
K-mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. K-mer Analysis
2.2. Local k-mer Analysis
2.3. Relative Spectra
2.4. Mapping
2.5. Correlation Heatmaps and Mean Correlations
2.6. Heatmap Summary Images
2.7. Software
3. Results
3.1. Viral Genomes
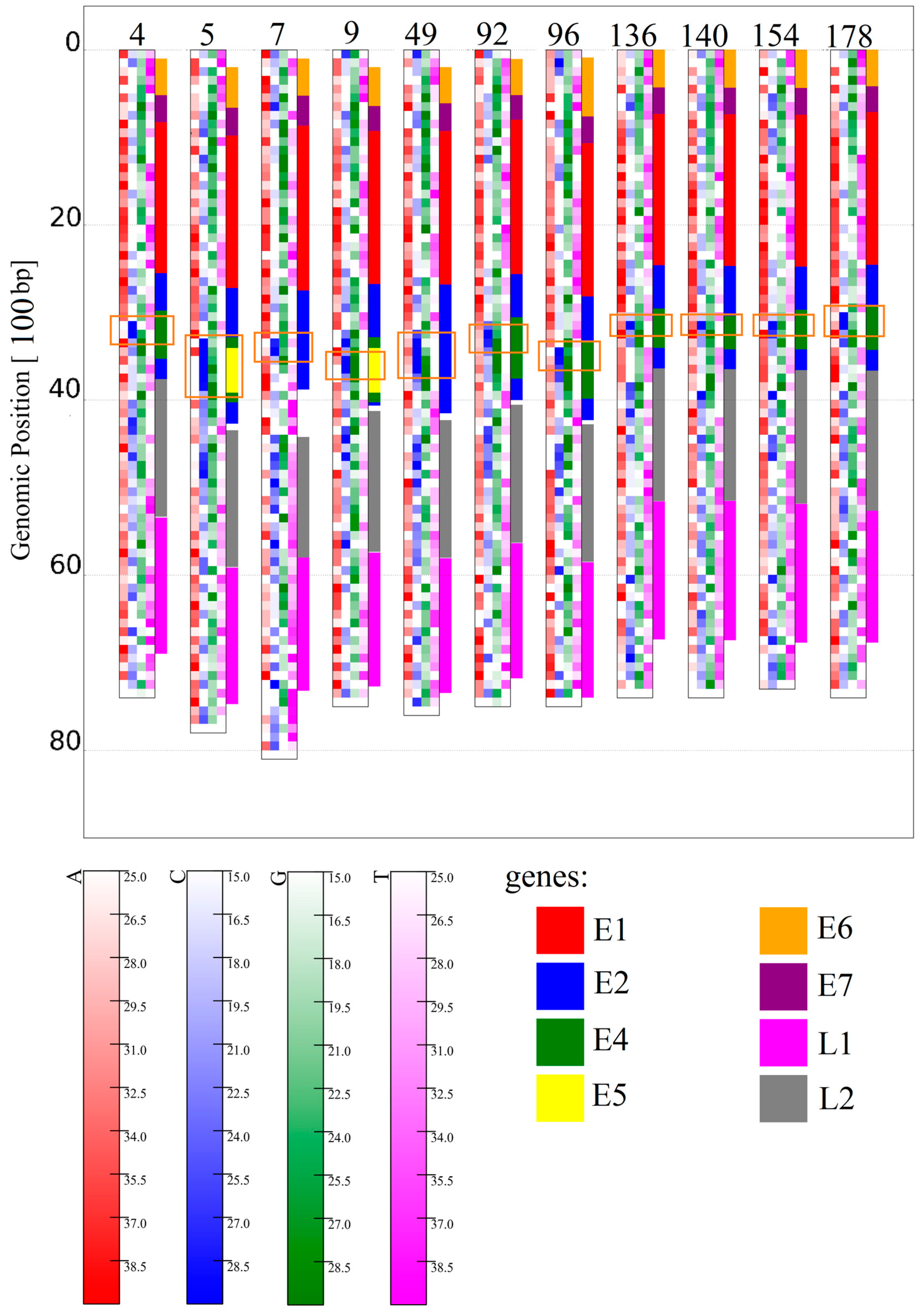
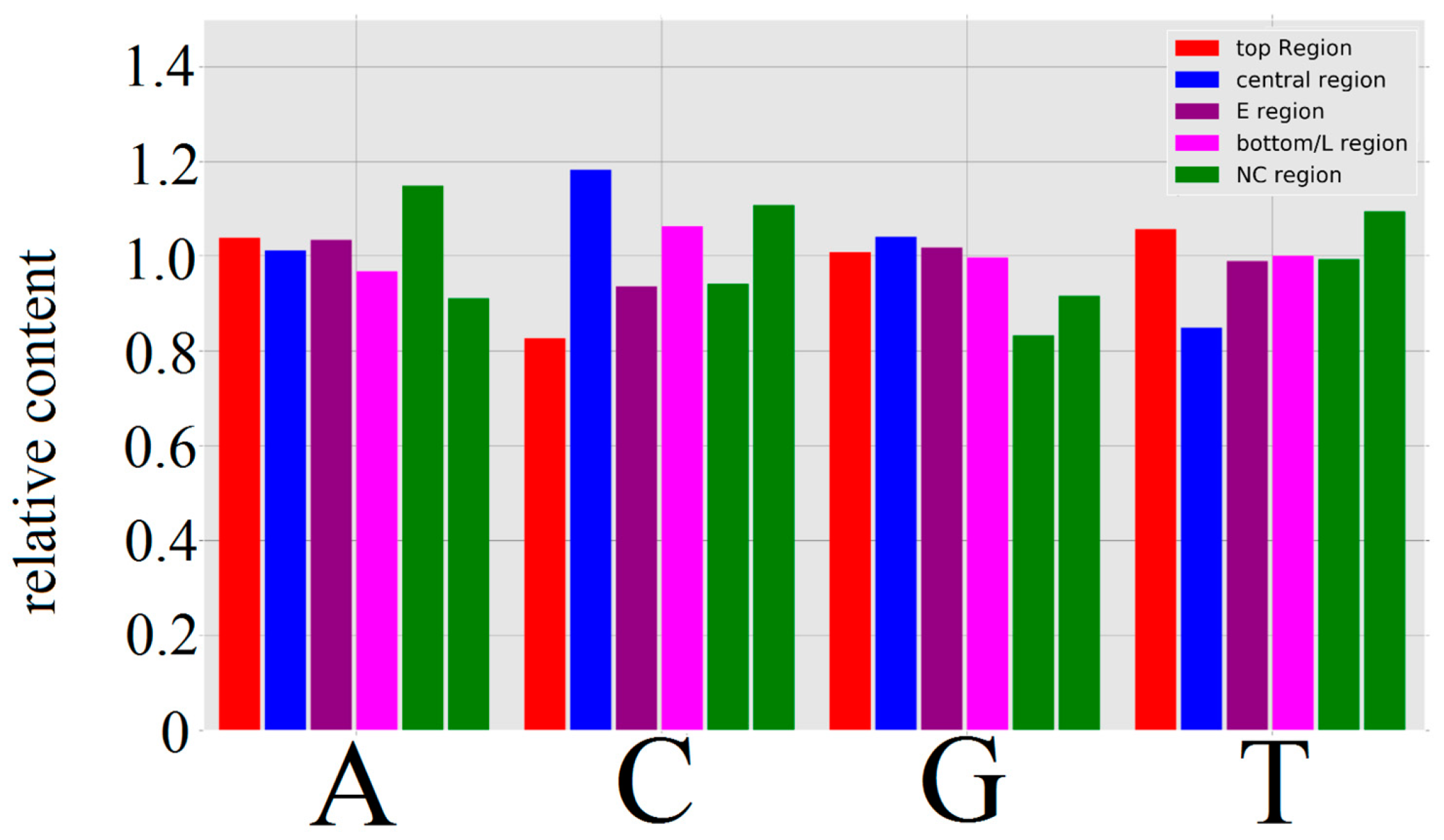
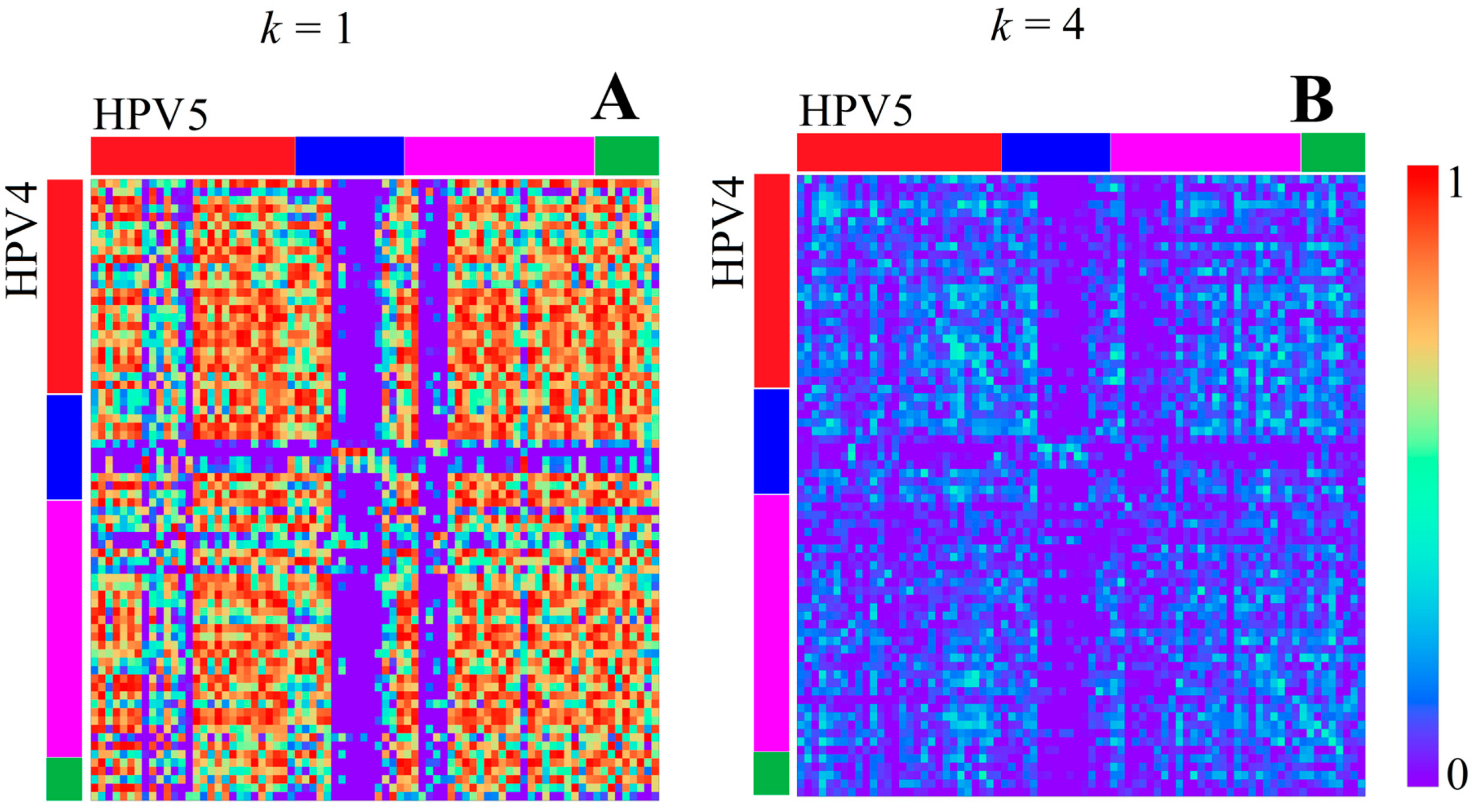
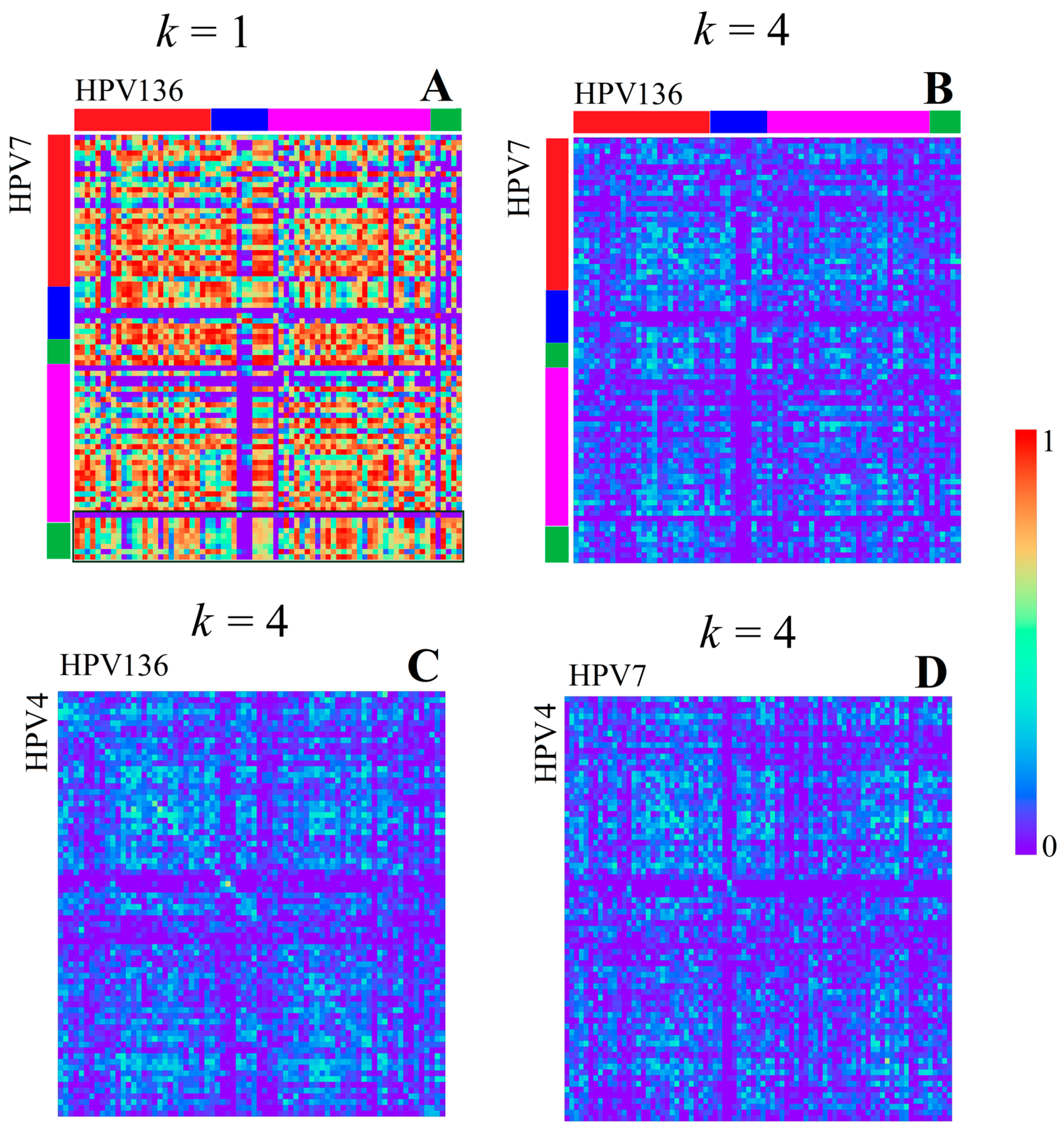
3.1.1. Papillomaviridae
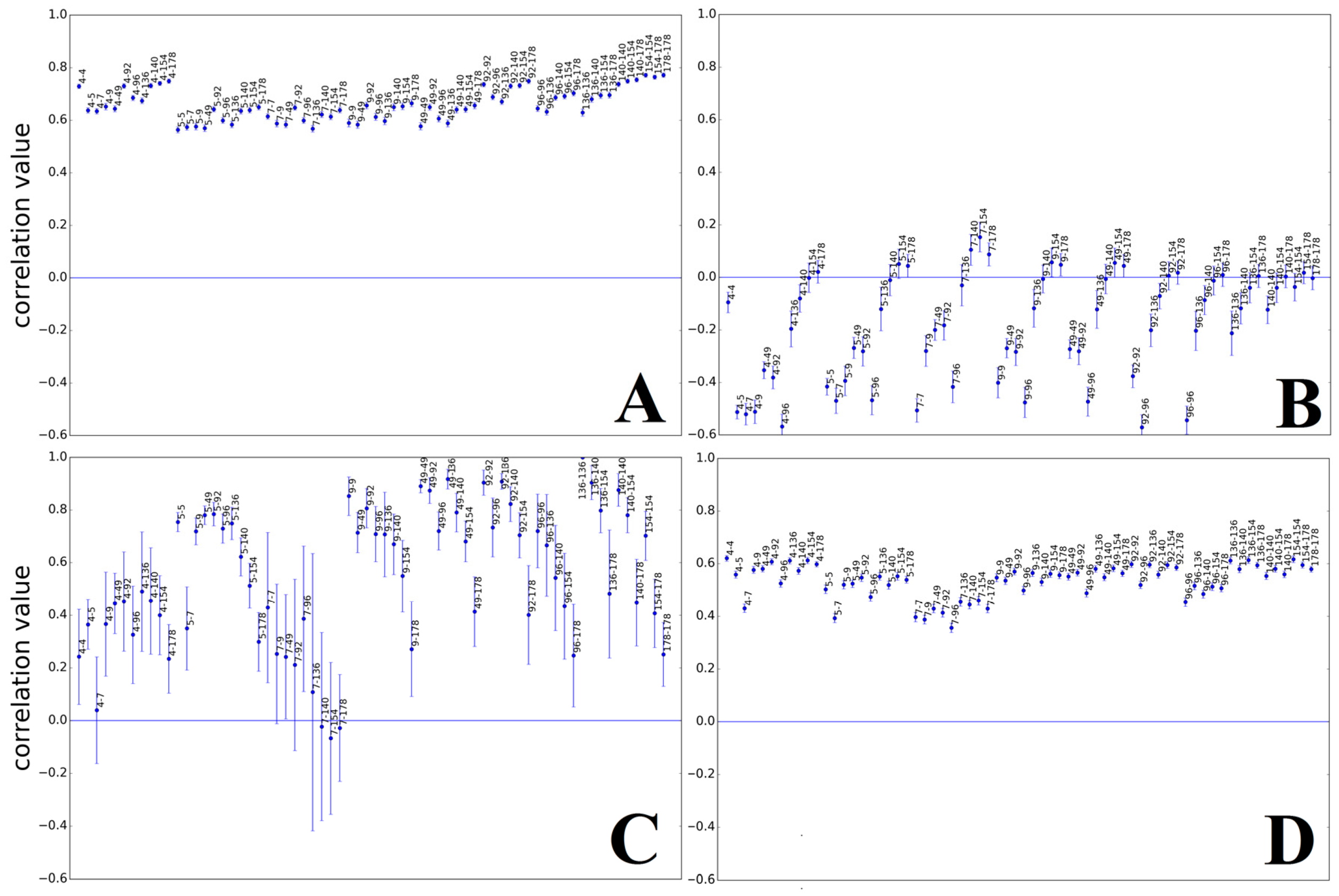
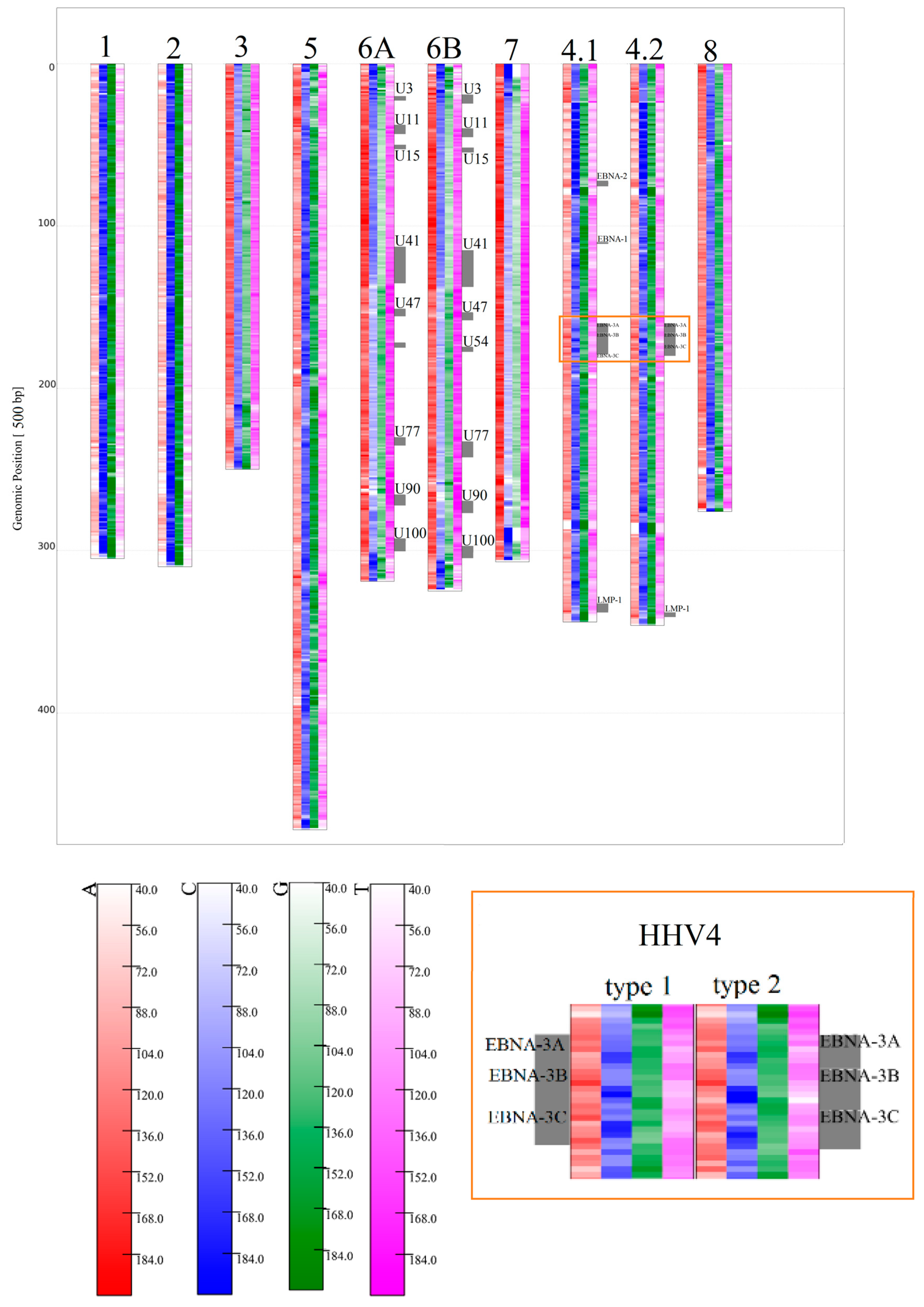
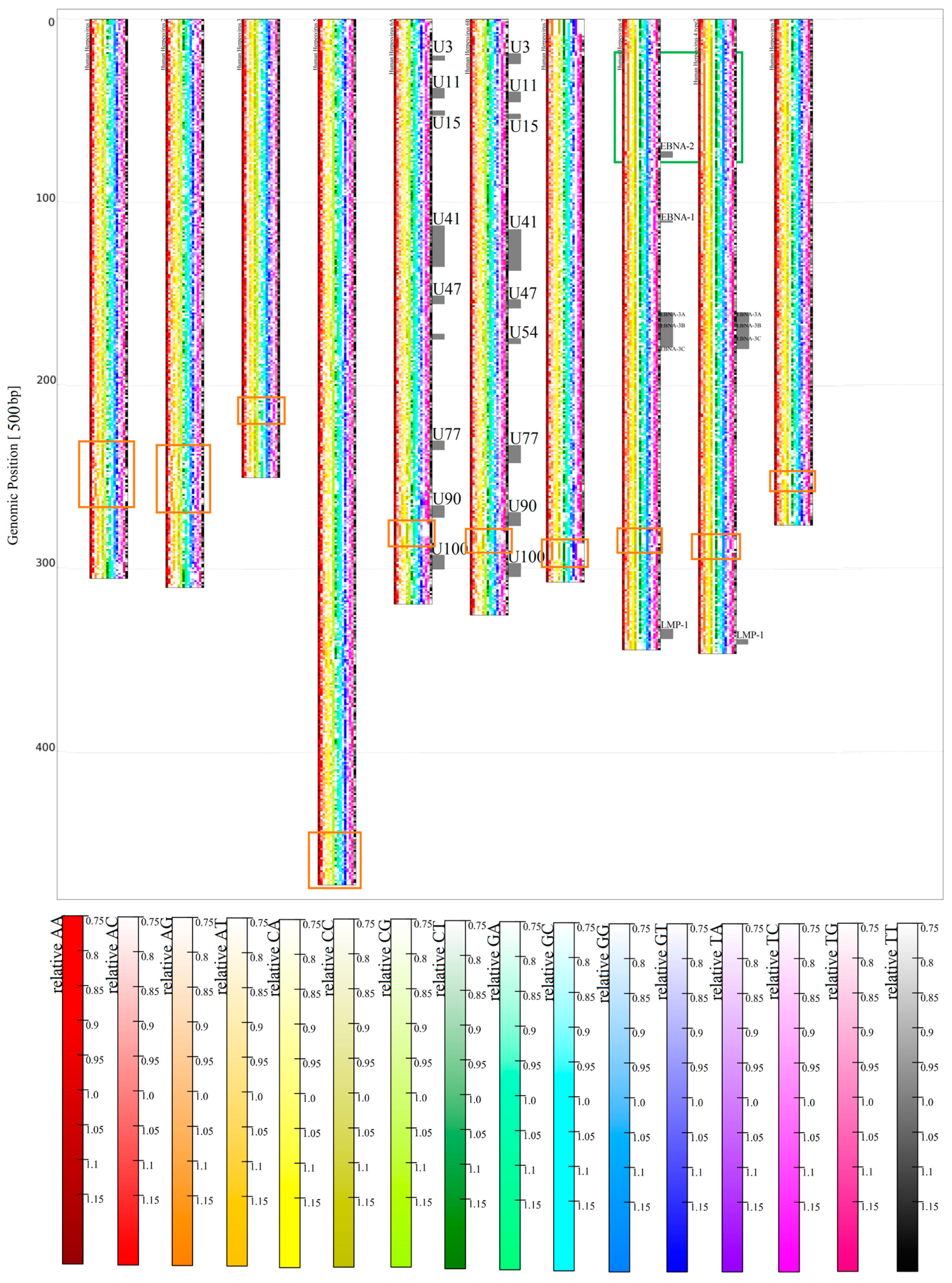
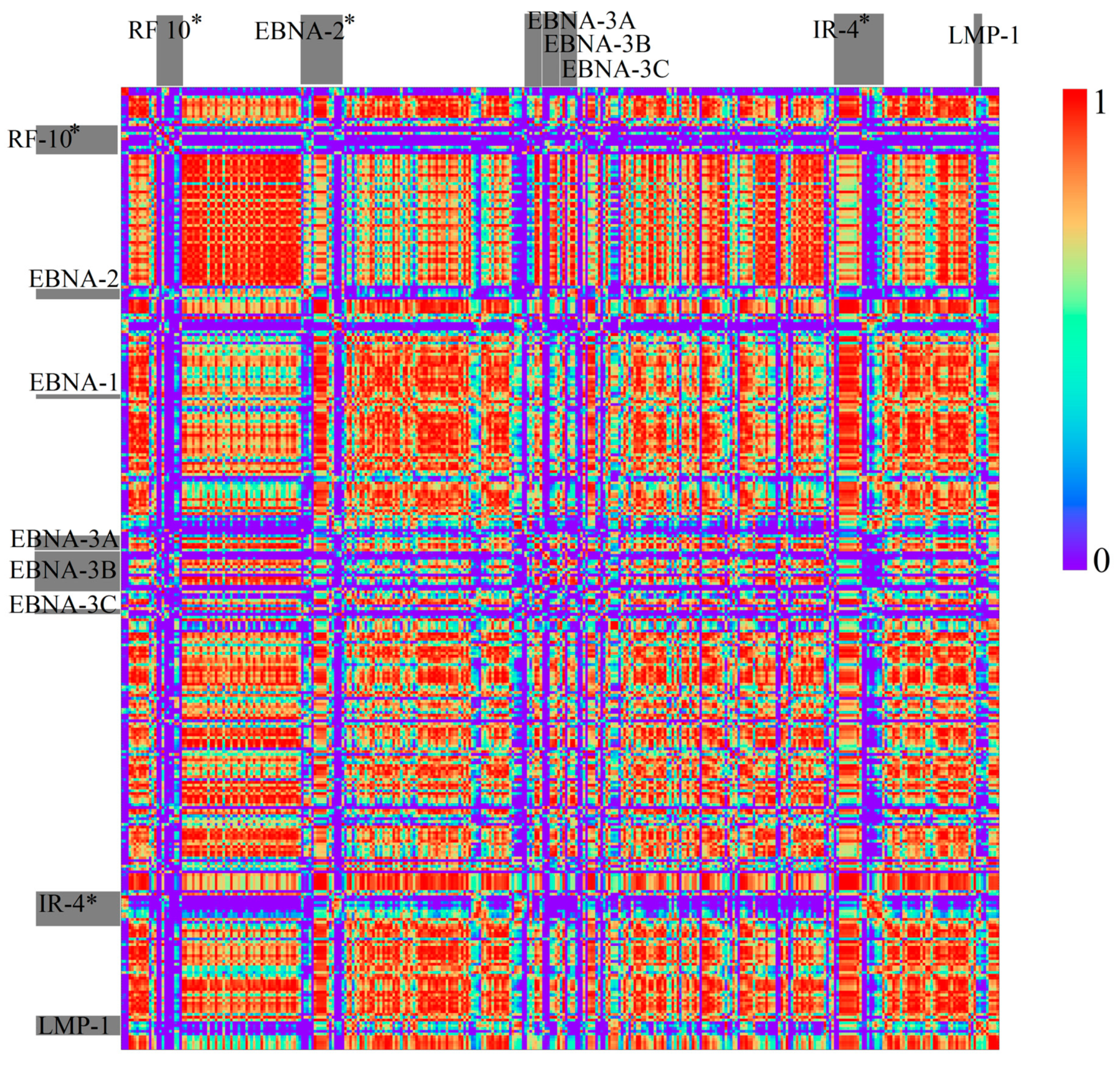
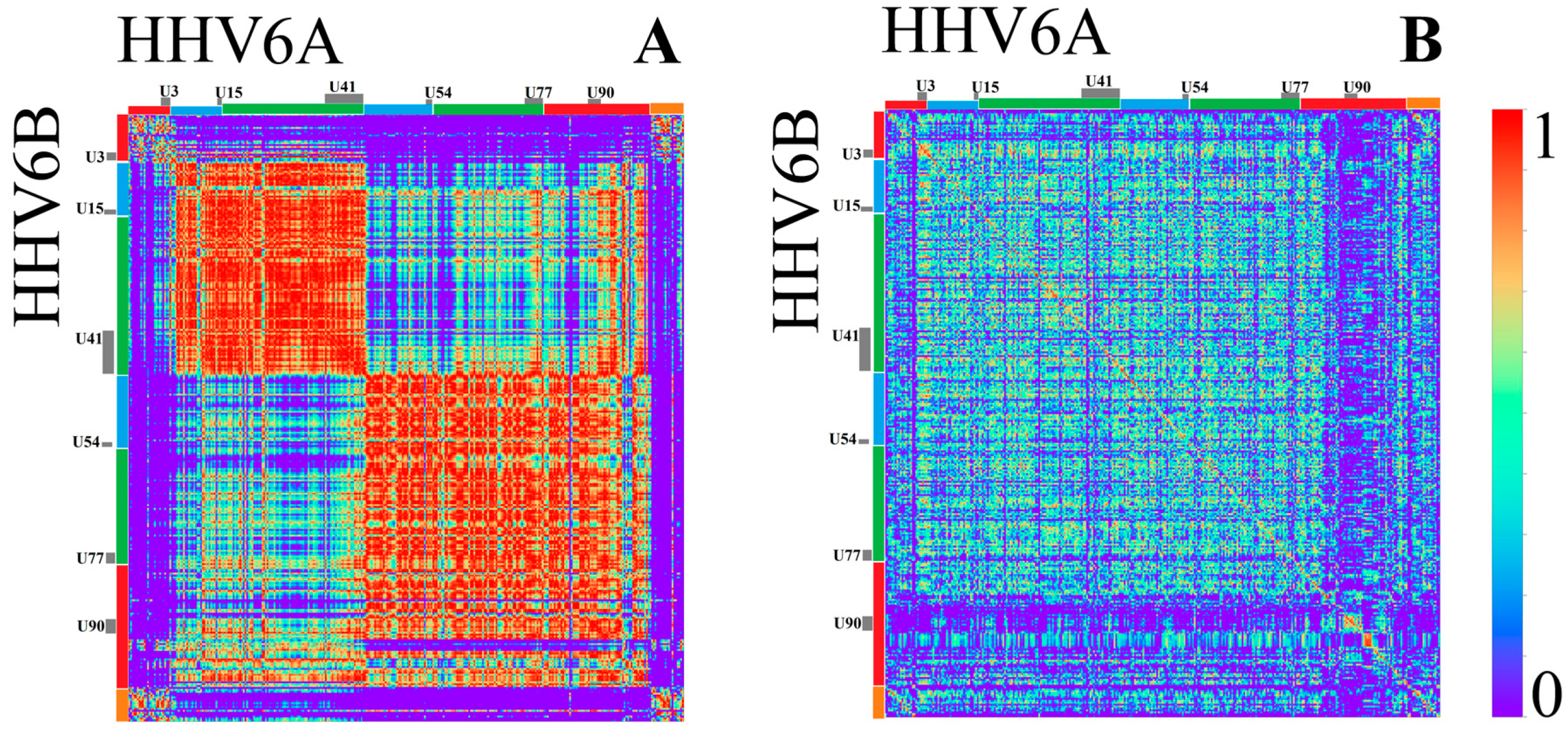
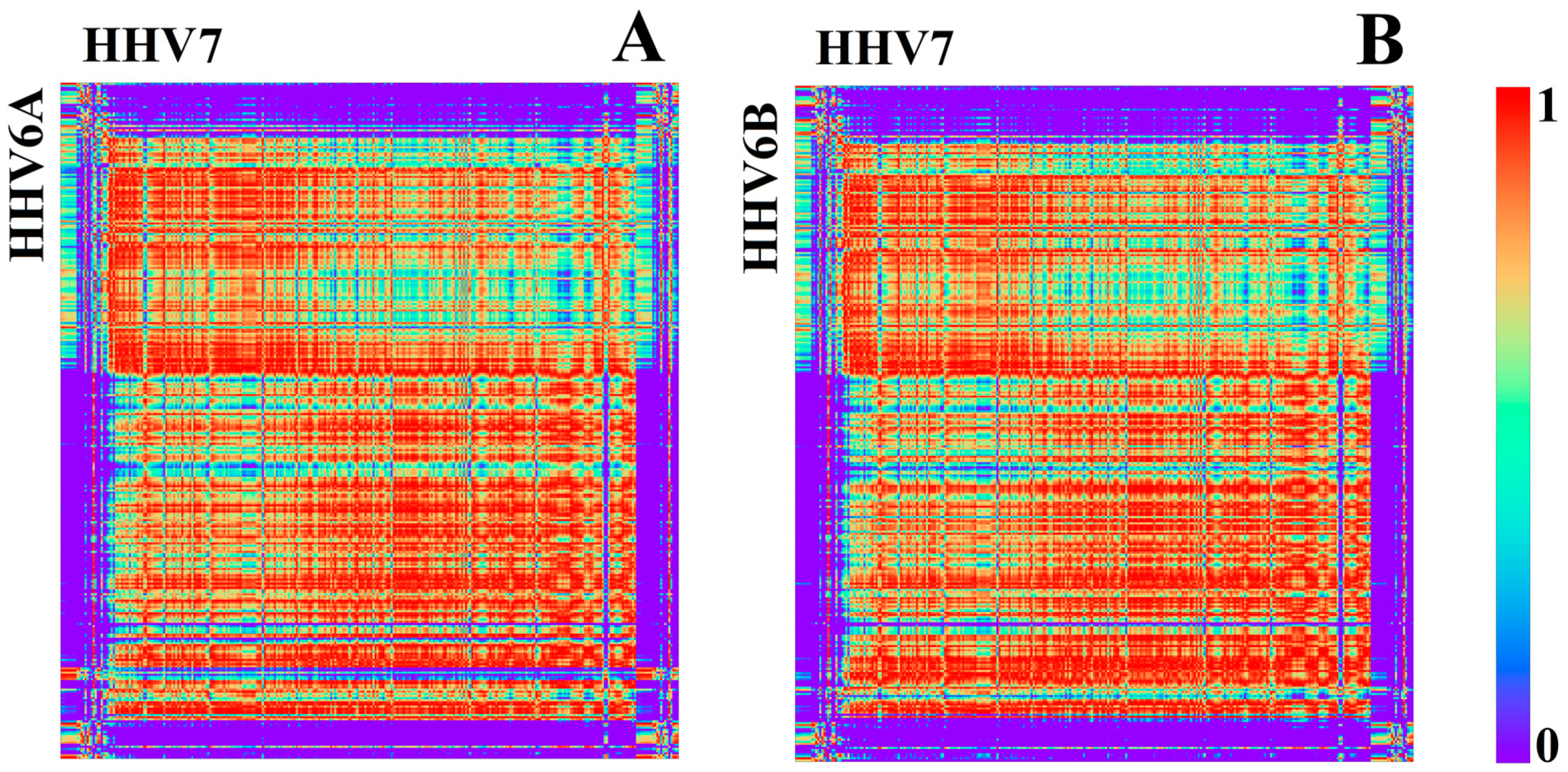
3.1.2. Herpesviridae
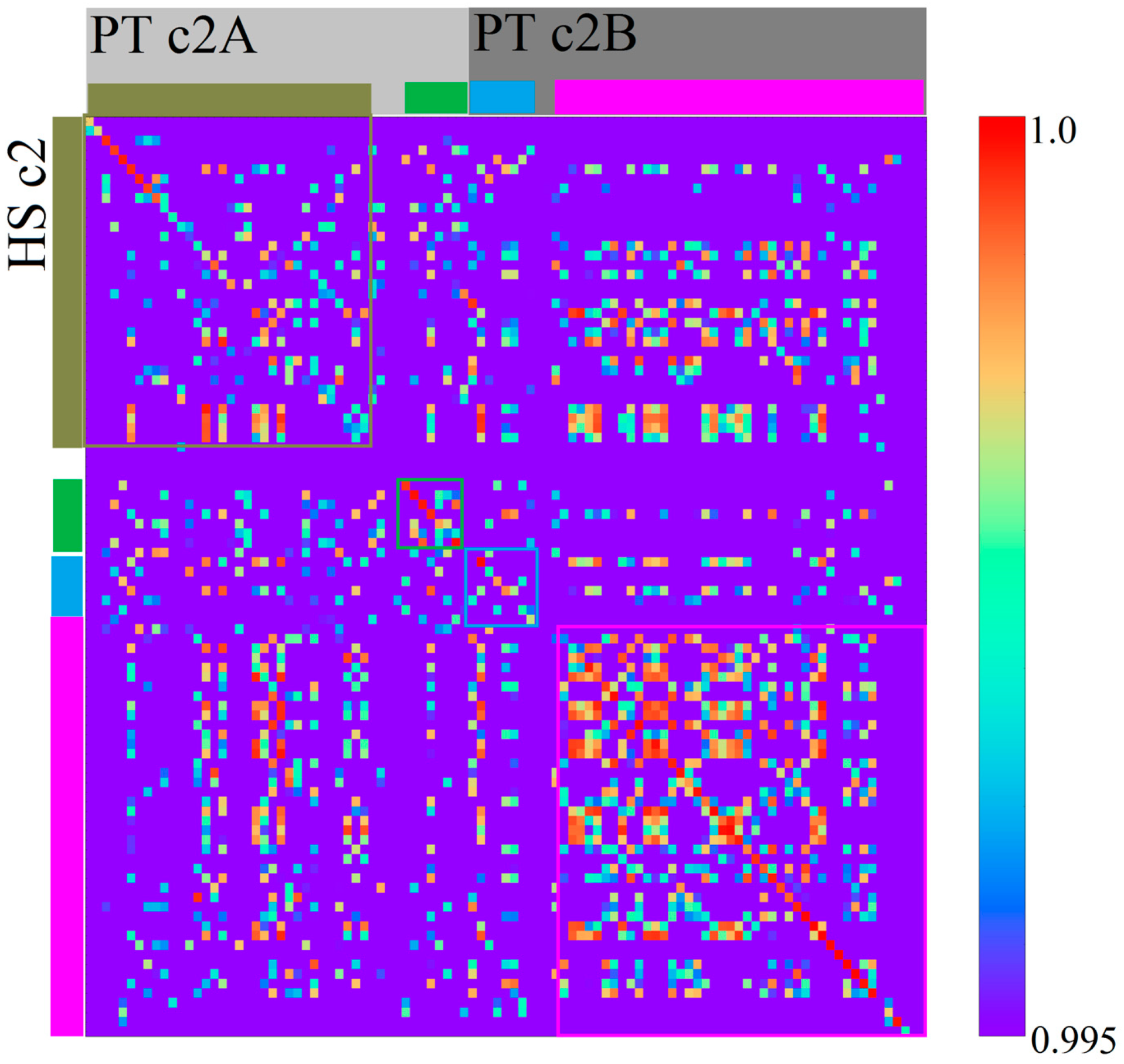
3.2. Homo sapiens
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chan, C.X.; Ragan, M.A. Next-generation phylogenetics. Biol. Direct 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Alsop, E.B.; Raymond, J. Resolving