
Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Replication Barriers Associated with Repeat DNA and Protein–DNA Complexes
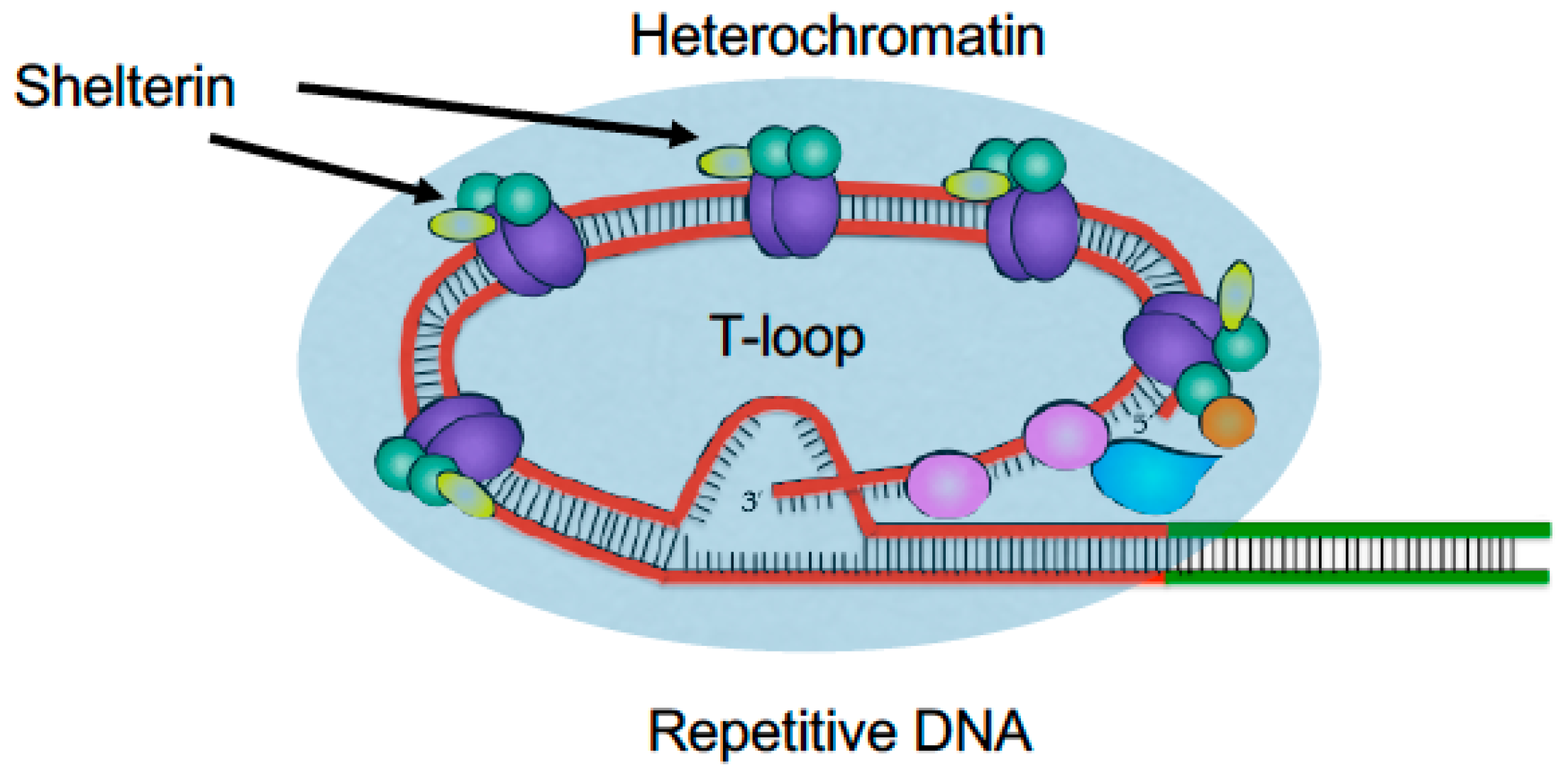
2.1. Telomeres
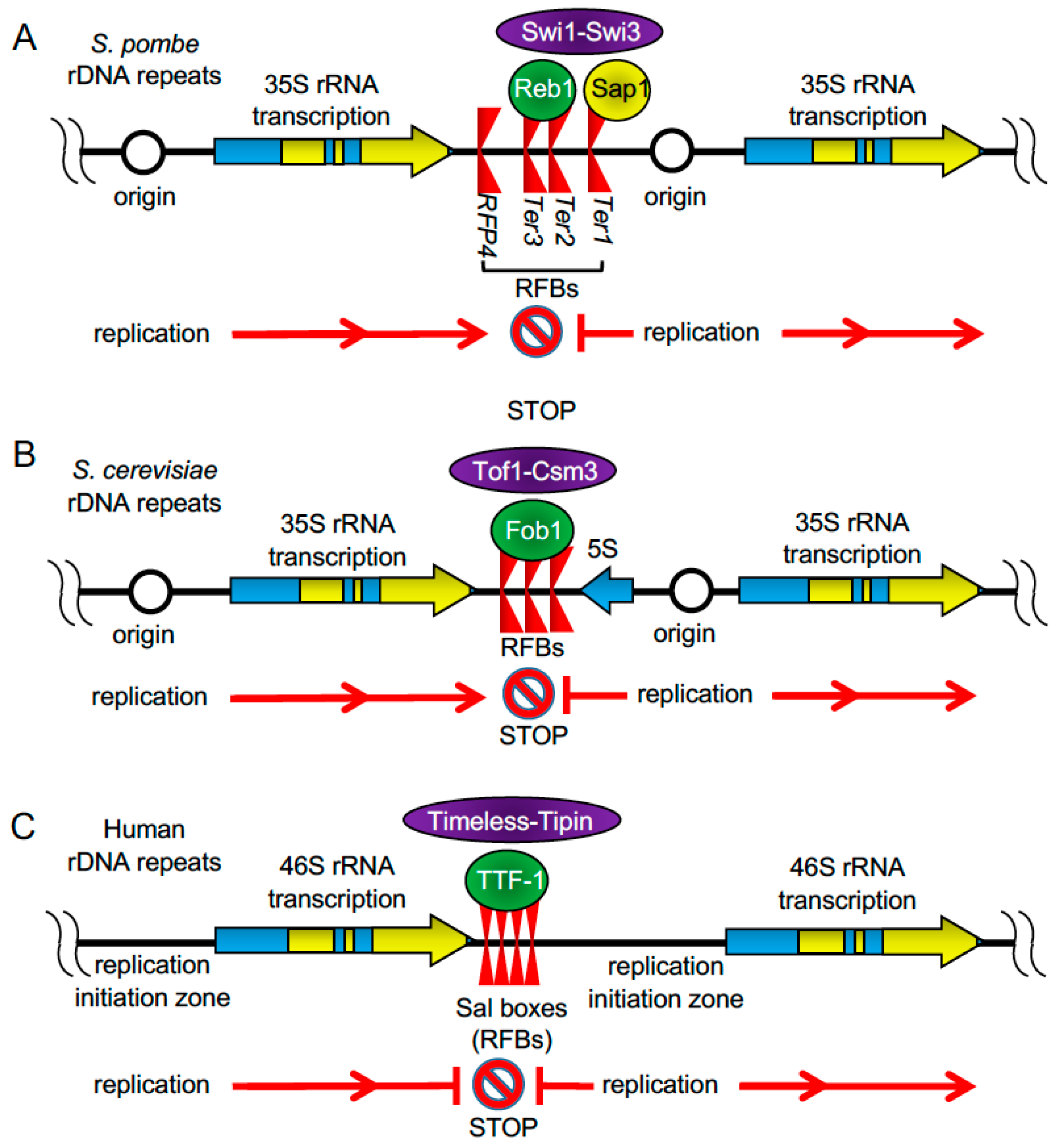
2.2. rDNA Repeats
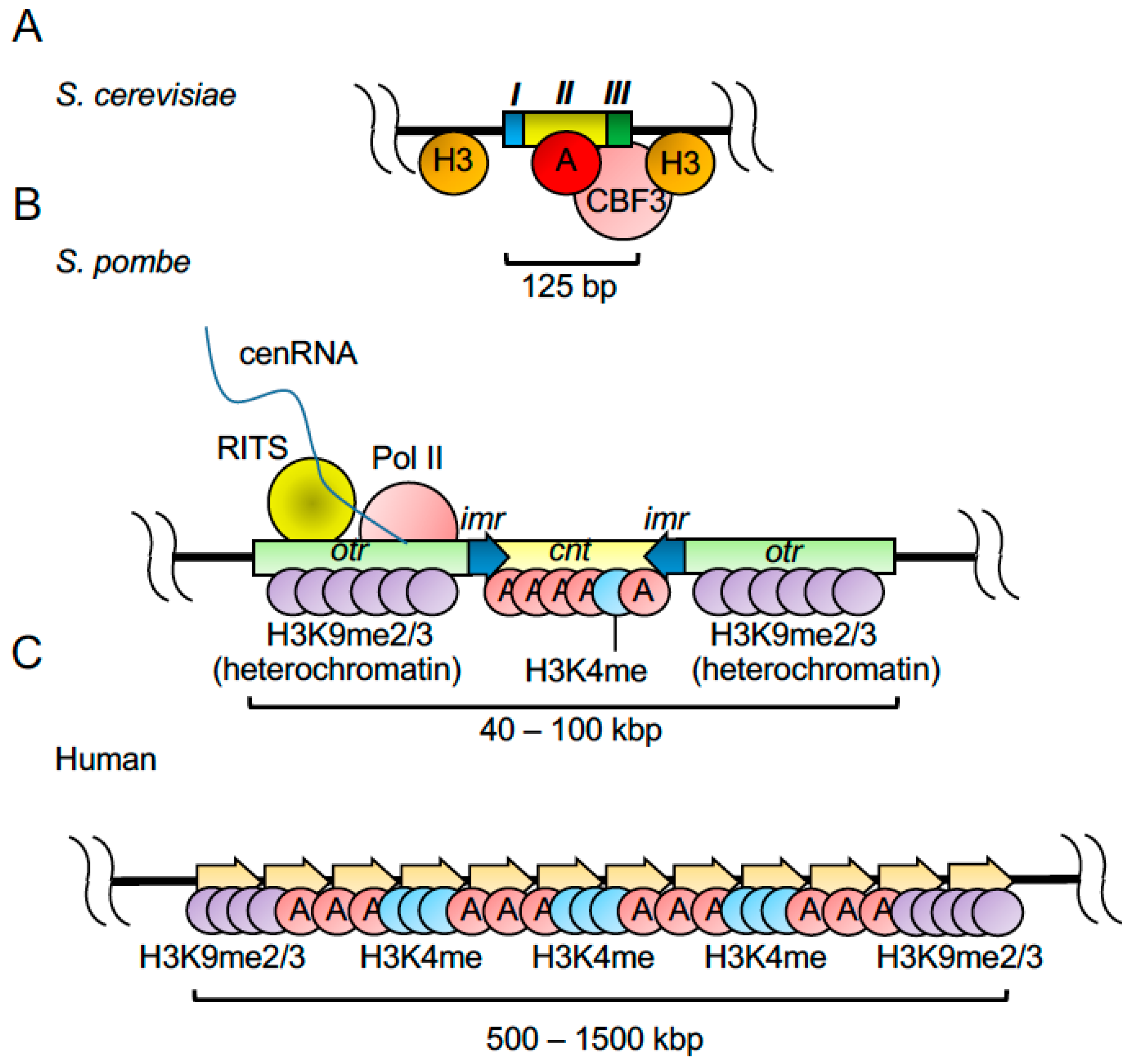
2.3. Centromeres
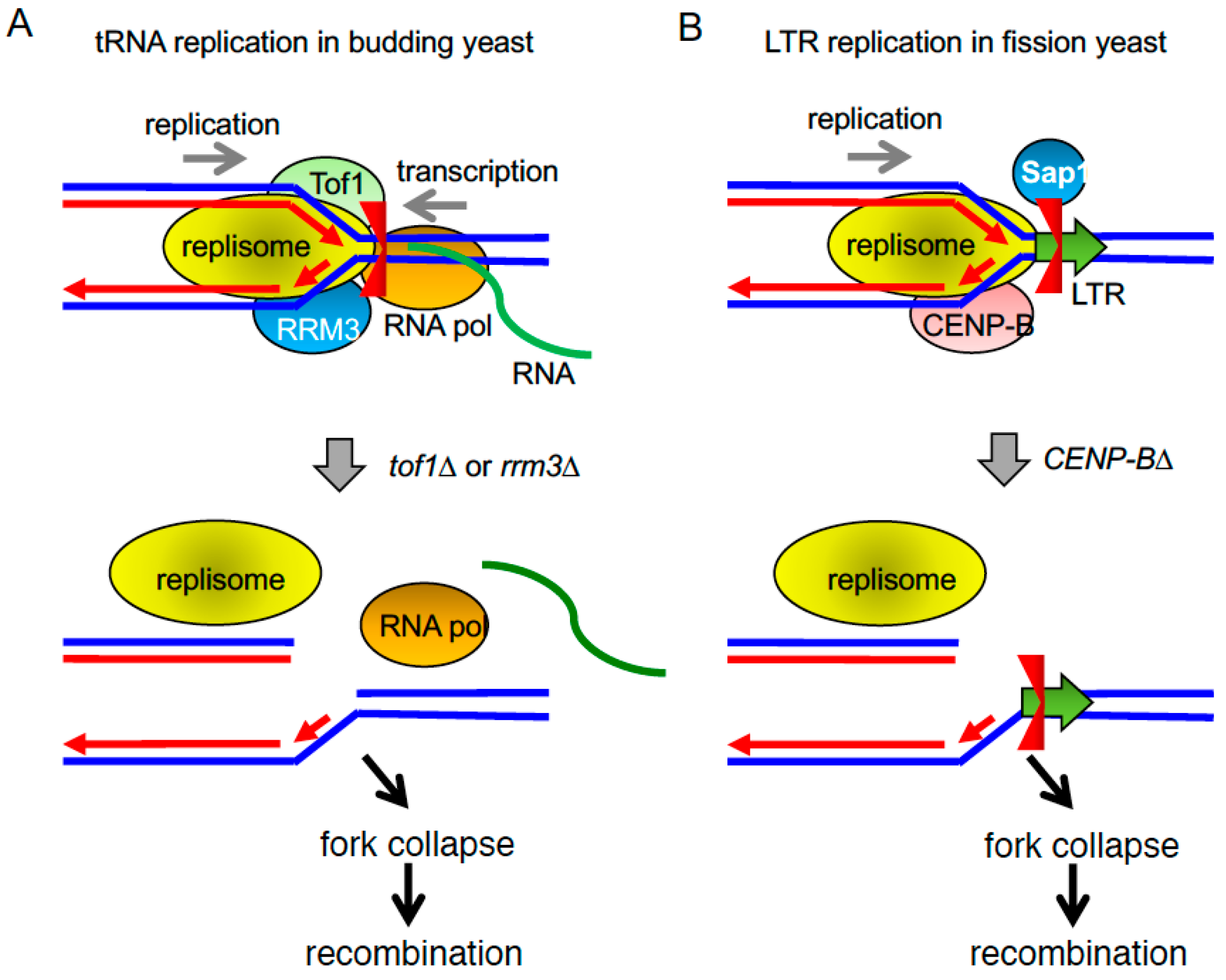
2.4. tRNA Genes and LTR Retrotransposons
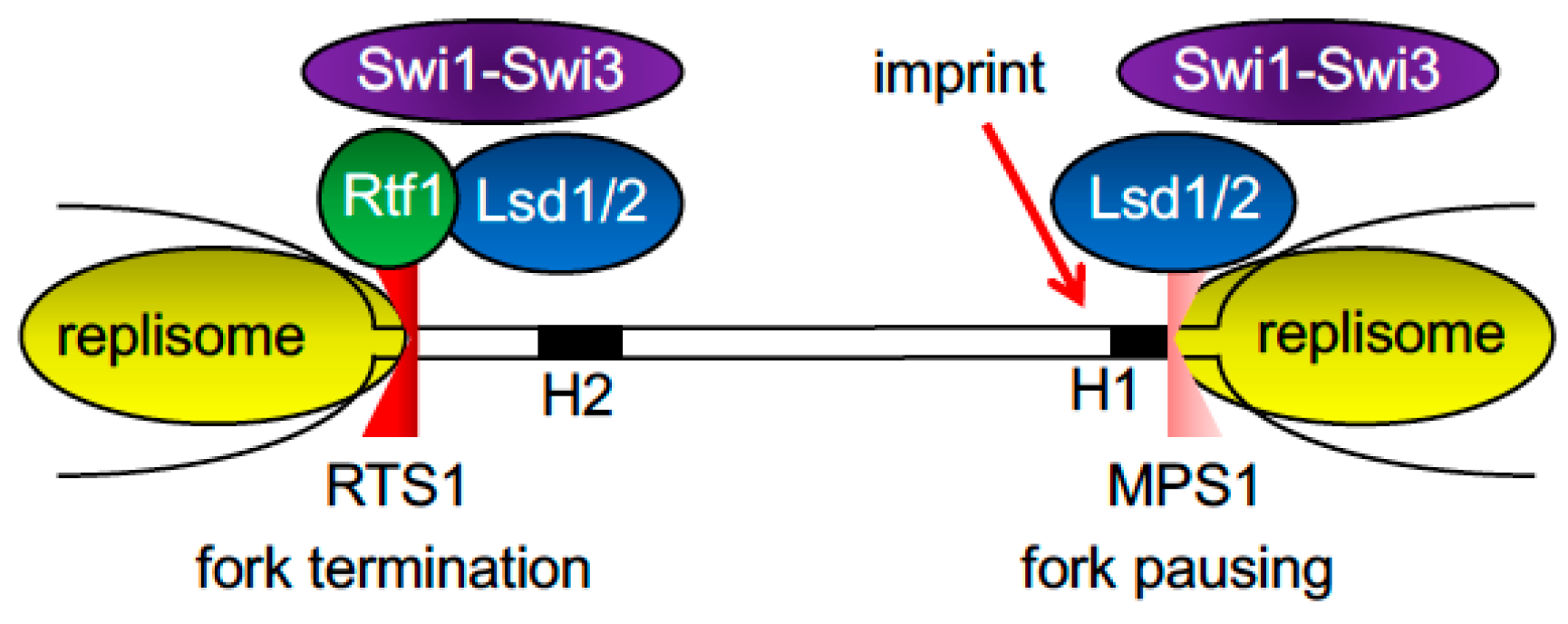
2.5. Fork Pausing and Termination at the Fission Yeast Mating-Type Locus
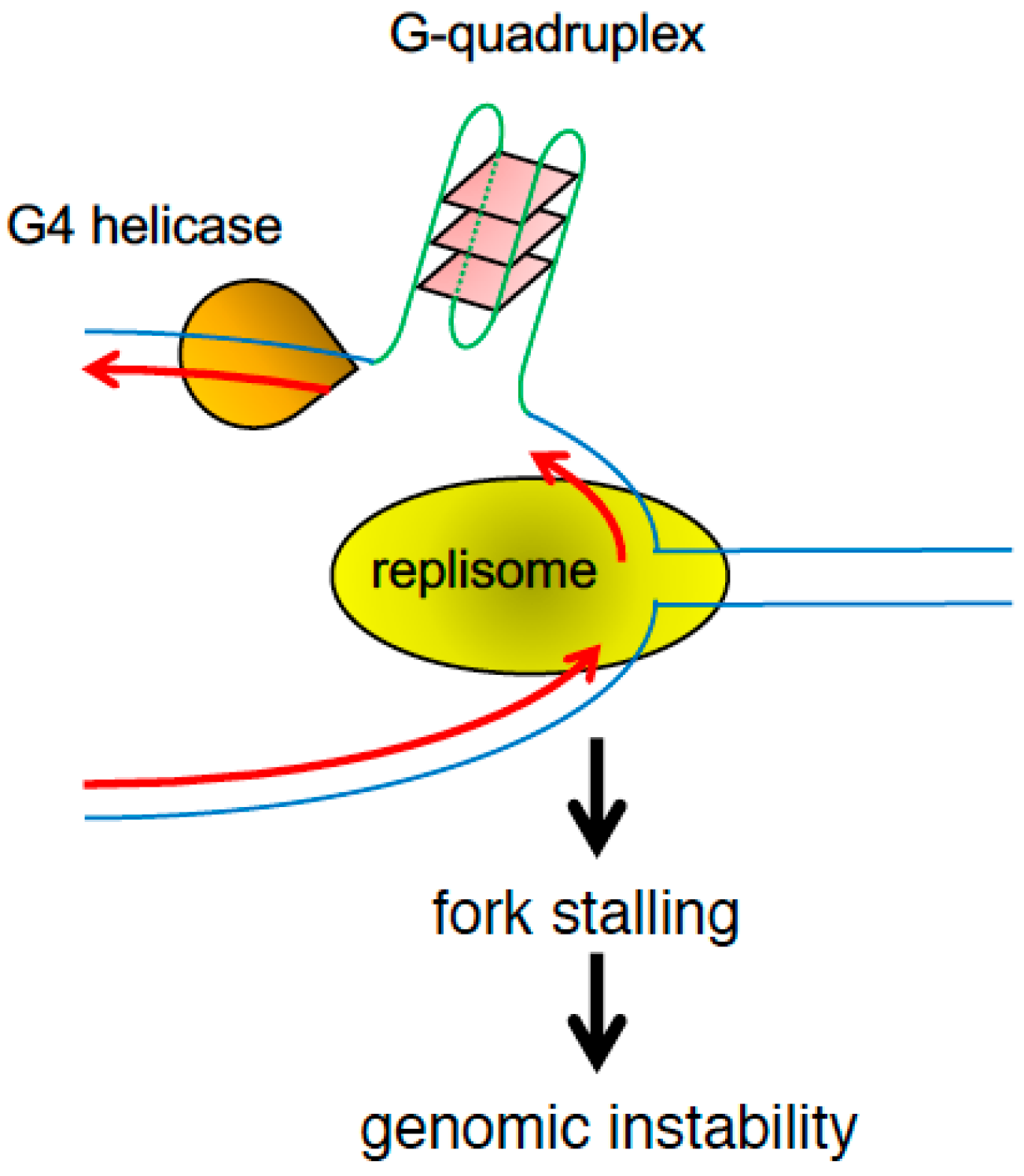
2.6. DNA Barriers Mediated by Repetitive DNA and Secondary Structures
3. Coordination between Transcription and Replication Machineries
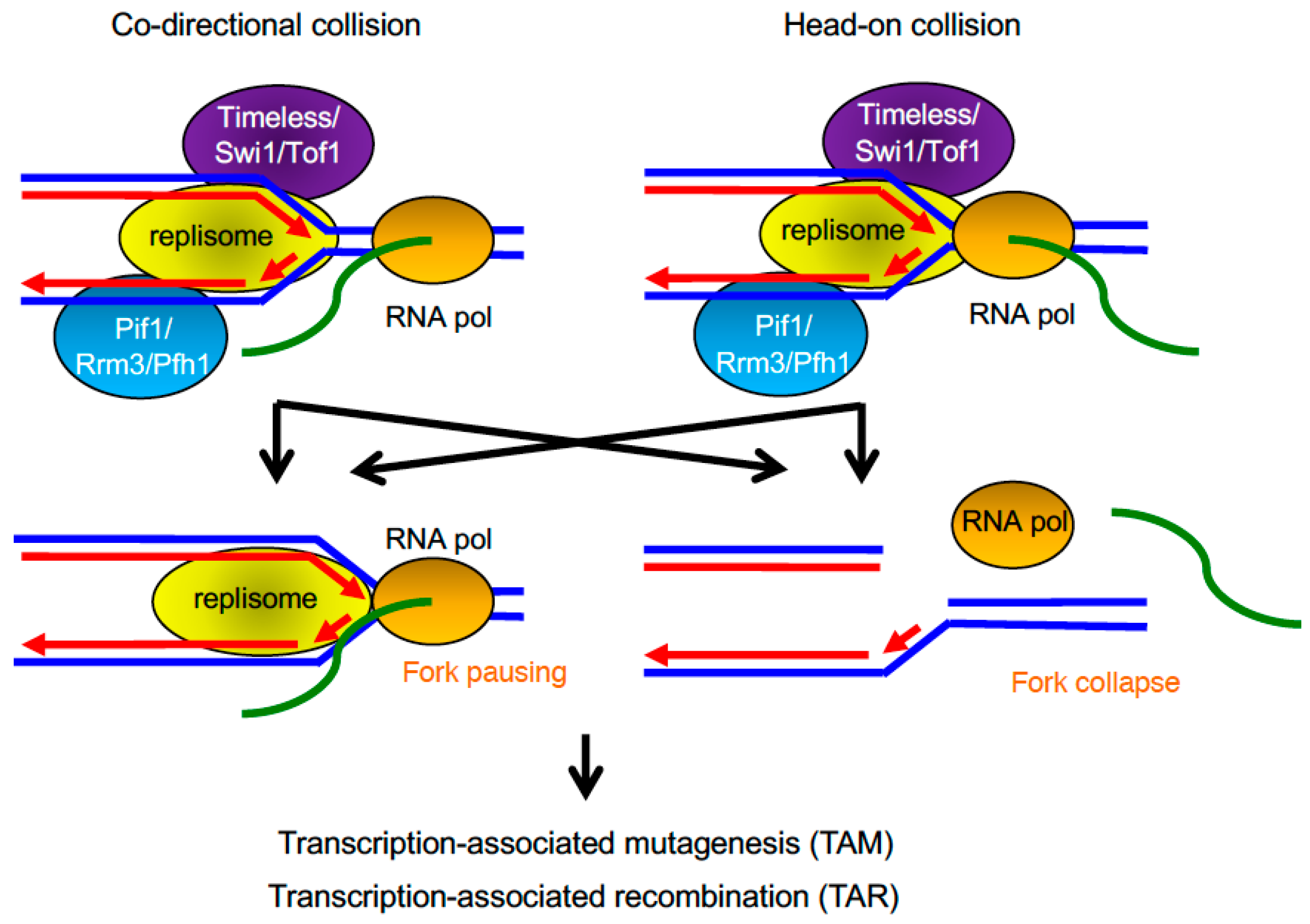
3.1. Transcription–Replication Encounters
3.2. Highly Transcribed Regions as Replicative Obstacles
3.3. Transcription-Associated Mutagenesis and Recombination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CBF3 | centromere binding factor 3 |
| CENP-A | centromere protein-A |
| CENP-B | centromere protein-B DSB: Double strand break |
| CMG | Cdc45-MCM-GINS |
| DTR | direct tandem repeat |
| FA | Fanconi Anemia |
| FPC | fork protection complex |
| IR | inverted repeat |
| LTR | long terminal repeat |
| MPS1 | mat1 pausing site 1 |
| MR | mirror repeat |
| RFB | replication fork barrier |
| RNA Pol II | RNA polymerase II |
| RNA Pol III | RNA polymerase III |
| RTS1 | replication termination site 1 |
| ssDNA | single-stranded DNA |
| TAM | transcription-associated mutagenesis |
| TAR | transcription-associated recombination |
| TTF-1 | transcription termination factor-I |
| TTS | transcription termination site |
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef]
- Lambert, S.; Carr, A.M. Replication stress and genome rearrangements: Lessons from yeast models. Curr. Opin. Genet. Dev. 2013, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Egel, R. Fission yeast mating-type switching: Programmed damage and repair. DNA Repair 2005, 4, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gaillard, H. Transcription and recombination: When RNA meets DNA. Cold Spring Harb. Perspect. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Rog, O.; Cooper, J.P. Semi-conservative DNA replication through telomeres requires Taz1. Nature 2006, 440, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Segurado, M.; de Luis, A.; Antequera, F. Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 2003, 4, 1048–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosopoulos, W.C.; Kosiyatrakul, S.T.; Yan, Z.; Calderano, S.G.; Schildkraut, C.L. Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J. Cell Biol. 2012, 197, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Kurth, I.; Gautier, J. Origin-dependent initiation of DNA replication within telomeric sequences. Nucleic Acids Res. 2010, 38, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ivessa, A.S.; Zhou, J.Q.; Schulz, V.P.; Monson, E.K.; Zakian, V.A. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 2002, 16, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.C.; Das, M.M.; Tanizawa, H.; Chang, Y.T.; Noma, K.; Nakamura, T.M.; Noguchi, E. Swi1timeless prevents repeat instability at fission yeast telomeres. PLoS Genet. 2016, 12, e1005943. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Noguchi, E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 2012, 11, 3945–3955. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, E.; Noguchi, C.; McDonald, W.H.; Yates, J.R., 3rd; Russell, P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 2004, 24, 8342–8355. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.C.; González-Medina, A.; Noguchi, E. Timeless protection of telomeres. Curr. Genet. 2016, 62, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Urtishak, K.A.; Smith, K.D.; Chanoux, R.A.; Greenberg, R.A.; Johnson, F.B.; Brown, E.J. Timeless maintains genomic stability and suppresses sister chromatid exchange during unperturbed DNA replication. J. Biol. Chem. 2009, 284, 8777–8785. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Dheekollu, J.; Deng, Z.; Lee, S.W.; Das, M.M.; Lieberman, P.M.; Noguchi, E. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle 2012, 11, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; Nimmo, E.R.; Allshire, R.C.; Cech, T.R. Regulation of telomere length and function by a Myb-domain protein in fission yeast. Nature 1997, 385, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Ishikawa, F. Telomere-bound TRF1 and TRF2 stall the replication fork at telomeric repeats. Nucleic Acids Res. 2004, 32, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F. Portrait of replication stress viewed from telomeres. Cancer Sci. 2013, 104, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Makovets, S.; Herskowitz, I.; Blackburn, E.H. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol. Cell. Biol. 2004, 24, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Kedziora, S.; Donaldson, A.D. At short telomeres Tel1 directs early replication and phosphorylates Rif1. PLoS Genet. 2014, 10, e1004691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Kibe, T.; Kabir, S.; de Lange, T. TRF1 negotiates ttaggg repeat-associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes Dev. 2014, 28, 2477–2491. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Wu, W.Q.; Duan, X.L.; Liu, N.N.; Li, H.H.; Fu, J.; Dou, S.X.; Li, M.; Xi, X.G. Molecular mechanism of G-quadruplex unwinding helicase: Sequential and repetitive unfolding of G-quadruplex by Pif1 helicase. Biochem. J. 2015, 466, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, N.; Capra, J.A.; Zakian, V.A. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Estrano, C.; Schvartzman, J.B.; Krimer, D.B.; Hernandez, P. Characterization of the pea rDNA replication fork barrier: Putative cis-acting and trans-acting factors. Plant Mol. Biol. 1999, 40, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Martin-Parras, L.; Martinez-Robles, M.L.; Schvartzman, J.B. Conserved features in the mode of replication of eukaryotic ribosomal RNA genes. EMBO J. 1993, 12, 1475–1485. [Google Scholar] [PubMed]
