Whole Genome Sequencing Revealed Mutations in Two Independent Genes as the Underlying Cause of Retinal Degeneration in an Ashkenazi Jewish Pedigree

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
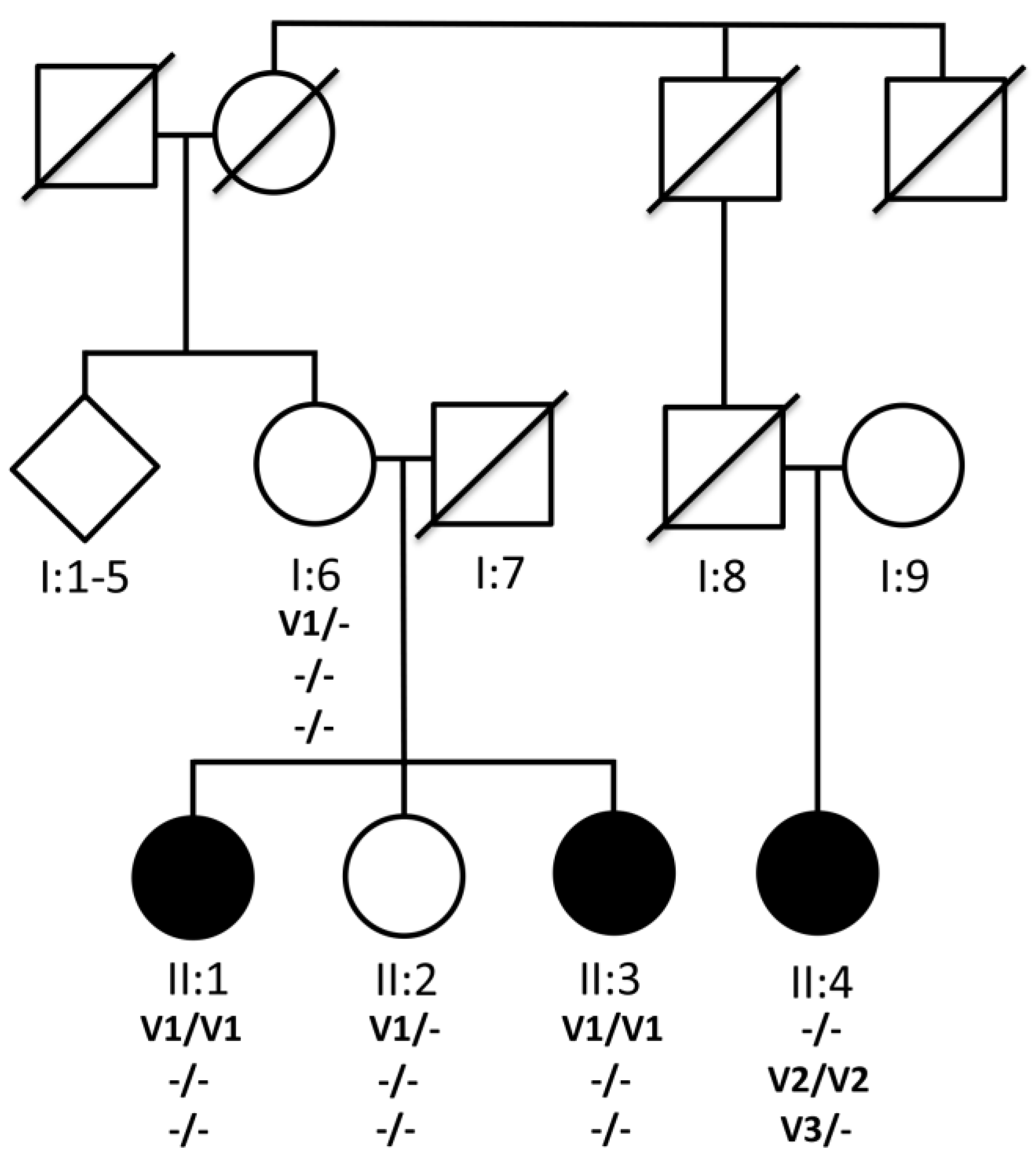
3.1. Pedigree
3.2. Clinical Information
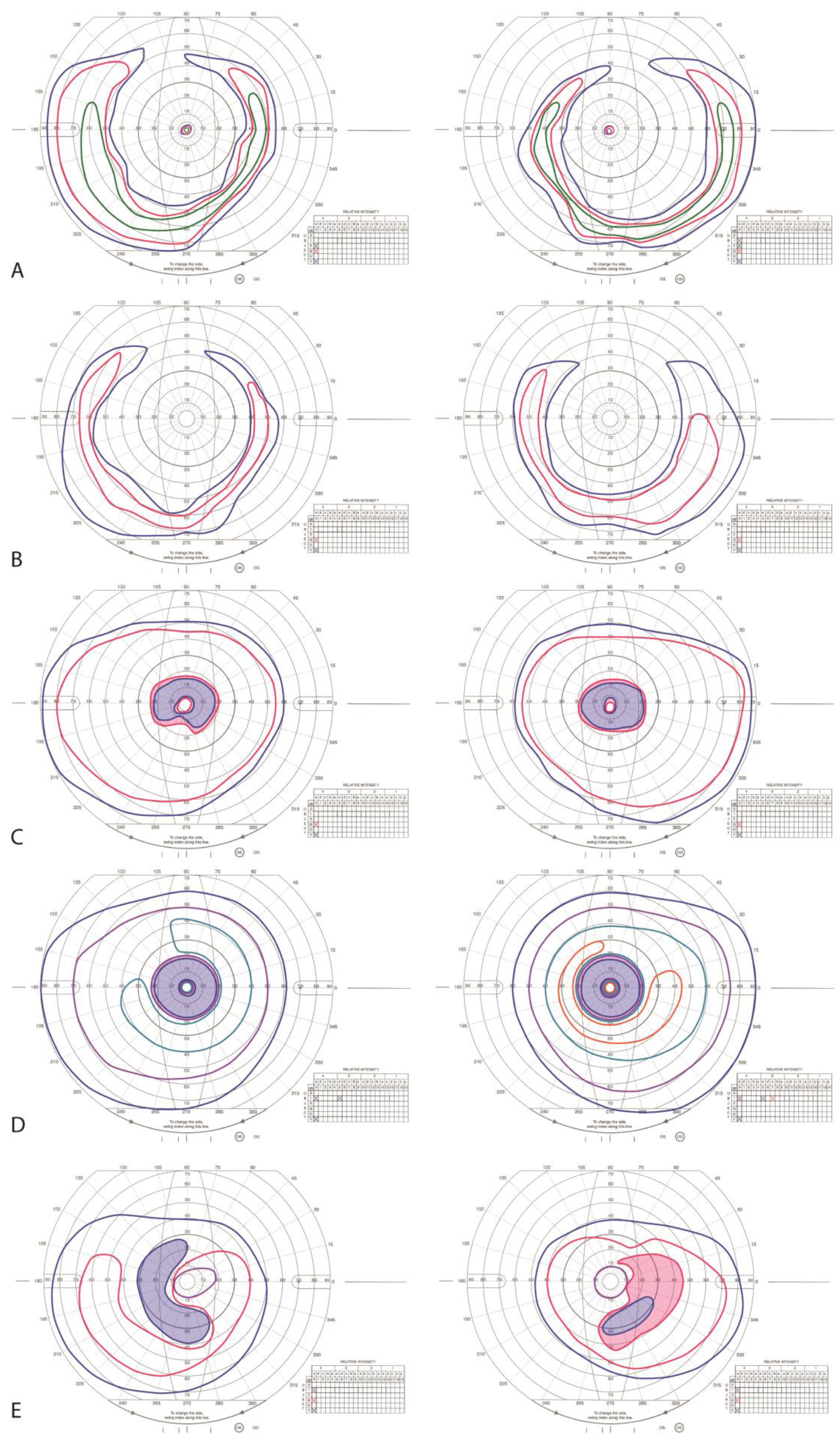
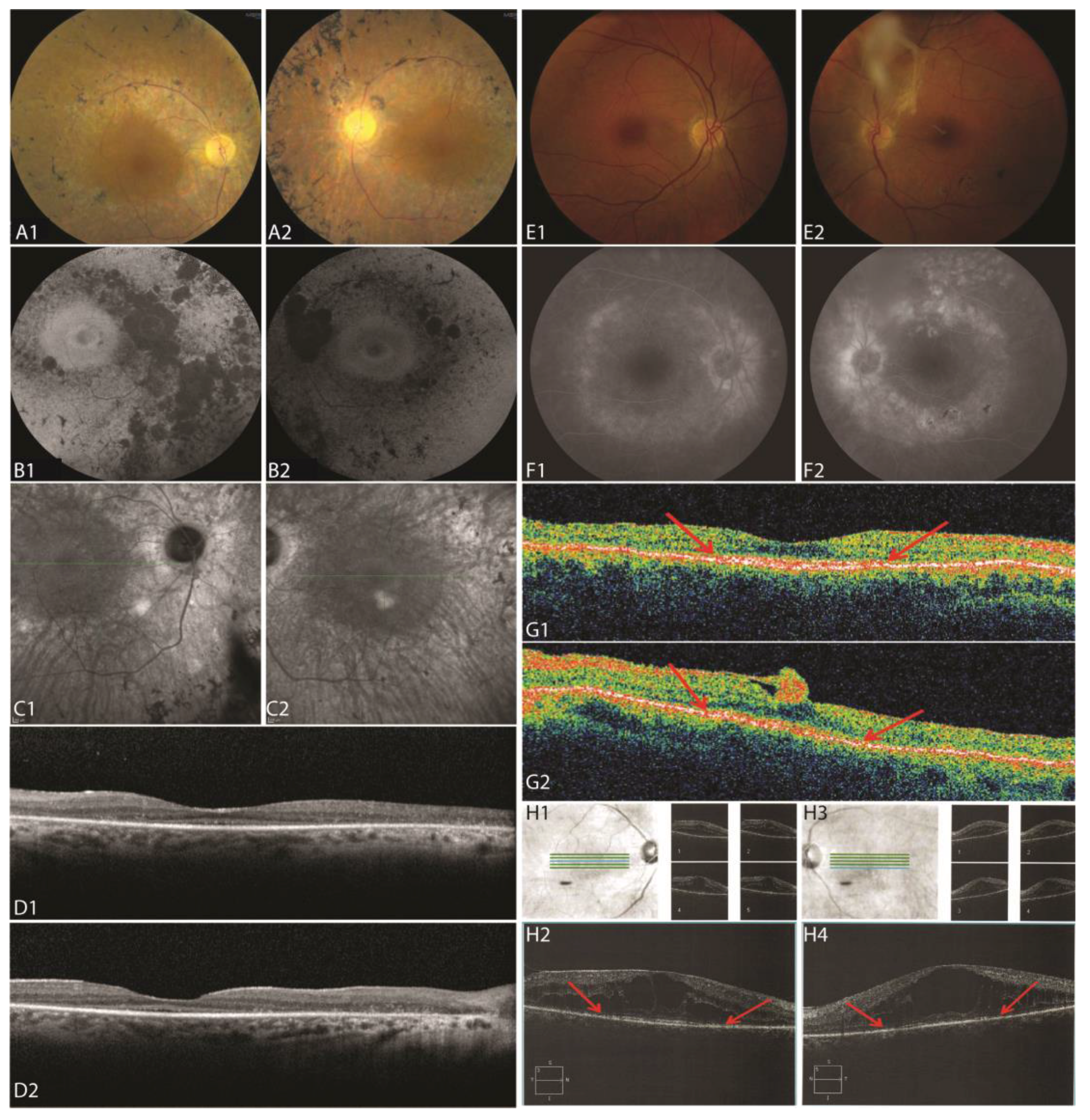
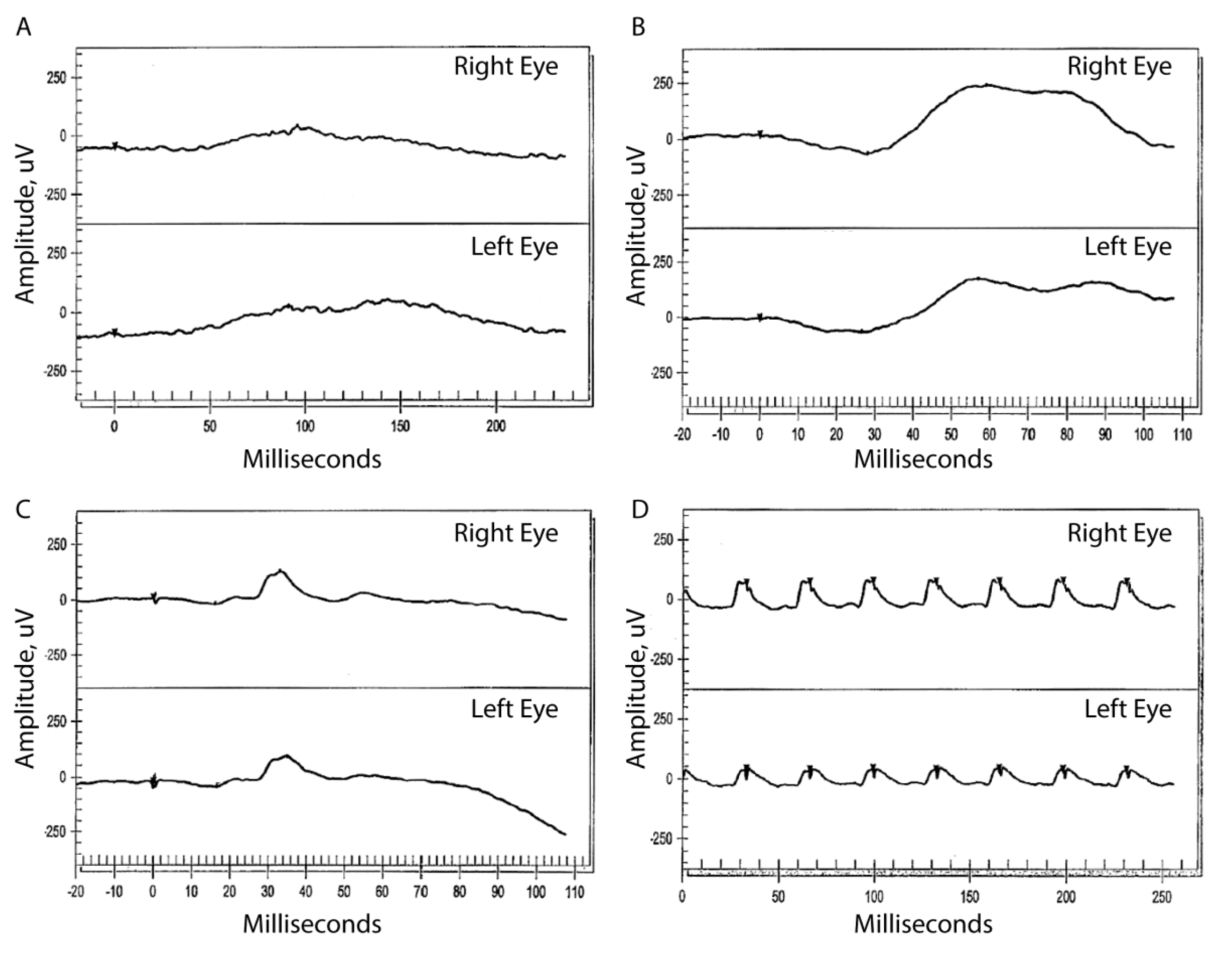
3.2.1. Patient II:1
3.2.2. Patient II:3
3.2.3. Patient II:4
3.3. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Travis, G.H. Mechanisms of cell death in the inherited retinal degenerations. Am. J. Hum. Genet. 1998, 62, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet. J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Retnet. Available online: www.sph.uth.tmc.edu/RetNet/ (accessed on 12 May 2017).
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. Iscev standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischler, G.; Leonard, S. Biobambam: Tools for read pair collation based algorithms on bam files. Source Code Biol. Med. 2014, 9, 13. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 111011–111033. [Google Scholar]