prokaryotic taxonomy without rRNA: Longer oligonucleotide word lengths improve genome and metagenome taxonomic classification. PLoS ONE 2013, 8, e67337. [Google Scholar] [CrossRef] [PubMed]
- Brendel, V.; Beckmann, J.S.; Trifonov, E.N. Linguistics of nucleotide sequences: morphology and comparison of vocabularies. J. Biomol. Struct. Dyn. 1986, 4, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Olman, V.; Xu, Y. Barcodes for genomes and applications. BMC Bioinform. 2008, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Bultrini, E.; Pizzi, E.; Del Giudice, P.; Frontali, C. Pentamer vocabularies characterizing introns and intron-like intergenic tracts from Caenorhabditis elegans and Drosophila melanogaster. Gene 2003, 304, 183–192. [Google Scholar] [CrossRef]
- Pizzi, E.; Frontali, C. Low-complexity regions in Plasmodium falciparum proteins. Genome Res. 2001, 11, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Hacker, J.; Kaper, J.B. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000, 54, 641–679. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Porwollik, S.; Wang, Y.; McClelland, M.; Rosen, H.; Libby, S.J.; Fang, F.C. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 2006, 313, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, E.; Frontali, C. Divergence of noncoding sequences and of insertions encoding nonglobular domains at a genomic region well conserved in plasmodia. J. Mol. Evolut. 2000, 50, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Pozzoli, U.; Menozzi, G.; Fumagalli, M.; Cereda, M.; Comi, G.P.; Cagliani, R.; Bresolin, N.; Sironi, M. Both selective and neutral processes drive GC content evolution in the human genome. BMC Evolut. Biol. 2008, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Jinwoo, P.; Seong-Whan, L.; Kenneth, P.N.; Sun, K. Comparative analysis