Comparative Transcriptome Analysis Identifies Putative Genes Involved in Steroid Biosynthesis in Euphorbia tirucalli

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Extraction
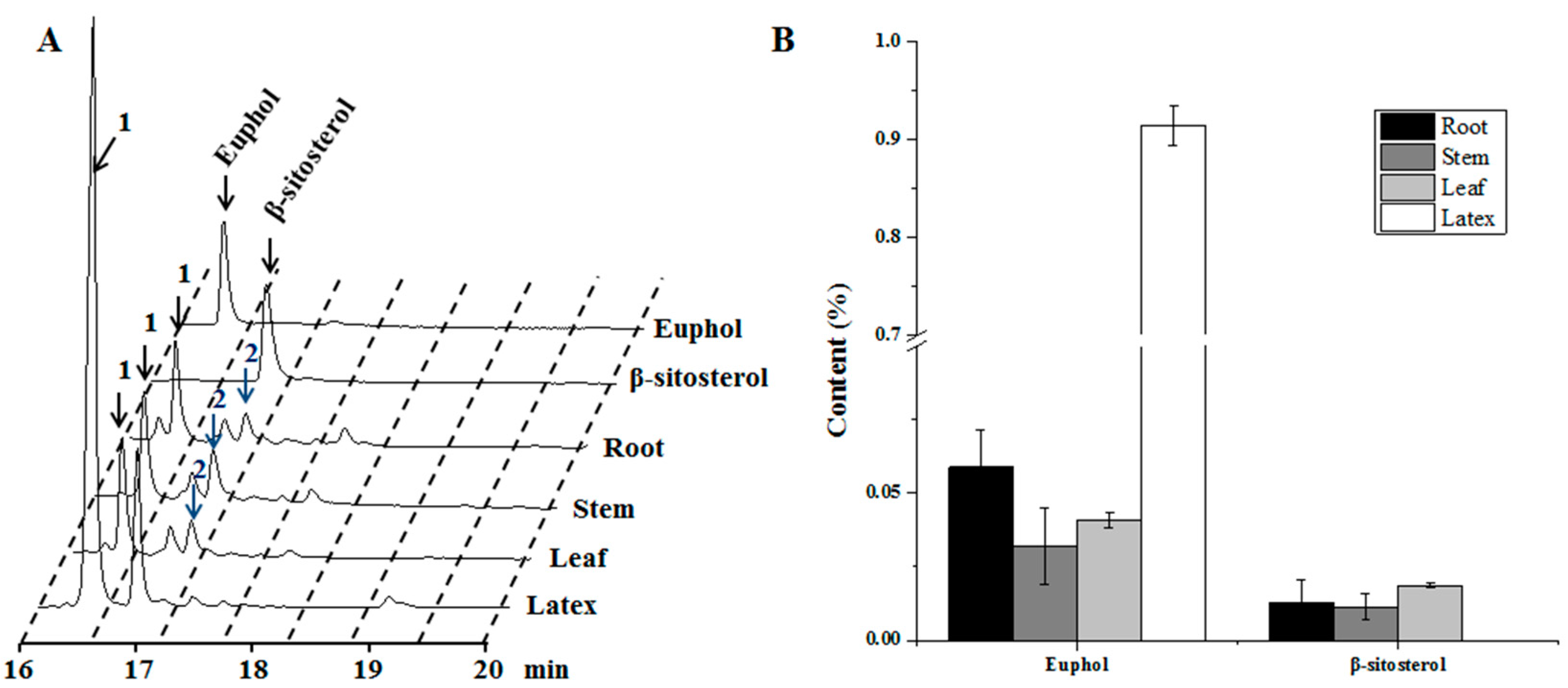
2.2. Phytochemical Analysis
2.3. Transcriptome Sequencing and de Novo Assembly
2.4. Functional Annotation
2.5. Differential Expression Analysis of Unigenes
2.6. Real-Time PCR Analysis
3. Results
3.1. Tissue-Specificity for the Biosynthesis of Sterol Compounds in E. tirucalli
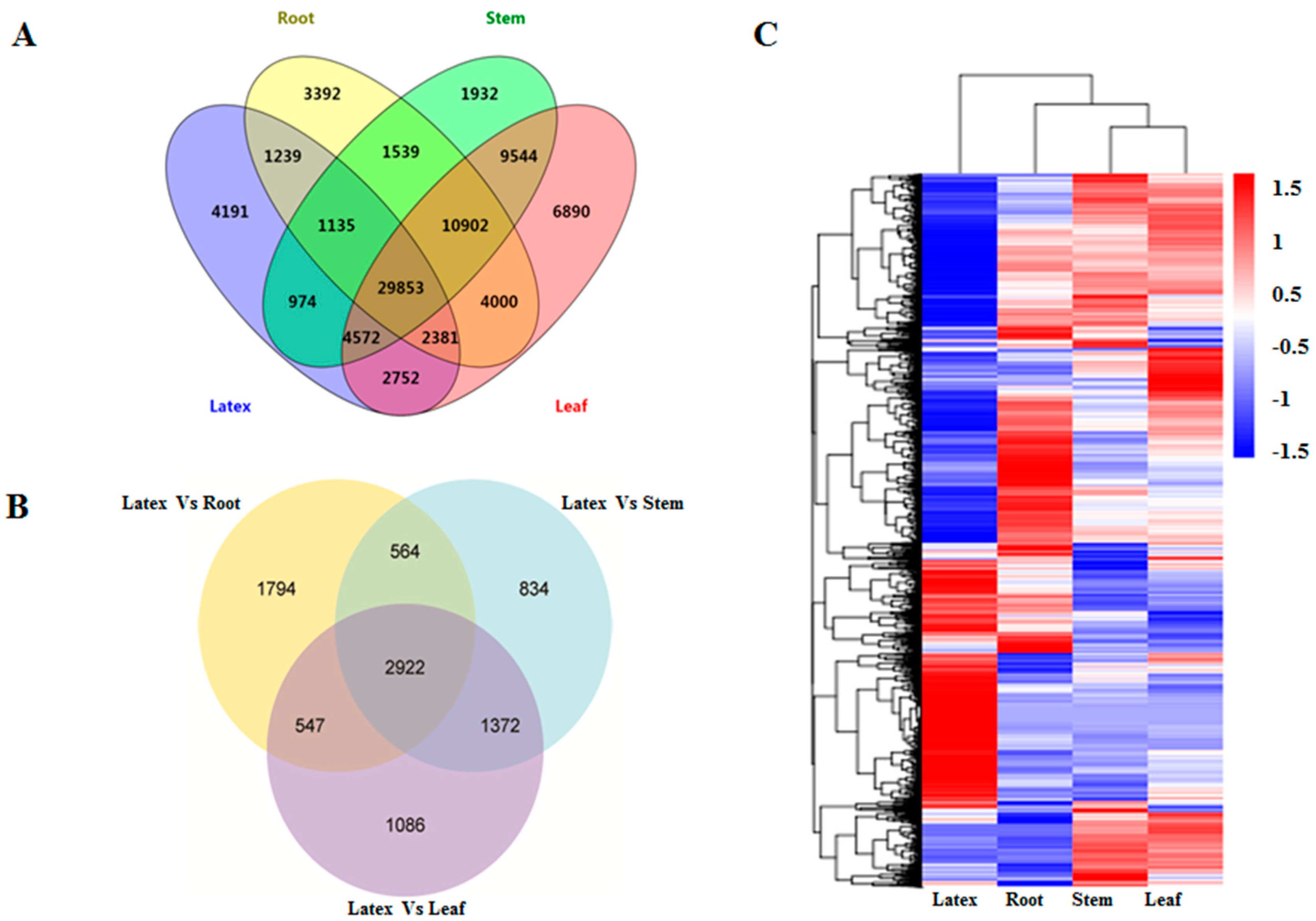
3.2. Construction of the E. tirucalli Transcriptome Integrated from Different Tissues Including the Latex
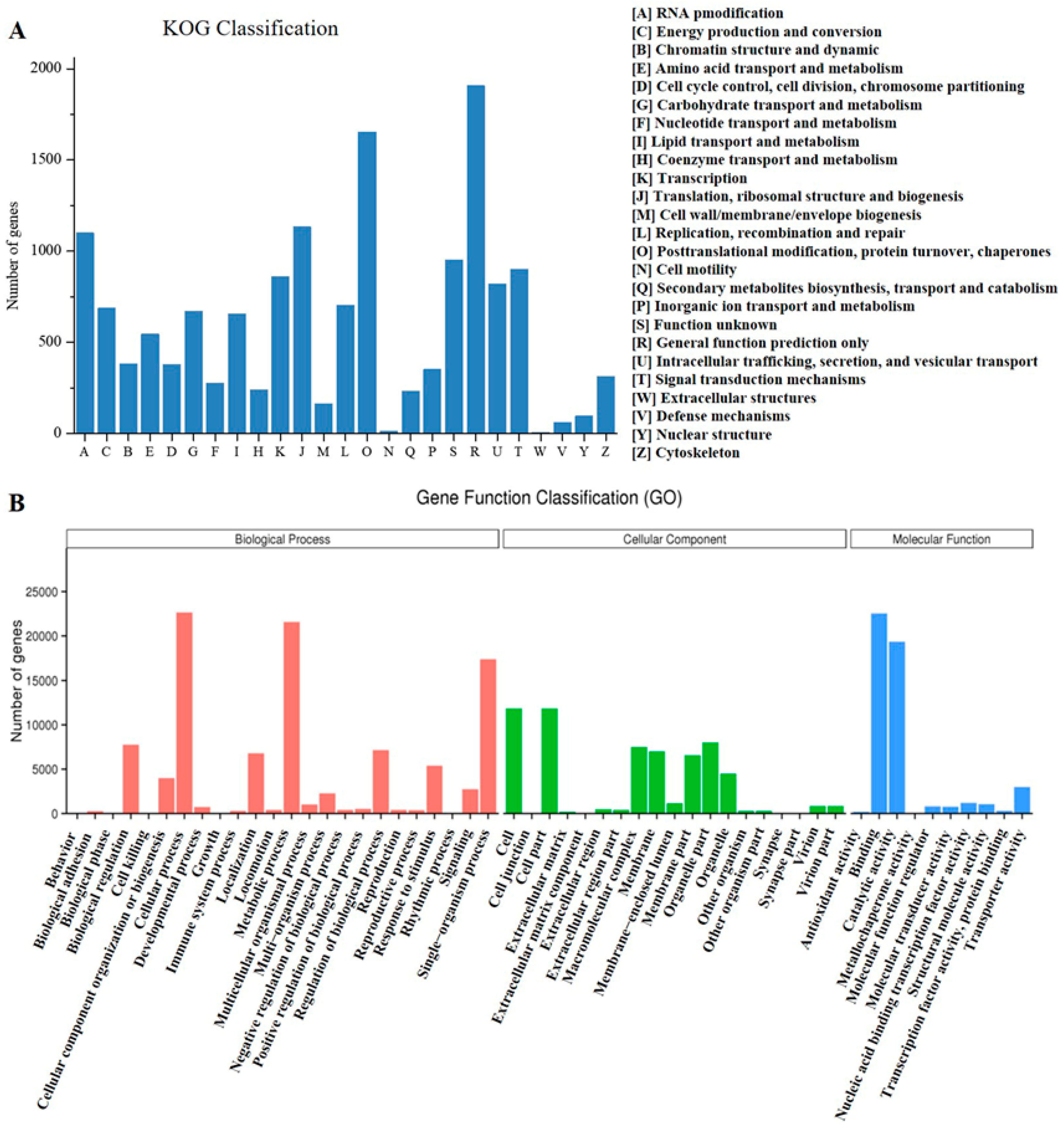
3.3. Functional Annotation and Classification of Unigenes
3.4. Gene Expression Analysis across Different Tissues
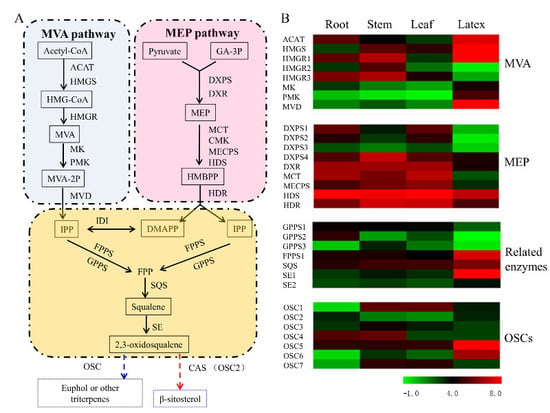
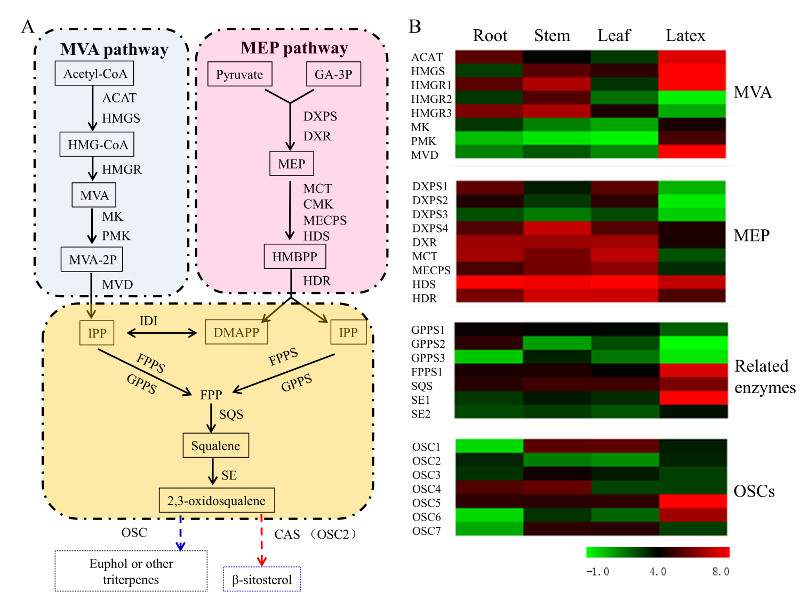
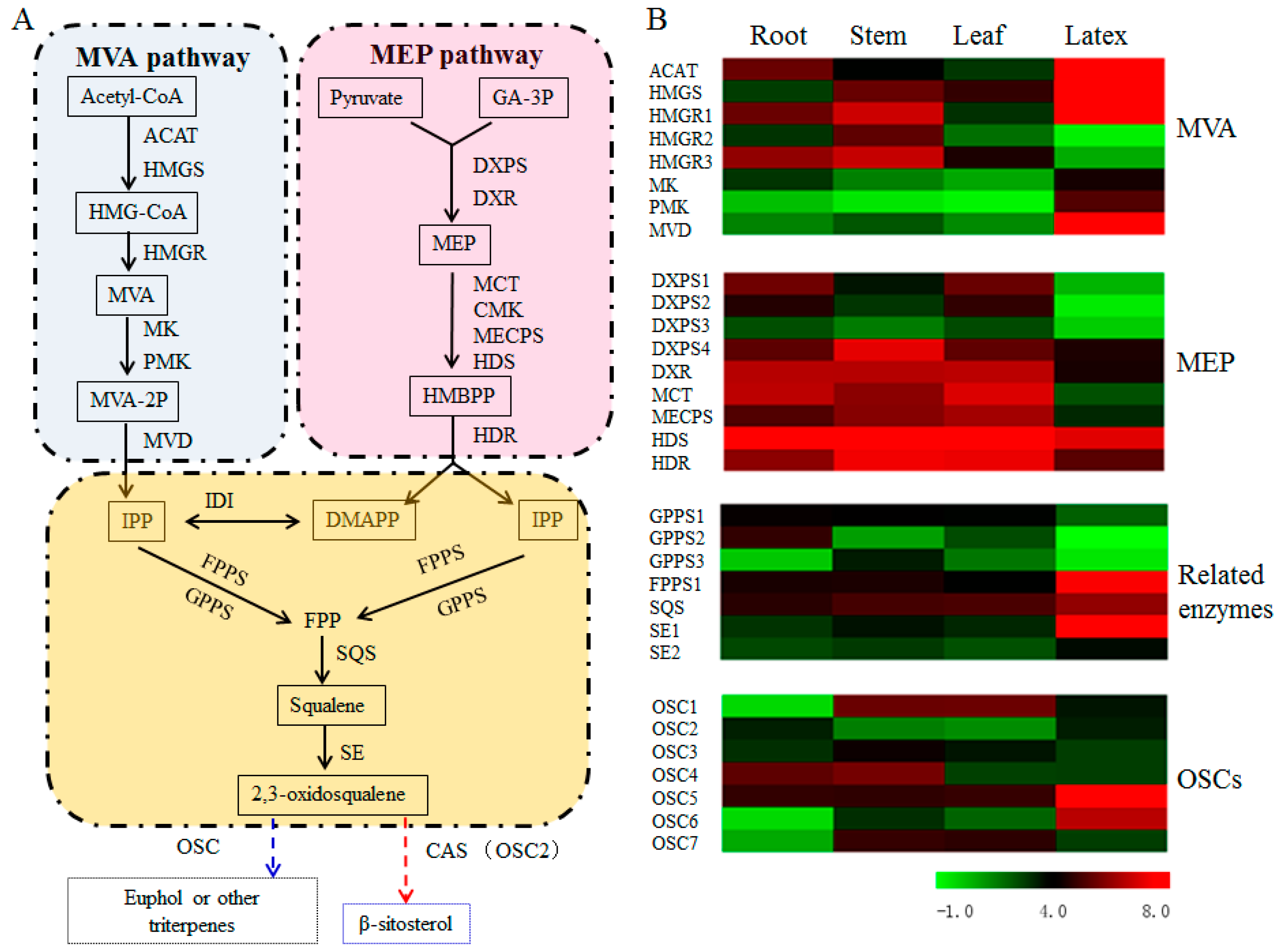
3.5. Transcript Abundance of the Upstream Pathway Genes Supported the Tissue Specificity for Euphol Biosynthesis in E. tirucalli
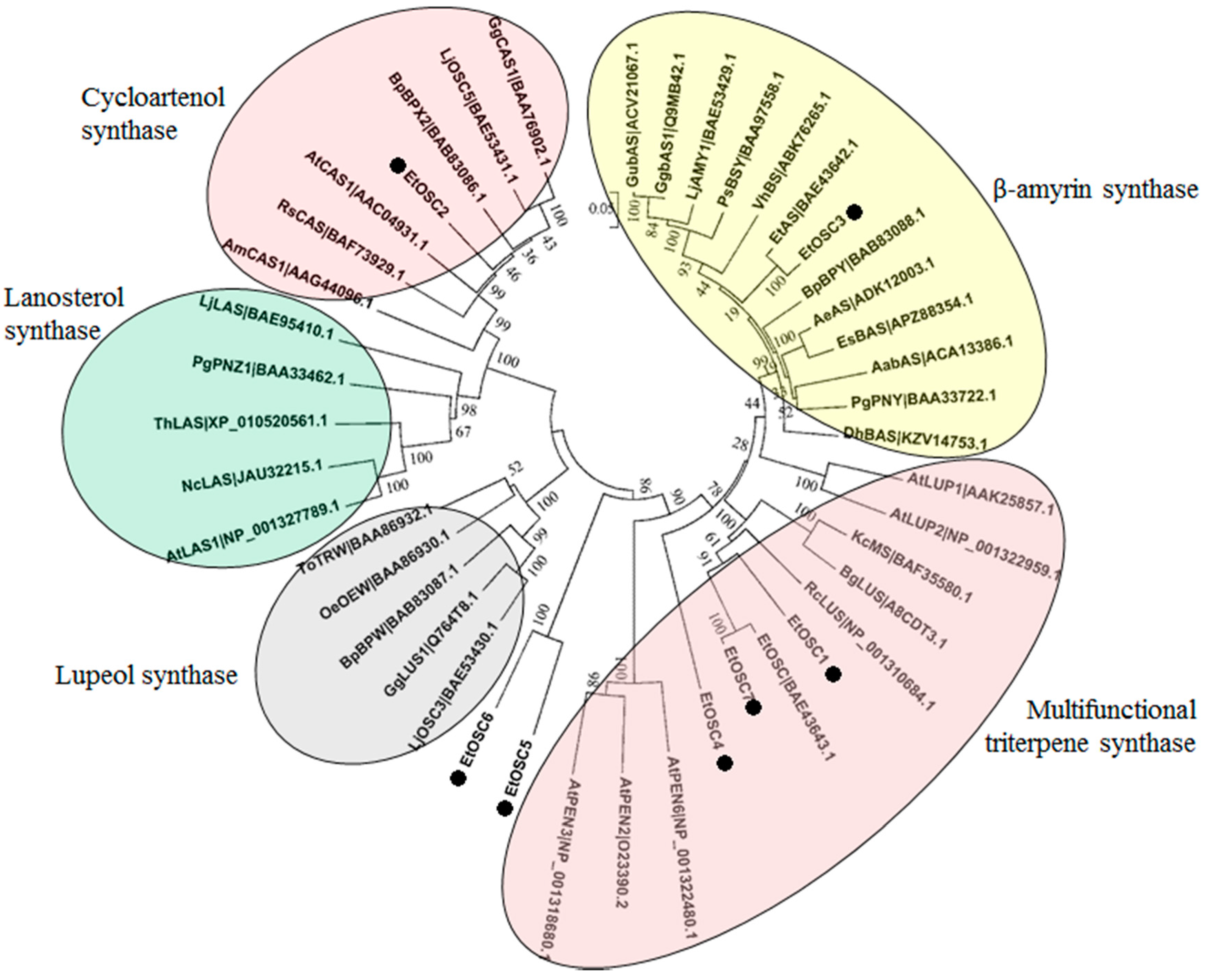
3.6. Identification of OSCs in the Transcriptome of E. tirucalli
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tokuoka, T.; Tobe, H. Embryology and systematics of Euphorbiaceaesens. lat.: A review and perspective. J. Plant Res. 1995, 108, 97–106. [Google Scholar] [CrossRef]
- Osato, T.; Mizuno, F.; Imai, S.; Aya, T.; Koizumi, S.; Kinoshita, T.; Tokuda, H.; Ito, Y.; Hirai, N.; Hirota, M. African Burkitt’s lymphoma and an Epstein-Barr virus-enhancing plant Euphorbia tirucalli. Lancet 1987, 1, 1257–1258. [Google Scholar] [CrossRef]
