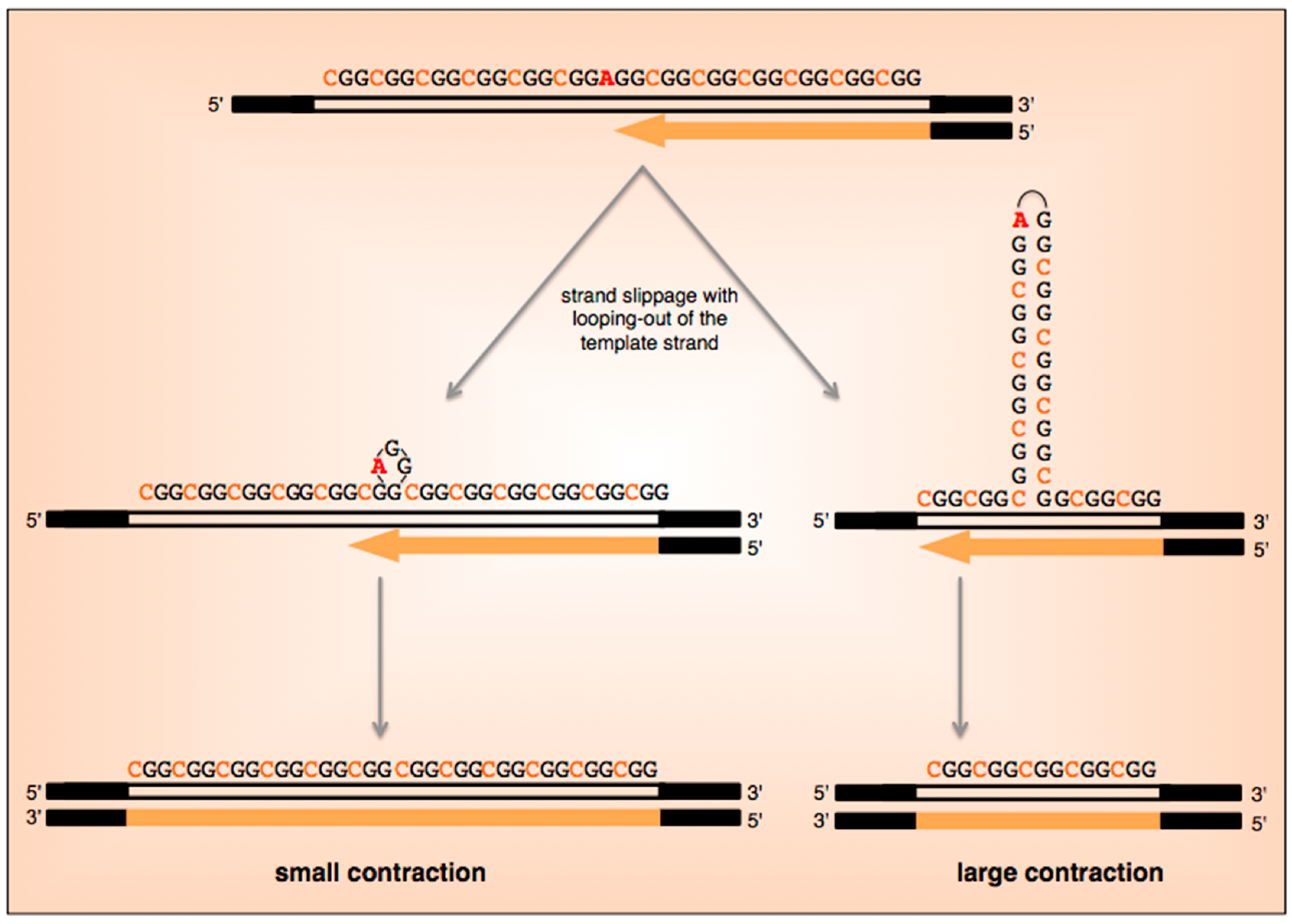
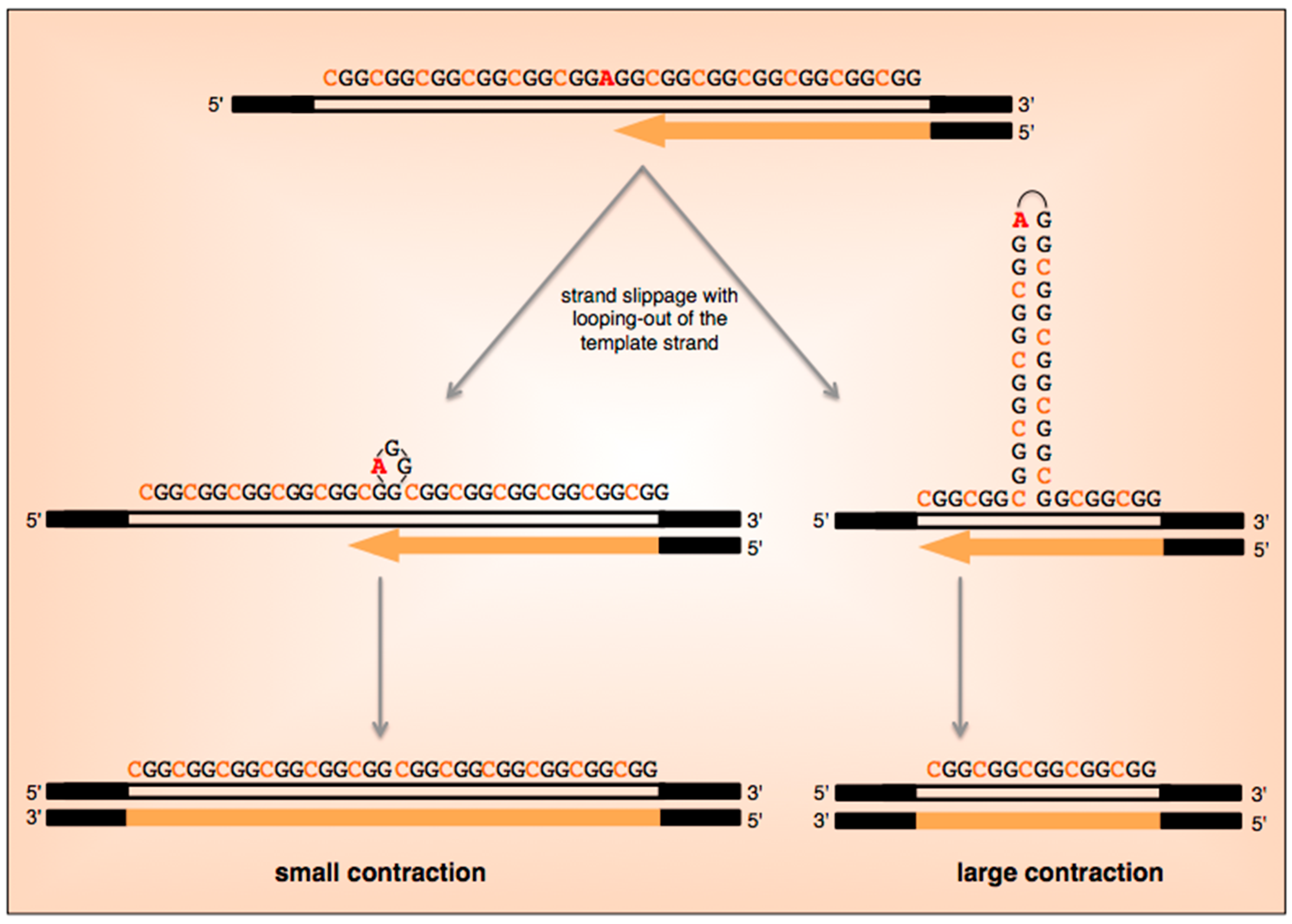
5. What Are the Current Models for Expansion?
6. Are Expansions and Contractions Reciprocal Events?
7. What Is Known about Contractions and the Potential for Error-Free Repair?
8. What Can Be Done to Block Expansions or Promote Error-Free Repair or Contractions?
9. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, C.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; van der Linde, H.; Lans, H.; de Lange, D.; Hirst, M.C.; Oostra, B.A. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum. Mol. Genet. 2001, 10, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A.J.; Jorens, H.Z.; Darby, J.K.; Oostra, B.; et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 1993, 4, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.R.; Schildkraut, C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 2014, 53, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. The dynamics of CGG-repeat instability and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. In revision.
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, D.; Hayward, B.E.; Aladjem, M.I.; Kumari, D.; Usdin, K. Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 2014, 23, 2940–2952. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Entezam, A.; Kumari, D.; Yudkin, D.; Qin, M.; Smith, C.B.; Usdin, K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013, 34, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Gonitel, R.; Moffitt, H.; Sathasivam, K.; Woodman, B.; Detloff, P.J.; Faull, R.L.; Bates, G.P. DNA instability in postmitotic neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 3467–3472. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.M.; Dragileva, E.; Kirby, A.; Lloret, A.; Lopez, E.; St Claire, J.; Panigrahi, G.B.; Hou, C.; Holloway, K.; Gillis, T.; et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet 2013, 9, e1003930. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.; Manley, K.; Simard, J.P.; Clark, G.W.; Slean, M.M.; Swami, M.; Shelbourne, P.F.; Tillier, E.R.; Monckton, D.G.; Messer, A.; et al. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013, 9, e1003280. [Google Scholar]
- Morales, F.; Vasquez, M.; Santamaria, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst) 2016, 40, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Pearson, C.E.; Coutinho, P.; Provost, S.; Amorim, A.; Dube, M.P.; Sequeiros, J.; Rouleau, G.A. Modifiers of (CAG)(n) instability in Machado-Joseph disease (MJD/SCA3) transmissions: an association study with DNA replication, repair and recombination genes. Hum. Genet. 2014, 133, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar]