- MacAlpine, D.M.; Zhang, Z.; Kapler, G.M. Type I elements mediate replication fork pausing at conserved upstream sites in the tetrahymena thermophila ribosomal DNA minichromosome. Mol. Cell. Biol. 1997, 17, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Hyrien, O.; Mechali, M. Chromosomal replication initiates and terminates at random sequences but at regular intervals in the ribosomal DNA of Xenopus early embryos. EMBO J. 1993, 12, 4511–4520. [Google Scholar] [PubMed]
- Lopez-estrano, C.; Schvartzman, J.B.; Krimer, D.B.; Hernandez, P. Co-localization of polar replication fork barriers and rRNA transcription terminators in mouse rDNA. J. Mol. Biol. 1998, 277, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Brewer, B.J.; Fangman, W.L. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 1988, 55, 637–643. [Google Scholar] [CrossRef]
- Linskens, M.H.; Huberman, J.A. Organization of replication of ribosomal DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1988, 8, 4927–4935. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Platt, T.H.; Schildkraut, C.L. Initiation and termination of DNA replication in human rRNA genes. Mol. Cell. Biol. 1993, 13, 6600–6613. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.K.; Gogel, E.; Berger, C.; Wallisch, M.; Muller, F.; Grummt, I.; Grummt, F. Termination of mammalian rDNA replication: Polar arrest of replication fork movement by transcription termination factor TTF-I. Cell 1997, 90, 559–567. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Kim, S.M.; Huberman, J.A. Ribosomal DNA replication in the fission yeast, Schizosaccharomyces pombe. Exp. Cell Res. 1998, 238, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Krings, G.; Bastia, D. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 2004, 101, 14085–14090. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gorostiaga, A.; Lopez-Estrano, C.; Krimer, D.B.; Schvartzman, J.B.; Hernandez, P. Transcription termination factor Reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol. Cell. Biol. 2004, 24, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Krings, G.; Bastia, D. Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J. Biol. Chem. 2005, 280, 39135–39142. [Google Scholar] [CrossRef] [PubMed]
- Arcangioli, B.; Klar, A.J. A novel switch-activating site (SAS1) and its cognate binding factor (SAP1) required for efficient mat1 switching in Schizosaccharomyces pombe. EMBO J. 1991, 10, 3025–3032. [Google Scholar] [PubMed]
- Ghazvini, M.; Ribes, V.; Arcangioli, B. The essential DNA-binding protein Sap1 of schizosaccharomyces pombe contains two independent oligomerization interfaces that dictate the relative orientation of the DNA-binding domain. Mol. Cell. Biol. 1995, 15, 4939–4946. [Google Scholar] [CrossRef] [PubMed]
- de Lahondes, R.; Ribes, V.; Arcangioli, B. Fission yeast Sap1 protein is essential for chromosome stability. Eukaryot Cell 2003, 2, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.; Noguchi, E. Sap1 promotes the association of the replication fork protection complex with chromatin and is involved in the replication checkpoint in Schizosaccharomyces pombe. Genetics 2007, 175, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Ramirez, E.; Sanchez-Gorostiaga, A.; Krimer, D.B.; Schvartzman, J.B.; Hernandez, P. The mating type switch-activating protein Sap1 is required for replication fork arrest at the rRNA genes of fission yeast. Mol. Cell. Biol. 2005, 25, 8755–8761. [Google Scholar] [CrossRef] [PubMed]
- Zaratiegui, M.; Vaughn, M.W.; Irvine, D.V.; Goto, D.; Watt, S.; Bahler, J.; Arcangioli, B.; Martienssen, R.A. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 2011, 469, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Krings, G.; Bastia, D. Molecular architecture of a eukaryotic DNA replication terminus-terminator protein complex. Mol. Cell. Biol. 2006, 26, 8061–8074. [Google Scholar] [CrossRef]
- Singh, S.K.; Sabatinos, S.; Forsburg, S.; Bastia, D. Regulation of replication termination by Reb1 protein-mediated action at a distance. Cell 2010, 142, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Bastia, D. Mechanistic insights into replication termination as revealed by investigations of the Reb1-Ter3 complex of Schizosaccharomyces pombe. Mol. Cell. Biol. 2008, 28, 6844–6857. [Google Scholar] [CrossRef] [PubMed]
- Melekhovets, Y.F.; Shwed, P.S.; Nazar, R.N. In vivo analyses of RNA polymerase I termination in Schizosaccharomyces pombe. Nucleic Acids Res. 1997, 25, 5103–5109. [Google Scholar] [CrossRef] [PubMed]
- Bastia, D.; Singh, S.K. “Chromosome kissing” and modulation of replication termination. Bioarchitecture 2011, 1, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sanchez, L.; Rodriguez-Lopez, M.; Garcia, Z.; Tenorio-Gomez, M.; Schvartzman, J.B.; Krimer, D.B.; Hernandez, P. The fission yeast rDNA-binding protein Reb1 regulates G1 phase under nutritional stress. J. Cell Sci. 2011, 124, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Zaman, S.; Jiang, J.C.; Jazwinski, S.M.; Bastia, D. Mechanism of regulation of ‘chromosome kissing’ induced by Fob1 and its physiological significance. Genes Dev. 2015, 29, 1188–1201. [Google Scholar] [CrossRef]