- Ernst, M.; Grace, O.M.; Saslislagoudakis, C.H.; Nilsson, N.; Simonsen, H.T.; Rønsted, N. Global medicinal uses of Euphorbia L. (Euphorbiaceae). J. Ethnopharmacol. 2015, 176, 90–101. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, L.F.; Fuentefria, A.M.; Klein Fda, S.; Machado, M.M. Antifungal activity against Cryptococcus neoformans strains and genotoxicity assessment in human leukocyte cells of Euphorbia tirucalli L. Braz. J. Microbiol. 2014, 45, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Bani, S.; Kaul, A.; Khan, B.; Gupta, V.K.; Satti, N.K.; Suri, K.A.; Qazi, G.N. Anti-arthritic activity of a biopolymeric fraction from Euphorbia tirucalli. J. Ethnopharmacol. 2007, 110, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Srivastava, V.K.; Chandra, R.; Singh, A. Larvicidal activity of latex and stem bark of Euphorbia tirucalli plant on the mosquito Culex quinquefasciatus. J. Commun. Dis. 2002, 34, 264–269. [Google Scholar] [PubMed]
- Betancur-Galvis, L.A.; Morales, G.E.; Forero, J.E.; Roldan, J. Cytotoxic and antiviral activities of Colombian medicinal plant extracts of the Euphorbia genus. Mem. Inst. Oswaldo Cruz 2002, 97, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Jurberg, P.; Cabral Neto, J.B.; Schall, V.T. Molluscicide activity of the “avelos” plant (Euphorbia tirucalli, L.) on Biomphalaria glabrata, the mollusc vector of schistosomiasis. Mem. Inst. Oswaldo Cruz 1985, 80, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Ahmed, Z.; Kazml, N.U.; Malik, A. Further triterpenes from the stem bark of Euphorbia tirucalli. Planta Med. 1987, 53, 577. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Kazmi, S.N.; Ahmed, Z.; Malik, A. Euphorcinol: A new pentacyclic triterpene from Euphorbia tirucalli. Planta Med. 1989, 55, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Fürstenberger, G.; Hecker, E. New highly irritant euphorbia factors from latex of Euphorbia tirucalli L. Experientia 1977, 33, 986–988. [Google Scholar] [CrossRef]
- Lin, M.W.; Lin, A.S.; Wu, D.C.; Wang, S.S.W.; Chang, F.R.; Wu, Y.C.; Huang, Y.B. Euphol from Euphorbia tirucalli selectively inhibits human gastric cancer cell growth through the induction of ERK1/2-mediated apoptosis. Food Chem. Toxicol. 2012, 50, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- Hastilestari, B.R.; Mudersbach, M.; Tomala, F.; Vogt, H.; Biskupekkorell, B.; Damme, P.V.; Guretzki, S.; Papenbrock, J. Euphorbia tirucalli L.—Comprehensive characterization of a drought tolerant plant with a potential as biofuel source. PLoS ONE 2013, 8, e63501. [Google Scholar] [CrossRef] [PubMed]
- Vriet, C.; Lemmens, K.; Vandepoele, K.; Reuzeau, C.; Russinova, E. Evolutionary trails of plant steroid genes. Trends Plant Sci. 2015, 20, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, K.; Suzuki, M.; Kikuchi, J.; Saito, K.; Muranaka, T. Dual biosynthetic pathways to phytosterol via cycloartenol and lanosterol in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Xiang, T.; Ohyama, K.; Seki, H.; Saito, K.; Muranaka, T.; Hayashi, H.; Katsube, Y.; Kushiro, T.; Shibuya, M. Lanosterol synthase in dicotyledonous plants. Plant Cell Physiol. 