- Entezam, A.; Lokanga, R.A.; Le, W.; Hoffman, G.; Usdin, K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2010, 31, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Zhao, X.N.; Entezam, A.; Usdin, K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum. Mol. Genet. 2014, 23, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.; Sah, S.; Filipovic-Sadic, S.; Krosting, J.; Sekinger, E.; Pan, R.; Hagerman, P.J.; Stenzel, T.T.; Tassone, F.; et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J. Mol. Diagn. 2010, 12, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Loeb, L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. USA 1994, 91, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Nadel, Y.; Weisman-Shomer, P.; Fry, M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 1995, 270, 28970–28977. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Mitas, M.; Yu, A.; Dill, J.; Haworth, I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 1995, 34, 12803–12811. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Sanz, L.A.; Chedin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Renciuk, D.; Kypr, J.; Vorlickova, M. CGG repeats associated with fragile X chromosome form left-handed Z-DNA structure. Biopolymers 2011, 95, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Jarem, D.A.; Wilson, N.R.; Schermerhorn, K.M.; Delaney, S. Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair (Amst) 2011, 10, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, S.V.; Catasti, P.; Chen, X.; Ratliff, R.; Moyzis, R.K.; Bradbury, E.M.; Gupta, G. Solution structures of the individual single strands of the fragile X DNA triplets (GCC)n.(GGC)n. Nucleic Acids Res. 1996, 24, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Eichler, E.E.; Lorenzetti, D.; Kramer, S.F.; Zoghbi, H.Y.; Nelson, D.L.; Sinden, R.R. Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation. Biochem 1998, 37, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; Bhavesh, N.S.; Hosur, R.V. Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of Fragile X Syndrome: NMR observations. Biochem. Biophys. Res. Commun. 2000, 278, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Fojtik, P.; Vorlickova, M. The fragile X chromosome (GCC) repeat folds into a DNA tetraplex at neutral pH. Nucleic Acids Res. 2001, 29, 4684–4690. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Spiro, C.; Pelletier, R.; Rolfsmeier, M.L.; Dixon, M.J.; Lahue, R.S.; Gupta, G.; Park, M.S.; Chen, X.; Mariappan, S.V.; McMurray, C.T. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol. Cell 1999, 4, 1079–1085. [Google Scholar] [CrossRef]