- Handsaker, R.E.; Korn, J.M.; Nemesh, J.; McCarroll, S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011, 43, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O'Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SNPEFF: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 20. [Google Scholar] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O'Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Chavali, V.R.; Agnello, G.; Stone, E.; Chakarova, C.; Duncan, J.L.; Kannabiran, C.; Homsher, M.; Bhattacharya, S.S.; Naeem, M.A.; et al. A missense mutation in ASRGL1 is involved in causing autosomal recessive retinal degeneration. Hum. Mol. Genet. 2016, 25, 2483–2497. [Google Scholar] [PubMed]
- Duncan, J.L.; Roorda, A.; Navani, M.; Vishweswaraiah, S.; Syed, R.; Soudry, S.; Ratnam, K.; Gudiseva, H.V.; Lee, P.; Gaasterland, T.; et al. Identification of a novel mutation in the CDHR1 gene in a family with recessive retinal degeneration. Arch. Ophthalmol. 2012, 130, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. 2017, 19, 643–651. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Neuille, M.; Terray, A.; Orhan, E.; Condroyer, C.; Demontant, V.; Michiels, C.; Antonio, A.; Boyard, F.; Lancelot, M.E.; et al. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am. J. Hum. Genet. 2014, 94, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl, H.P.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 368rv6. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Ohsugi, M.; Yamamoto, T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006, 8, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Eisenberger, T.; Nagel-Wolfrum, K.; Wolfrum, U.; Bolz, H.J. C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol. 2015, 99, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Suga, A.; Mizota, A.; Kato, M.; Kuniyoshi, K.; Yoshitake, K.; Sultan, W.; Yamazaki, M.; Shimomura, Y.; Ikeo, K.; Tsunoda, K.; et al. Identification of novel mutations in the LRR-Cap domain of c21orf2 in Japanese patients with retinitis pigmentosa and cone-rod dystrophy. Invest. Ophth. Vis. Sci. 2016, 57, 4255–4263. [Google Scholar] [CrossRef] [PubMed]
- McInerney-Leo, A.M.; Wheeler, L.; Marshall, M.S.; Anderson, L.K.; Zankl, A.; Brown, M.A.; Leo, P.J.; Wicking, C.; Duncan, E.L. Homozygous variant in c21orf2 in a case of jeune syndrome with severe thoracic involvement: Extending the phenotypic spectrum. Am. J. Med. Genet. A 2017, 173, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Iida, A.; Miyake, N.; Nishiguchi, K.M.; Fujita, K.; Nakazawa, T.; Alswaid, A.; Albalwi, M.A.; Kim, O.H.; Cho, T.J.; et al. Axial spondylometaphyseal dysplasia is caused by c21orf2 mutations. PLoS ONE 2016, 11, e0150555. [Google Scholar]