2006, 47, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, M.D.; Xiong, Q.; Lodeiro, S.; Hua, L.; Matsuda, S.P. Lanosterol biosynthesis in plants. Arch. Biochem. Biophys. 2006, 447, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Yamashita, H.; Kajikawa, M.; Ohyama, K.; Nakayachi, O.; Sugiyama, R.; Yamato, K.T.; Muranaka, T.; Fukuzawa, H.; Takemura, M. Cloning and characterization of a squalene synthase gene from a petroleum plant, Euphorbia tirucalli L. Planta 2009, 229, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Sugiyama, R.; Nakayachi, O.; Takemura, M.; Ohyama, K. Expression of the gene for sterol-biosynthesis enzyme squalene epoxidase in parenchyma cells of the oil plant, Euphorbia tirucalli. Planta 2007, 226, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, M.; Yamato, K.T.; Fukuzawa, H.; Sakai, Y.; Uchida, H.; Ohyama, K. Cloning and characterization of a cDNA encoding β-amyrin synthase from petroleum plant Euphorbia tirucalli L. Phytochemistry 2005, 66, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Lu, F.; Wang, S.; An, Y.; Su, X.; Li, X.; Ma, L.; Han, G. De novo sequencing and transcriptome analysis reveal key genes regulating steroid metabolism in leaves, roots, adventitious roots and calli of Periploca sepium bunge. Front. Plant Sci. 2017, 8, 594. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gou, J.; Chen, F.; Li, C.; Zhang, Y. Comparative transcriptome analysis identifies putative genes involved in the biosynthesis of xanthanolides in Xanthium strumarium L. Front. Plant Sci. 2016, 7, 1317. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Miura, K.; Aoki, W.; Yamato, K.T.; Taira, T.; Murakami, R.; Aburaya, S. Transcriptome and proteome analyses provide insight into laticifer’s defense of Euphorbia tirucalli against pests. Plant Physiol. Biochem. 2016, 108, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopaedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Mao, T. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; Garcíagómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.H.; Mercer, E.I.; Goodwin, T.W. The stereospecific biosynthesis of plant sterols and alpha- and beta-amyrin. Biochem. J. 1966, 99, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.Z.; Zhao, X.Y.; Deng, K.Q.; Xu, L.I.; Liu, X.M. Development on the molecular biology of oxidosqualene cyclase-the key enzyme in sterols biosynthesis. Life Sci. Res. 2007, 11, 10–15. [Google Scholar]
- Kushiro, T.; Shibuya, M.; Ebizuka, Y. Mutational studies on triterpene synthases: Engineering lupeol synthase into β-amyrin synthase. J. Am. Chem. Soc. 2000, 122, 6816–6824. [Google Scholar] [CrossRef]
- Morlacchi, P.; Wilson, W.K.; Xiong, Q.; Bhaduri, A.; Sttivend, D.; Kolesnikova, M.D.; Matsuda, S.P.T. Product profile of PEN3: The last unexamined oxidosqualene cyclase in Arabidopsis thaliana. Org. Lett. 2009, 11, 2627–2630. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Vishnoi, G.; Wal, A.; Wal, P. Medicinal value of Euphorbia tirucalli. Int. J. Pharm. Biol. Arch. 2013, 4, 31–40. [Google Scholar] [CrossRef]