- Gacy, A.M.; Goellner, G.; Juranic, N.; Macura, S.; Mcmurray, C.T. Trinucleotide Repeats That Expand in Human-Disease Form Hairpin Structures in-Vitro. Cell 1995, 81, 533–540. [Google Scholar] [CrossRef]
- Paiva, A.M.; Sheardy, R.D. Influence of sequence context and length on the structure and stability of triplet repeat DNA oligomers. Biochemistry 2004, 43, 14218–14227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Kumari, D.; Gupta, S.; Wu, D.; Evanitsky, M.; Yang, W.; Usdin, K. MutSbeta generates both expansions and contractions in a mouse model of the Fragile X-associated Disorders. Hum. Mol. Genet. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Lokanga, R.; Allette, K.; Gazy, I.; Wu, D.; Usdin, K. A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice? PLoS Genet 2016, 12, e1006190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.N.; Usdin, K. Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 2014, 35, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. The transcription-coupled repair protein ERCC6/CSB also protects against repeat expansion in a mouse model of the fragile X premutation. Hum. Mutat. 2015, 36, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Usdin, K. ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008, 36, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Usdin, K. ATM and ATR protect the genome against two different types of tandem repeat instability in Fragile X premutation mice. Nucleic Acids Res. 2009, 37, 6371–6377. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. To switch or not to switch: at the origin of repeat expansion disease. Mol. Cell 2014, 53, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.; Murray, A.; Brightwell, G.; Morton, N.E.; Jacobs, P.A. Closely linked cis-acting modifier of expansion of the CGG repeat in high risk FMR1 haplotypes. Hum. Mutat. 2007, 28, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Zaninovic, N.; Zhan, Q.; Madireddy, A.; Nolin, S.L.; Ersalesi, N.; Yan, Z.; Rosenwaks, Z.; Schildkraut, C.L. Cis-acting DNA sequence at a replication origin promotes repeat expansion to fragile X full mutation. J. Cell Biol. 2014, 206, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Foiry, L.; Dong, L.; Savouret, C.; Hubert, L.; te Riele, H.; Junien, C.; Gourdon, G. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum. Genet. 