using k-mer and k-flank patterns provides evidence for CpG island sequence evolution in mammalian genomes. Nucleic Acids Res. 2013, 41, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Ilene, K.M.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2005, 33, D34–D38. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. Note on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Compt. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Acland, A.; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bryant, S.H.; Canese, K.; Church, D.M.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 40, D13–D25. [Google Scholar]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-trascriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 2011, 1230, E37–E49. [Google Scholar] [CrossRef] [PubMed]
- Elson, D.; Chargaff, E. On the desoxyribonucleic acid content of sea urchin gametes. Expertientia 1952, 8, 143–145. [Google Scholar] [CrossRef]
- Dominguez, G.; Dambaugh, T.R.; Stamey, F.R.; Dewhurst, S.N.; Inoue, S.; Pellett, P.E. Human herpesvirus 6B genome sequence: Coding content and comparison with human herpesvirus 6A. J. Vorol. 1999, 73, 8040–8052. [Google Scholar]
- Dolan, A.; Addison, C.; Gatherer, D.; Davison, A.J.; McGeoch, D.J. The genome of Epstein-Barr virus type 2 strain AG876. J. Virol. 2006, 350, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Megaw, A.G.; Rapaport, D.; Avidor, B.; Frenkel, N.; Davison, A.J. The DNA sequence of the RK strain of human herpesvirus 7. J. Virol. 1998, 244, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Yunis, J.J.; Sawyer, J.R. The Striking Resemblance of high-resolution G-banded chromosomes of man and chimpanzee. Science 1980, 208, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Pratas, D.; Silva, R.M.; Pinho, A.J.; Ferreira, P.J.S.G. An alignment-free method to find and visualise rearrangements between pairs of DNA sequences. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A. Malaria research in the post-genomic era. Nature 2008, 455, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Clay, O.; Caccio, S.; Zoubak, S.; Mouchiroud, D.; Bernardi, G. Human coding and noncoding DNA: Compositional correlations. Mol. Phylogenet. Evolut. 1996, 5, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Mouchiroud, D.; Gautier, C. Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J. Mol. Evolut. 1995, 40, 308–317. [Google Scholar] [CrossRef]
- Fullerton, S.M.; Carvalho, A.B.; Clark, A.G. Local Rates of Recombination Are Positively Correlated with GC Content in the Human Genom. Mol. Biol. Evolut. 2001, 8, 1139–1142. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Accession Number |
|---|---|
| Human Papillomavirus 4 (HPV4) | NC_001457.1 |
| Human Papillomavirus 5 (HPV5) | NC_001531.1 |
| Human Papillomavirus 7 (HPV7) | NC_001595.1 |
| Human Papillomavirus 9 (HPV9) | NC_001596.1 |
| Human Papillomavirus 49 (HPV49) | NC_001591.1 |
| Human Papillomavirus 92 (HPV92) | NC_004500.1 |
| Human Papillomavirus 96 (HPV96) | NC_005134.2 |
| Human Papillomavirus 136 (HPV136) | NC_017994.1 |
| Human Papillomavirus 140 (HPV140) | NC_017996.1 |
| Human Papillomavirus 154 (HPV154) | NC_021483.1 |
| Human Papillomavirus 178 (HPV178) | NC_023891.1 |
| Human Herpesvirus 1 (HHV1) | NC_001806.2 |
| Human Herpesvirus 2 (HHV2) | NC_001798.2 |
| Human Herpesvirus 3 (HHV3) | NC_001348.1 |
| Human Herpesvirus 4 type1 (HHV4 type1) | NC_007605.1 |
| Human Herpesvirus 4 type2 (HHV4 type2) | NC_009334.1 |
| Human Herpesvirus 5 (HHV5) | NC_006273.2 |
| Human Herpesvirus 6A (HHV6A) | NC_001664.2 |
| Human Herpesvirus 6B (HHV6B) | NC_000898.1 |
| Human Herpesvirus 7 (HHV7) | NC_001716.2 |
| Human Herpesvirus 8 (HHV8) | NC_009333.1 |
| Homo Sapiens Chromosome 2 | NC_000002.12 |
| Pan Troglodytes Chromosome 2A | NC_006469.3 |
| Pan Troglodytes Chromosome 2B | NC_006470.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sievers, A.; Bosiek, K.; Bisch, M.; Dreessen, C.; Riedel, J.; Froß, P.; Hausmann, M.; Hildenbrand, G. K-mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features. Genes 2017, 8, 122. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8040122
Sievers A, Bosiek K, Bisch M, Dreessen C, Riedel J, Froß P, Hausmann M, Hildenbrand G. K-mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features. Genes. 2017; 8(4):122. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8040122
Chicago/Turabian StyleSievers, Aaron, Katharina Bosiek, Marc Bisch, Chris Dreessen, Jascha Riedel, Patrick Froß, Michael Hausmann, and Georg Hildenbrand. 2017. "K-mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features" Genes 8, no. 4: 122. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8040122