- Rosenwasser, S.; Mausz, M.A.; Schatz, D.; Sheyn, U.; Malitsky, S.; Aharoni, A.; Weinstock, E.; Tzfadia, O.; Ben-Dor, S.; Feldmesser, E. Rewiring host lipid metabolism by large viruses determines the fate of Emiliania huxleyi, a bloom-forming alga in the ocean. Plant Cell 2014, 26, 2689–2707. [Google Scholar] [CrossRef] [PubMed]
- Dhar, N.; Rana, S.; Razdan, S.; Bhat, W.W.; Hussain, A.; Dhar, R.S.; Vaishnavi, S.; Hamid, A.; Vishwakarma, R.; Lattoo, S.K. Cloning and functional characterization of three branch point oxidosqualene cyclases from Withania somnifera (L.) dunal. J. Biol. Chem. 2014, 289, 17249–17267. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Sample | Number (n) | Total Nucleotides (G nt) | Valid Ratio (%) | GC (%) | Q30 (%) | N50 (bp) | Average Length (bp) |
|---|---|---|---|---|---|---|---|---|
| Raw reads | Latex | 19,838,038 | 1.98 | — | — | — | — | |
| Root | 44,125,666 | 6.61 | — | — | — | — | ||
| Stem | 54,363,252 | 8.15 | — | — | — | — | ||
| Leaf | 54,091,184 | 8.11 | — | — | — | — | ||
| Clean reads | Latex | 19,590,464 | 1.95 | 98.48 | 42.71 | 96.34 | — | — |
| Root | 42,271,760 | 6.34 | 95.92 | 43.15 | 89.50 | — | — | |
| Stem | 52,432,630 | 7.86 | 96.44 | 42.98 | 90.12 | — | — | |
| Leaf | 50,429,140 | 7.56 | 93.22 | 43.53 | 87.22 | — | — | |
| Total | Unigenes | 91,619 | 0.12 | — | — | — | 2099 | 1351 |
| Database | Number of Unigenes | Percentage (%) b |
|---|---|---|
| Nr a | 46,804 | 51.08 |
| SwissProt | 36,923 | 40.30 |
| KOG | 13,292 | 14.50 |
| GO | 38,652 | 42.18 |
| KEGG | 17,447 | 19.04 |
| Annotated in all databases | 7835 | 8.55 |
| Annotated in at least one database | 52,448 | 57.24 |
| Pathway Term | Number | Pathway Term | Number | Pathway Term | Number | |