- Huang, J.; Moazed, D. Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev. 2003, 17, 2162–2176. [Google Scholar] [CrossRef] [PubMed]
- Shou, W.; Seol, J.H.; Shevchenko, A.; Baskerville, C.; Moazed, D.; Chen, Z.W.; Jang, J.; Shevchenko, A.; Charbonneau, H.; Deshaies, R.J. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell 1999, 97, 233–244. [Google Scholar] [CrossRef]
- Straight, A.F.; Shou, W.; Dowd, G.J.; Turck, C.W.; Deshaies, R.J.; Johnson, A.D.; Moazed, D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 1999, 97, 245–256. [Google Scholar] [CrossRef]
- Kobayashi, T.; Horiuchi, T.; Tongaonkar, P.; Vu, L.; Nomura, M. Sir2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 2004, 117, 441–453. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ganley, A.R. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 2005, 309, 1581–1584. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, N.K.; Zzaman, S.; Mohanty, B.K.; Bastia, D. Replication fork arrest and rDNA silencing are two independent and separable functions of the replication terminator protein Fob1 of Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 12612–12619. [Google Scholar] [CrossRef] [PubMed]
- Jakociunas, T.; Domange Jordo, M.; Ait Mebarek, M.; Bunner, C.M.; Verhein-Hansen, J.; Oddershede, L.B.; Thon, G. Subnuclear relocalization and silencing of a chromosomal region by an ectopic ribosomal DNA repeat. Proc. Natl. Acad. Sci. USA 2013, 110, E4465–E4473. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I.; Maier, U.; Ohrlein, A.; Hassouna, N.; Bachellerie, J.P. Transcription of mouse rDNA terminates downstream of the 3′ end of 28S RNA and involves interaction of factors with repeated sequences in the 3′ spacer. Cell 1985, 43, 801–810. [Google Scholar] [CrossRef]
- Bartsch, I.; Schoneberg, C.; Grummt, I. Evolutionary changes of sequences and factors that direct transcription termination of human and mouse ribsomal genes. Mol. Cell. Biol. 1987, 7, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Grummt, I.; Rosenbauer, H.; Niedermeyer, I.; Maier, U.; Ohrlein, A. A repeated 18 bp sequence motif in the mouse rDNA spacer mediates binding of a nuclear factor and transcription termination. Cell 1986, 45, 837–846. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Kobayashi, T. The human RNA polymerase I transcription terminator complex acts as a replication fork barrier that coordinates the progress of replication with rRNA transcription activity. Mol. Cell. Biol. 2015, 35, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Bensimon, A. DNA replication origin plasticity and perturbed fork progression in human inverted repeats. Mol. Cell. Biol. 2005, 25, 6789–6797. [Google Scholar] [CrossRef] [PubMed]
- Bastia, D.; Srivastava, P.; Zaman, S.; Choudhury, M.; Mohanty, B.K.; Bacal, J.; Langston, L.D.; Pasero, P.; O’Donnell, M.E. Phosphorylation of CMG helicase and Tof1 is required for programmed fork arrest. Proc. Natl. Acad. Sci. USA 2016, 113, E3639–E3648. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.K.; Bairwa, N.K.; Bastia, D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2006, 103, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.K.; Bairwa, N.K.; Bastia, D. Contrasting roles of checkpoint proteins as recombination modulators at Fob1-Ter complexes with or without fork arrest. Eukaryot Cell 2009, 8, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.H.; Kang, Y.H.; An, Y.Y.; Tappin, I.; Hurwitz, J.; Lee, J.K. Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. Proc. Natl. Acad. Sci. USA 2013, 110, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Calzada, A.; Hodgson, B.; Kanemaki, M.; Bueno, A.; Labib, K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005, 19, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Katou, Y.; Kanoh, Y.; Bando, M.; Noguchi, H.; Tanaka, H.; Ashikari, T.; Sugimoto, K.; Shirahige, K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 2003, 424, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Zech, J.; Godfrey, E.L.; Masai, H.; Hartsuiker, E.; Dalgaard, J.Z. The DNA-binding domain of S. pombe Mrc1 (claspin) acts to enhance stalling at replication barriers. PLoS ONE 2015, 10, e0132595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, S.E.; Ren, J.; Bhattacharjee, S.; Chang, A.Y.; Sanchez, M.; Valbuena, A.; Antequera, F.; Martienssen, R.A. Dicer promotes transcription termination at sites of replication stress to maintain genome stability. Cell 2014, 159, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Zaratiegui, M.; Castel, S.E.; Irvine, D.V.; Kloc, A.; Ren, J.; Li, F.; de Castro, E.; Marin, L.; Chang, A.Y.; Goto, D.; et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA pol II. Nature 2011, 479, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.F.; Mackay, A.M.; Ainsztein, A.M.; Goldberg, I.G.; Earnshaw, W.C. The centromere: Hub of chromosomal activities. Science 1995, 270, 1591–1594. [Google Scholar] [CrossRef] [PubMed]
- Amor, D.J.; Kalitsis, P.; Sumer, H.; Choo, K.H. Building the centromere: From foundation proteins to 3D organization. Trends Cell Biol. 2004, 14, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A.; Moazed, D. Centromere assembly and propagation. Cell 2007, 128, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Aneuploidy: Instigator and inhibitor of tumorigenesis. Cancer Res. 2007, 67, 10103–10105. [Google Scholar] [CrossRef] [PubMed]