- Mockel, A.; Perdomo, Y.; Stutzmann, F.; Letsch, J.; Marion, V.; Dollfus, H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011, 30, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Huang, F.; Wu, K.C.; Wu, J.; Chen, J.; Pang, C.P.; Lu, F.; Qu, J.; Jin, Z.B. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 2015, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Wycisk, K.A.; Zeitz, C.; Feil, S.; Wittmer, M.; Forster, U.; Neidhardt, J.; Wissinger, B.; Zrenner, E.; Wilke, R.; Kohl, S.; et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 2006, 79, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Ba-Abbad, R.; Arno, G.; Carss, K.; Stirrups, K.; Penkett, C.J.; Moore, A.T.; Michaelides, M.; Raymond, F.L.; Webster, A.R.; Holder, G.E. Mutations in CACNA2D4 cause distinctive retinal dysfunction in humans. Ophthalmology 2016, 123, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID/Gender | Age (Years) | Age at First Sx (Years) | BCVA | Slit-Lamp Exam | Dilated Fundus Exam | Genetics |
|---|---|---|---|---|---|---|
| OD, OS | ||||||
| II:1/F | 60 | 26 | 20/800 20/800 | OU: mild NS cataract | OU: mild disc pallor, cellophane ERM, bone spicules, no CME | Gene: KIZ |
| Chr20: 21136463 | ||||||
| c.226C>T (p.Arg76*) | ||||||
| II:3/F | 54 | 44 | 20/25 20/25 | OU: normal | OD: normal | Gene: KIZ |
| OS: dense ERM with fibrosis | c.226C>T (p.Arg76*) | |||||
| II:4/F | 66 | 39 | 20/100 20/200 | OU: normal | OU: bone spicules, CME | Gene: C21orf2 |