|---|---|---|---|---|---|---|
| Latex vs. Leaf | Latex vs. Root | Latex vs. Stem | ||||
| Up-regulated DEGs in latex compared to other tissues | Sesquiterpenoid and triterpenoid biosynthesis | 19 | Sesquiterpenoid and triterpenoid biosynthesis | 19 | Sesquiterpenoid and triterpenoid biosynthesis | 17 |
| Endocytosis | 32 | Photosynthesis—antenna proteins | 11 | Terpenoid backbone biosynthesis | 15 | |
| Regulation of autophagy | 15 | Photosynthesis | 14 | Peroxisome | 25 | |
| Terpenoid backbone biosynthesis | 15 | Galactose metabolism | 17 | Endocytosis | 29 | |
| Galactose metabolism | 17 | Regulation of autophagy | 14 | Galactose metabolism | 15 | |
| Steroid biosynthesis | 11 | Butanoate metabolism | 8 | Synthesis and degradation of ketone bodies | 3 | |
| Ether lipid metabolism | 8 | Pyruvate metabolism | 23 | Regulation of autophagy | 12 | |
| Fatty acid biosynthesis | 13 | Terpenoid backbone biosynthesis | 13 | Protein processing in endoplasmic reticulum | 33 | |
| Synthesis and degradation of ketone bodies | 3 | Steroid biosynthesis | 10 | Fatty acid biosynthesis | 12 | |
| Peroxisome | 22 | Peroxisome | 22 | Steroid biosynthesis | 9 | |
| Down-regulated DEGs in latex compared to other tissues | Photosynthesis | 41 | Ribosome | 143 | Photosynthesis | 39 |
| Oxidative phosphorylation | 54 | Plant hormone signal transduction | 57 | Oxidative phosphorylation | 56 | |
| Photosynthesis - antenna proteins | 14 | Oxidative phosphorylation | 43 | Carbon fixation in photosynthetic organisms | 34 | |