- Greenfeder, S.A.; Newlon, C.S. Replication forks pause at yeast centromeres. Mol. Cell. Biol. 1992, 12, 4056–4066. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Caddle, M.S.; Bulboaca, G.H.; Wohlgemuth, J.G.; Baum, M.; Clarke, L.; Calos, M.P. Replication of centromere II of Schizosaccharomyces pombe. Mol. Cell. Biol. 1995, 15, 5165–5172. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.R.; Grishchuk, E.L.; West, R.R. Chromosome-microtubule interactions during mitosis. Annu. Rev. Cell Dev. Biol. 2002, 18, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Yeh, E. Tension management in the kinetochore. Curr. Biol. 2010, 20, R1040–R1048. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, T.; Earnshaw, W.C. The centromere: Chromatin foundation for the kinetochore machinery. Dev. Cell 2014, 30, 496–508. [Google Scholar] [CrossRef]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Hodgson, B.; Calzada, A.; Labib, K. Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol. Biol. Cell 2007, 18, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- McAinsh, A.D.; Tytell, J.D.; Sorger, P.K. Structure, function, and regulation of budding yeast kinetochores. Annu. Rev. Cell Dev. Biol. 2003, 19, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Gomez-Raja, J.; Larriba, G.; Dubey, D.D.; Sanyal, K. Rad51-Rad52 mediated maintenance of centromeric chromatin in candida albicans. PLoS Genet. 2014, 10, e1004344. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.W.; Straight, A.F. Centromere formation: From epigenetics to self-assembly. Trends Cell Biol. 2006, 16, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Leman, A.R.; Noguchi, E. The replication fork: Understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes 2013, 4, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Dubey, D.D.; Huberman, J.A. Early-replicating heterochromatin. Genes Dev. 2003, 17, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Takahashi, T.S.; Nakagawa, T.; Nakayama, J.; Masukata, H. The heterochromatin protein Swi6/Hp1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat. Cell Biol. 2009, 11, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Chretien, L.; Cote, J.; Kelly, T.J.; Forsburg, S.L. S. pombe replication protein Cdc18 (Cdc6) interacts with Swi6 (HP1) heterochromatin protein: Region specific effects and replication timing in the centromere. Cell Cycle 2011, 10, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, M.; Seth, M.; Lee, H.C.; Gu, W.; Ishidate, T.; Conte, D., Jr.; Mello, C.C. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell 2012, 150, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Petreaca, R.C.; Jensen, A.; Yuan, J.P.; Green, M.D.; Forsburg, S.L. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 2013, 3, 638–645. [Google Scholar] [CrossRef]
- Errico, A.; Aze, A.; Costanzo, V. Mta2 promotes Tipin-dependent maintenance of replication fork integrity. Cell Cycle 2014, 13, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Bloom, M.S.; Solanki-Patil, T.; Smith, S.; Sipahimalani, P.; Li, Z.; Kofoed, M.; Ben-Aroya, S.; Myung, K.; Hieter, P. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011, 7, e1002057. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Vizeacoumar, F.J.; Bahr, S.; Li, J.; Warringer, J.; Vizeacoumar, F.S.; Min, R.; Vandersluis, B.; Bellay, J.; Devit, M.; et al. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotechnol. 2011, 29, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.; Vaisica, J.A.; Ou, J.; Baryshnikova, A.; Lu, Y.; Roth, F.P.; Brown, G.W. Genome rearrangements caused by depletion of essential DNA replication proteins in Saccharomyces cerevisiae. Genetics 2012, 192, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, F.J.; Degtyareva, N.P.; Lobachev, K.; Petes, T.D. Chromosomal translocations in yeast induced by low levels of DNA polymerase: A model for chromosome fragile sites. Cell 2005, 120, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, F.J.; Degtyareva, N.P.; Kokoska, R.J.; Petes, T.D. Reduced levels of DNA polymerase δ induce chromosome fragile site instability in yeast. Mol. Cell. Biol. 2008, 28, 5359–5368. [Google Scholar] [CrossRef] [PubMed]
- Admire, A.; Shanks, L.; Danzl, N.; Wang, M.; Weier, U.; Stevens, W.; Hunt, E.; Weinert, T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006, 20, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Roeder, G.S.; Fink, G.R. DNA rearrangements associated with a transposable element in yeast. Cell 1980, 21, 239–249. [Google Scholar] [CrossRef]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.M.; Newlon, C.S. DNA replication fork pause sites dependent on transcription. Science 1996, 272, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Soragni, E.; Kassavetis, G.A. Absolute gene occupancies by RNA polymerase III, TFIIIB, and TFIIIC in Saccharomyces cerevisiae. J. Biol. Chem. 2008, 283, 26568–26576. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. Trnascan-se: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M. Genomic tRNA Database. Available online: http://lowelab.ucsc.edu/GtRNAdb/ (accessed on 20 Novermber 2016).
- McFarlane, R.J.; Whitehall, S.K. tRNA genes in eukaryotic genome organization and reorganization. Cell Cycle 2009, 8, 3102–3106. [Google Scholar] [CrossRef] [PubMed]
- Haldar, D.; Kamakaka, R.T. tRNA genes as chromatin barriers. Nat. Struct. Mol. Biol. 2006, 13, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Pryce, D.W.; Ramayah, S.; Jaendling, A.; McFarlane, R.J. Recombination at DNA replication fork barriers is not universal and is differentially regulated by Swi1. Proc. Natl. Acad. Sci. USA 2009, 106, 4770–4775. [Google Scholar] [CrossRef]