2006, 119, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Wesoly, J.; Agarwal, S.; Sigurdsson, S.; Bussen, W.; Van Komen, S.; Qin, J.; van Steeg, H.; van Benthem, J.; Wassenaar, E.; Baarends, W.M.; et al. Differential contributions of mammalian Rad54 paralogs to recombination, DNA damage repair, and meiosis. Mol. Cell. Biol. 2006, 26, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, T.; Van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.; Lohman, P.H.; Pastink, A. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Buerstedde, J.M.; Bezzubova, O.; Morrison, C.; Takata, M.; Shinohara, A.; Takeda, S. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef] [PubMed]
- Manley, K.; Shirley, T.L.; Flaherty, L.; Messer, A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 1999, 23, 471–473. [Google Scholar] [PubMed]
- Savouret, C.; Garcia-Cordier, C.; Megret, J.; te Riele, H.; Junien, C.; Gourdon, G. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol. Cell. Biol. 2004, 24, 629–637. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, W.J.; Nelen, M.R.; Wansink, D.G.; Coerwinkel, M.M.; te Riele, H.; Groenen, P.J.; Wieringa, B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 2002, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Lebel, L.A.; Vrbanac, V.; Teed, A.; te Riele, H.; MacDonald, M.E. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum. Mol. Genet. 2003, 12, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.A.; Yang, Z.; Lai, M.; Gajec, M.; Badger, J.D., 2nd; Hayes, J.J.; Edelmann, W.; Kucherlapati, R.; Wilson, T.M.; McMurray, C.T. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat. Struct. Mol. Biol. 2005, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Halabi, A.; Ditch, S.; Wang, J.; Grabczyk, E. DNA mismatch repair complex MutSbeta promotes GAA.TTC repeat expansion in human cells. J. Biol. Chem. 2012, 287, 29958–29967. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Campau, E.; Soragni, E.; Ku, S.; Puckett, J.W.; Dervan, P.B.; Gottesfeld, J.M. Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J. Biol. Chem. 2012, 287, 29861–29872. [Google Scholar] [CrossRef] [PubMed]
- Gannon, A.M.; Frizzell, A.; Healy, E.; Lahue, R.S. MutSbeta and histone deacetylase complexes promote expansions of trinucleotide repeats in human cells. Nucleic Acids Res. 2012, 40, 10324–10333. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Senejani, A.G.; Sweasy, J.B.; Usdin, K. Heterozygosity for a hypomorphic polbeta mutation reduces the expansion frequency in a mouse model of the fragile X-related disorders. PLoS Genet. 2015, 11, e1005181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Møllersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar]