| Porphyrin and chlorophyll metabolism | 20 | Citrate cycle (TCA cycle) | 19 | Photosynthesis - antenna proteins | 14 | |
| Carbon fixation in photosynthetic organisms | 28 | Carotenoid biosynthesis | 15 | Pentose phosphate pathway | 28 | |
| Carotenoid biosynthesis | 17 | Flavonoid biosynthesis | 12 | Porphyrin and chlorophyll metabolism | 20 | |
| Limonene and pinene degradation | 15 | Stilbenoid, diarylheptanoid and gingerol biosynthesis | 9 | Glycolysis/Gluconeogenesis | 46 | |
| Ribosome | 70 | Phenylpropanoid biosynthesis | 33 | Ribosome | 72 | |
| Cell cycle—caulobacter | 12 | Spliceosome | 46 | Plant hormone signal transduction | 47 | |
| nitrogen metabolism | 15 | Cysteine and methionine metabolism | 25 | Glyoxylate and dicarboxylate metabolism | 25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, W.; Li, C.; Mosongo, I.; Liang, Q.; Liu, M.; Wang, X. Comparative Transcriptome Analysis Identifies Putative Genes Involved in Steroid Biosynthesis in Euphorbia tirucalli. Genes 2018, 9, 38. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9010038
Qiao W, Li C, Mosongo I, Liang Q, Liu M, Wang X. Comparative Transcriptome Analysis Identifies Putative Genes Involved in Steroid Biosynthesis in Euphorbia tirucalli. Genes. 2018; 9(1):38. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9010038
Chicago/Turabian StyleQiao, Weibo, Changfu Li, Isidore Mosongo, Qin Liang, Mengdi Liu, and Xin Wang. 2018. "Comparative Transcriptome Analysis Identifies Putative Genes Involved in Steroid Biosynthesis in Euphorbia tirucalli" Genes 9, no. 1: 38. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9010038