- Usmanova MN, T.N. Bioinformatic analysis of retroelement-associated sequences in human and mouse promoters. Proc. World Acad. Sci. Eng. Technol. 2008, 34, 553–561. [Google Scholar]
- Bochman, M.L.; Judge, C.P.; Zakian, V.A. The Pif1 family in prokaryotes: What are our helicases doing in your bacteria? Mol. Biol. Cell 2011, 22, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- de la Loza, M.C.; Wellinger, R.E.; Aguilera, A. Stimulation of direct-repeat recombination by RNA polymerase III transcription. DNA Repair 2009, 8, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Dunaway, S.; Torres, J.Z.; Bessler, J.B.; Zakian, V.A. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006, 20, 3104–3116. [Google Scholar] [CrossRef] [PubMed]
- Steinacher, R.; Osman, F.; Dalgaard, J.Z.; Lorenz, A.; Whitby, M.C. The DNA helicase Pfh1 promotes fork merging at replication termination sites to ensure genome stability. Genes Dev. 2012, 26, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, K.R.; Guise, A.J.; Pourbozorgi-Langroudi, P.; Cristea, I.M.; Zakian, V.A.; Capra, J.A.; Sabouri, N. Pfh1 is an accessory replicative helicase that interacts with the replisome to facilitate fork progression and preserve genome integrity. PLoS Genet. 2016, 12, e1006238. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Haeusler, R.A.; Good, P.D.; Engelke, D.R. Nucleolar clustering of dispersed tRNA genes. Science 2003, 302, 1399–1401. [Google Scholar] [CrossRef] [PubMed]
- Rozenzhak, S.; Mejia-Ramirez, E.; Williams, J.S.; Schaffer, L.; Hammond, J.A.; Head, S.R.; Russell, P. Rad3 decorates critical chromosomal domains with γH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet. 2010, 6, e1001032. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H.; Boeke, J.D. Human transposon tectonics. Cell 2012, 149, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Bowen, N.J.; Jordan, I.K.; Epstein, J.A.; Wood, V.; Levin, H.L. Retrotransposons and their recognition of pol II promoters: A comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res. 2003, 13, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.Z.; Rosado-Lugo, J.D.; Cranz-Mileva, S.; Ciccaglione, K.M.; Tournier, V.; Zaratiegui, M. Arrested replication forks guide retrotransposon integration. Science 2015, 349, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.G.; Esnault, C.; Guo, Y.; Hung, S.; McQueen, P.G.; Levin, H.L. Serial number tagging reveals a prominent sequence preference of retrotransposon integration. Nucleic Acids Res. 2014, 42, 8449–8460. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Levin, H.L. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 2010, 20, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Tsankov, A.; Yanagisawa, Y.; Rhind, N.; Regev, A.; Rando, O.J. Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res. 2011, 21, 1851–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cam, H.P.; Noma, K.; Ebina, H.; Levin, H.L.; Grewal, S.I. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature 2008, 451, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Leem, Y.E.; Levin, H.L. Transposon integration enhances expression of stress response genes. Nucleic Acids Res. 2013, 41, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Baller, J.A.; Gao, J.; Stamenova, R.; Curcio, M.J.; Voytas, D.F. A nucleosomal surface defines an integration hotspot for the Saccharomyces cerevisiae Ty1 retrotransposon. Genome Res. 2012, 22, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Bridier-Nahmias, A.; Tchalikian-Cosson, A.; Baller, J.A.; Menouni, R.; Fayol, H.; Flores, A.; Saib, A.; Werner, M.; Voytas, D.F.; Lesage, P. Retrotransposons. An RNA polymerase III subunit determines sites of retrotransposon integration. Science 2015, 348, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Mularoni, L.; Zhou, Y.; Bowen, T.; Gangadharan, S.; Wheelan, S.J.; Boeke, J.D. Retrotransposon Ty1 integration targets specifically positioned asymmetric nucleosomal DNA segments in tRNA hotspots. Genome Res. 2012, 22, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, J.Z.; Klar, A.J. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 2000, 102, 745–751. [Google Scholar] [CrossRef]
- Eydmann, T.; Sommariva, E.; Inagawa, T.; Mian, S.; Klar, A.J.; Dalgaard, J.Z. Rtf1-mediated eukaryotic site-specific replication termination. Genetics 2008, 180, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kaykov, A.; Holmes, A.M.; Arcangioli, B. Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast. EMBO J. 2004, 23, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Vengrova, S.; Dalgaard, J.Z. RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching. Genes Dev. 2004, 18, 794–804. [Google Scholar] [CrossRef]