- Muftuoglu, M.; de Souza-Pinto, N.C.; Dogan, A.; Aamann, M.; Stevnsner, T.; Rybanska, I.; Kirkali, G.; Dizdaroglu, M.; Bohr, V.A. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem. 2009, 284, 9270–9279. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Chen, C.; Zeng, X.; Christiansen, M.; Bohr, V.A. Functional crosstalk between hOGG1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst) 2002, 1, 913–927. [Google Scholar] [CrossRef]
- Wong, H.K.; Muftuoglu, M.; Beck, G.; Imam, S.Z.; Bohr, V.A.; Wilson, D.M., 3rd. Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007, 35, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Dragileva, E.; Hendricks, A.; Teed, A.; Gillis, T.; Lopez, E.T.; Friedberg, E.C.; Kucherlapati, R.; Edelmann, W.; Lunetta, K.L.; MacDonald, M.E.; et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol. Dis. 2009, 33, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Conca Dioguardi, C.; Uslu, B.; Haynes, M.; Kurus, M.; Gul, M.; Miao, D.Q.; De Santis, L.; Ferrari, M.; Bellone, S.; Santin, A.; et al. Granulosa cell and oocyte mitochondrial abnormalities in a mouse model of fragile X primary ovarian insufficiency. Mol. Hum. Reprod. 2016, 22, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef] [PubMed]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.-Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Pena, S.; McMurray, C.T. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet 2015. In Press. [Google Scholar] [CrossRef] [PubMed]
- Møllersen, L.; Moldestad, O.; Rowe, A.D.; Bjølgerud, A.; Holm, I.; Tveterås, L.; Klungland, A.; Retterstøl, L. Effects of Anthocyanins on CAG Repeat Instability and Behaviour in Huntington’s Disease R6/1 Mice. PLoS Curr. Huntingt. Dis. 2016. [Google Scholar] [CrossRef]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.-N.; Usdin, K. Ups and Downs: Mechanisms of Repeat Instability in the Fragile X-Related Disorders. Genes 2016, 7, 70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090070
Zhao X-N, Usdin K. Ups and Downs: Mechanisms of Repeat Instability in the Fragile X-Related Disorders. Genes. 2016; 7(9):70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090070
Chicago/Turabian StyleZhao, Xiao-Nan, and Karen Usdin. 2016. "Ups and Downs: Mechanisms of Repeat Instability in the Fragile X-Related Disorders" Genes 7, no. 9: 70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090070