| Homozygous 1.135 kb deletion | ||||||
| Chr21: 45,755,728–45,756,862 | ||||||
| Gene: CACNA2D4 | ||||||
| Heterozygous 30.651 kb deletion | ||||||
| Chr12: 1,949,399–1,980,050 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustafson, K.; Duncan, J.L.; Biswas, P.; Soto-Hermida, A.; Matsui, H.; Jakubosky, D.; Suk, J.; Telenti, A.; Frazer, K.A.; Ayyagari, R. Whole Genome Sequencing Revealed Mutations in Two Independent Genes as the Underlying Cause of Retinal Degeneration in an Ashkenazi Jewish Pedigree. Genes 2017, 8, 210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090210
Gustafson K, Duncan JL, Biswas P, Soto-Hermida A, Matsui H, Jakubosky D, Suk J, Telenti A, Frazer KA, Ayyagari R. Whole Genome Sequencing Revealed Mutations in Two Independent Genes as the Underlying Cause of Retinal Degeneration in an Ashkenazi Jewish Pedigree. Genes. 2017; 8(9):210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090210
Chicago/Turabian StyleGustafson, Kevin, Jacque L. Duncan, Pooja Biswas, Angel Soto-Hermida, Hiroko Matsui, David Jakubosky, John Suk, Amalio Telenti, Kelly A. Frazer, and Radha Ayyagari. 2017. "Whole Genome Sequencing Revealed Mutations in Two Independent Genes as the Underlying Cause of Retinal Degeneration in an Ashkenazi Jewish Pedigree" Genes 8, no. 9: 210. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090210