- Holmes, A.; Roseaulin, L.; Schurra, C.; Waxin, H.; Lambert, S.; Zaratiegui, M.; Martienssen, R.A.; Arcangioli, B. Lsd1 and Lsd2 control programmed replication fork pauses and imprinting in fission yeast. Cell Rep. 2012, 2, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. Lsd1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Lee, M.G.; Hakimi, M.A.; Cam, H.P.; Grewal, S.I.; Shiekhattar, R. Fission yeast homologs of human histone H3 lysine 4 demethylase regulate a common set of genes with diverse functions. J. Biol. Chem. 2006, 281, 35983–35988. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog Lsd1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; Holt, D.G.; Panigrahi, A.; Wilhelm, B.T.; Erdjument-Bromage, H.; Tempst, P.; Bahler, J.; Cairns, B.R. Genome-wide dynamics of SAPHIRE, an essential complex for gene activation and chromatin boundaries. Mol. Cell. Biol. 2007, 27, 4058–4069. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Zaratiegui, M.; Villen, J.; Vaughn, M.W.; Verdel, A.; Huarte, M.; Shi, Y.; Gygi, S.P.; Moazed, D.; Martienssen, R.A.; et al. S. pombe Lsd1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol. Cell 2007, 26, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.; Mirkin, S.M. Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(ccg)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- The huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Yamagata, T.; Burgess, D.L.; Rasmussen, A.; Grewal, R.P.; Watase, K.; Khajavi, M.; McCall, A.E.; Davis, C.F.; Zu, L.; et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000, 26, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Scott, H.S.; Buresi, C.; Rossier, C.; Bottani, A.; Morris, M.A.; Malafosse, A.; Antonarakis, S.E. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 1997, 386, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Hile, S.E.; Eckert, K.A. Positive correlation between DNA polymerase α-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J. Mol. Biol. 2004, 335, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Hoyne, P.R.; Maher, L.J., 3rd. Functional studies of potential intrastrand triplex elements in the Escherichia coli genome. J. Mol. Biol. 2002, 318, 373–386. [Google Scholar] [CrossRef]
- Krasilnikova, M.M.; Mirkin, S.M. Replication stalling at friedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Rey, L.; Wang, G.; Pillaire, M.J.; Puget, N.; Selves, J.; Biard, D.S.; Shin-ya, K.; Vasquez, K.M.; Cazaux, C.; et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol. Carcinog. 2009, 48, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Diviacco, S.; Rapozzi, V.; Xodo, L.; Helene, C.; Quadrifoglio, F.; Giovannangeli, C. Site-directed inhibition of DNA replication by triple helix formation. FASEB J. 2001, 15, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Myers, S.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J. Biol. Chem. 2012, 287, 33412–33423. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.P.; Lu, L.; Blaszak, R.T.; Bissler, J.J. PKD1 intron 21: Triplex DNA formation and effect on replication. Nucleic Acids Res. 2004, 32, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K.M. Models for chromosomal replication-independent non-B DNA structure-induced genetic instability. Mol. Carcinog. 2009, 48, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Paeschke, K.; Singh, M.; Zakian, V.A. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010, 6, e1000861. [Google Scholar] [CrossRef] [PubMed]
- Eddy, J.; Maizels, N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006, 34, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Hershman, S.G.; Chen, Q.; Lee, J.Y.; Kozak, M.L.; Yue, P.; Wang, L.S.; Johnson, F.B. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2008, 36, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Kudlicki, A.S. G-quadruplexes involving both strands of genomic DNA are highly abundant and colocalize with functional sites in the human genome. PLoS ONE 2016, 11, e0146174. [Google Scholar] [CrossRef] [PubMed]
- Kamath-Loeb, A.S.; Loeb, L.A.; Johansson, E.; Burgers, P.M.; Fry, M. Interactions between the Werner syndrome helicase and DNA polymerase δ specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001, 276, 16439–16446. [Google Scholar] [CrossRef] [PubMed]
- Woodford, K.J.; Howell, R.M.; Usdin, K. A novel K(+)-dependent DNA synthesis arrest site in a commonly occurring sequence motif in eukaryotes. J. Biol. Chem. 1994, 269, 27029–27035. [Google Scholar] [PubMed]
- Kruisselbrink, E.; Guryev, V.; Brouwer, K.; Pontier, D.B.; Cuppen, E.; Tijsterman, M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr. Biol. 2008, 18, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.D.; Fleetwood, S.; Berroyer, A.; Kim, N.; Larson, E.D. Sites of instability in the human TCF3 (E2A) gene adopt G-quadruplex DNA structures in vitro. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.N.; Machwe, A.; Wang, Z.; Orren, D.K. Intramolecular telomeric G-quadruplexes dramatically inhibit DNA synthesis by replicative and translesion polymerases, revealing their potential to lead to genetic change. PLoS ONE 2014, 9, e80664. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.Q.; Sinden, R.R. Preferential DNA secondary structure mutagenesis in the lagging strand of replication in E. coli. Nature 1991, 352, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, O.; Bourdoncle, A.; Boule, J.B.; Brosh, R.M., Jr.; Mergny, J.L. G-quadruplexes and helicases. Nucleic Acids Res. 2016, 44, 1989–2006. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Walter, J.C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 2015, 37, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shin-ya, K.; Brosh, R.M., Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; George, F.; Kuper, J.; Hamon, F.; Shin-ya, K.; Teulade-Fichou, M.P.; Kisker, C.; Brosh, R.M., Jr. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J. Biol. Chem. 2013, 288, 28217–28229. [Google Scholar] [CrossRef]
- Castillo Bosch, P.; Segura-Bayona, S.; Koole, W.; van Heteren, J.T.; Dewar, J.M.; Tijsterman, M.; Knipscheer, P. FANCJ promotes DNA synthesis through G-quadruplex structures. EMBO J. 2014, 33, 2521–2533. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, W.C.; Kosiyatrakul, S.T.; Schildkraut, C.L. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J. Cell Biol. 2015, 210, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Suhasini, A.N.; Rawtani, N.A.; Wu, Y.; Sommers, J.A.; Sharma, S.; Mosedale, G.; North, P.S.; Cantor, S.B.; Hickson, I.D.; Brosh, R.M., Jr. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011, 30, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Murat, P.; Phillips, L.G.; Patel, K.J.; Balasubramanian, S.; Sale, J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012, 40, 1485–1498. [Google Scholar] [CrossRef] [PubMed]
- Wallgren, M.; Mohammad, J.B.; Yan, K.P.; Pourbozorgi-Langroudi, P.; Ebrahimi, M.; Sabouri, N. G-rich telomeric and ribosomal DNA sequences from the fission yeast genome form stable G-quadruplex DNA structures in vitro and are unwound by the Pfh1 DNA helicase. Nucleic Acids Res. 2016, 44, 6213–6231. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.L.; Liu, N.N.; Yang, Y.T.; Li, H.H.; Li, M.; Dou, S.X.; Xi, X.G. G-quadruplexes significantly stimulate Pif1 helicase-catalyzed duplex DNA unwinding. J. Biol. Chem. 2015, 290, 7722–7735. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, S.; Hurley, L.H. The role of G-quadruplex/i-motif secondary structures as cis-acting regulatory elements. Pure Appl. Chem. 2010, 82, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Besnard, E.; Babled, A.; Lapasset, L.; Milhavet, O.; Parrinello, H.; Dantec, C.; Marin, J.M.; Lemaitre, J.M. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012, 19, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, F.; Schlumpf, T.; Schmid, V.; Rohs, R.; Beisel, C.; Paro, R. High-resolution profiling of Drosophila replication start sites reveals a DNA shape and chromatin signature of metazoan origins. Cell Rep. 2015, 11, 821–834. [Google Scholar] [CrossRef]
- Valton, A.L.; Hassan-Zadeh, V.; Lema, I.; Boggetto, N.; Alberti, P.; Saintome, C.; Riou, J.F.; Prioleau, M.N. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014, 33, 732–746. [Google Scholar] [CrossRef]
- Gaillard, H.; Herrera-Moyano, E.; Aguilera, A. Transcription-associated genome instability. Chem. Rev. 2013, 113, 8638–8661. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, A.; Tehranchi, A.; MacAlpine, D.M.; Wang, J.D. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010, 6, e1000810. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D.; Ehrlich, S.D.; Michel, B. Transcription-induced deletions in plasmid vectors: M13 DNA replication as a source of instability. Mol. Gen. Genet. 1996, 252, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Alberts, B.M. Head-on collision between a DNA replication apparatus and RNA polymerase transcription complex. Science 1995, 267, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell. Biol. 2005, 25, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Jinks-Robertson, S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science 1995, 268, 1616–1619. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Abdulovic, A.L.; Gealy, R.; Lippert, M.J.; Jinks-Robertson, S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair 2007, 6, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- French, S. Consequences of replication fork movement through transcription units in vivo. Science 1992, 258, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Berkmen, M.B.; Grossman, A.D. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Postow, L.; Ullsperger, C.; Keller, R.W.; Bustamante, C.; Vologodskii, A.V.; Cozzarelli, N.R. Positive torsional strain causes the formation of a four-way junction at replication forks. J. Biol. Chem. 2001, 276, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Guy, L.; Roten, C.A. Genometric analyses of the organization of circular chromosomes: A universal pressure determines the direction of ribosomal RNA genes transcription relative to chromosome replication. Gene 2004, 340, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Nicolay, S.; Touchon, M.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Thermes, C. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007, 17, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Vieira, K.F.; Levings, P.P.; Hill, M.A.; Crusselle, V.J.; Kang, S.H.; Engel, J.D.; Bungert, J. Recruitment of transcription complexes to the β-globin gene locus in vivo and in vitro. J. Biol. Chem. 2004, 279, 50350–50357. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, E.; Borkovec, J.; Kovacik, L.; Svidenska, S.; Schrofel, A.; Skalnikova, M.; Svindrych, Z.; Krizek, P.; Ovesny, M.; Hagen, G.M.; et al. Separation of replication and transcription domains in nucleoli. J. Struct. Biol. 2014, 188, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Pliss, A.; Koberna, K.; Vecerova, J.; Malinsky, J.; Masata, M.; Fialova, M.; Raska, I.; Berezney, R. Spatio-temporal dynamics at rDNA foci: Global switching between DNA replication and transcription. J. Cell. Biochem. 2005, 94, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S. DNA replication initiation patterns and spatial dynamics of the human ribosomal RNA gene loci. J. Cell Sci. 2011, 124, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Verdun, D.; Roussel, P.; Gebrane-Younes, J. Emerging concepts of nucleolar assembly. J. Cell Sci. 2002, 115, 2265–2270. [Google Scholar] [PubMed]
- Busch, H.; Smetana, K. The Nucleolus; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Cabal, G.G.; Genovesio, A.; Rodriguez-Navarro, S.; Zimmer, C.; Gadal, O.; Lesne, A.; Buc, H.; Feuerbach-Fournier, F.; Olivo-Marin, J.C.; Hurt, E.C.; et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature 2006, 441, 770–773. [Google Scholar] [CrossRef]
- Casolari, J.M.; Brown, C.R.; Komili, S.; West, J.; Hieronymus, H.; Silver, P.A. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 2004, 117, 427–439. [Google Scholar] [CrossRef]
- Elias-Arnanz, M.; Salas, M. Bacteriophage ϕ29 DNA replication arrest caused by codirectional collisions with the transcription machinery. EMBO J. 1997, 16, 5775–5783. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Garcia-Rubio, M.L.; Wellinger, R.E.; Luna, R.; Aguilera, A. An hpr1 point mutation that impairs transcription and mrnp biogenesis without increasing recombination. Mol. Cell. Biol. 2006, 26, 7451–7465. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. The connection between transcription and genomic instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Chavez, S.; Aguilera, A. The yeast hpr1 gene has a functional role in transcriptional elongation that uncovers a novel source of genome instability. Genes Dev. 1997, 11, 3459–3470. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.B.; Struhl, K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell 2005, 17, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Rondon, A.G.; Jimeno, S.; Garcia-Rubio, M.; Aguilera, A. Molecular evidence that the eukaryotic THO/TREX complex is required for efficient transcription elongation. J. Biol. Chem. 2003, 278, 39037–39043. [Google Scholar] [CrossRef] [PubMed]
- Strasser, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondon, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Zenklusen, D.; Vinciguerra, P.; Wyss, J.C.; Stutz, F. Stable mRNP formation and export require cotranscriptional recruitment of the mRNA export factors Yra1p and Sub2p by Hpr1p. Mol. Cell. Biol. 2002, 22, 8241–8253. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; Hodgson, B. Replication fork barriers: Pausing for a break or stalling for time? EMBO Rep. 2007, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Omont, N.; Kepes, F. Transcription/replication collisions cause bacterial transcription units to be longer on the leading strand of replication. Bioinformatics 2004, 20, 2719–2725. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428. [Google Scholar] [CrossRef] [PubMed]
- McAvoy, S.; Ganapathiraju, S.C.; Ducharme-Smith, A.L.; Pritchett, J.R.; Kosari, F.; Perez, D.S.; Zhu, Y.; James, C.D.; Smith, D.I. Non-random inactivation of large common fragile site genes in different cancers. Cytogenet. Genome Res. 2007, 118, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, N.; McDonald, K.R.; Webb, C.J.; Cristea, I.M.; Zakian, V.A. DNA replication through hard-to-replicate sites, including both highly transcribed RNA pol II and pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 2012, 26, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Versini, G.; Cordon-Preciado, V.; Alabert, C.; Pasero, P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol. Cell 2005, 19, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Jinks-Robertson, S.; Bhagwat, A.S. Transcription-associated mutagenesis. Annu. Rev. Genet. 2014, 48, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Pybus, C.; Pedraza-Reyes, M.; Ross, C.A.; Martin, H.; Ona, K.; Yasbin, R.E.; Robleto, E. Transcription-associated mutation in Bacillus subtilis cells under stress. J. Bacteriol. 2010, 192, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Burguiere-Slezak, G.; Van der Kemp, P.A.; Boiteux, S. Topoisomerase 1 provokes the formation of short deletions in repeated sequences upon high transcription in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Hicks, W.M.; Kim, M.; Haber, J.E. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010, 329, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Keil, R.L.; Roeder, G.S. Cis-acting, recombination-stimulating activity in a fragment of the ribosomal DNA of S. cerevisiae. Cell 1984, 39, 377–386. [Google Scholar] [CrossRef]
- Grimm, C.; Schaer, P.; Munz, P.; Kohli, J. The strong adh1 promoter stimulates mitotic and meiotic recombination at the ade6 gene of Schizosaccharomyces pombe. Mol. Cell. Biol. 1991, 11, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.J.; Rothstein, R. Elevated recombination rates in transcriptionally active DNA. Cell 1989, 56, 619–630. [Google Scholar] [CrossRef]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.; Huertas, P.; Gonzalez-Barrera, S.; Aguilera, A. Recombinogenic effects of DNA-damaging agents are synergistically increased by transcription in Saccharomyces cerevisiae. New insights into transcription-associated recombination. Genetics 2003, 165, 457–466. [Google Scholar] [PubMed]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem. 1985, 54, 665–697. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Mizuguchi, G.; Hamiche, A.; Wu, C. A chromatin remodelling complex involved in transcription and DNA processing. Nature 2000, 406, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Garcia-Rubio, M.L.; Gonzalez-Aguilera, C.; Luna, R.; Aguilera, A. A genome-wide function of THSC/TREX-2 at active genes prevents transcription-replication collisions. Nucleic Acids Res. 2014, 42, 12000–12014. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030098
Gadaleta MC, Noguchi E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes. 2017; 8(3):98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030098
Chicago/Turabian StyleGadaleta, Mariana C., and Eishi Noguchi. 2017. "Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome" Genes 8, no. 3: 98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030098