Assembly of a Complete Mitogenome of Chrysanthemum nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae Mitogenomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Sequencing Data
2.2. Assembly of the Mitogenome Using Three Different Strategies
2.3. Annotation of the Mitogenome
2.4. Identification of Repeat-Mediated Recombination
2.5. Assemblies and Annotations of Asteraceae Organelle Draft Genomes
2.6. Analysis of Asteraceae Mitogenomes Constitution
2.7. Phylogenetic Analysis of Asteraceae Organelle Genomes
2.8. Data Accessibility
3. Results
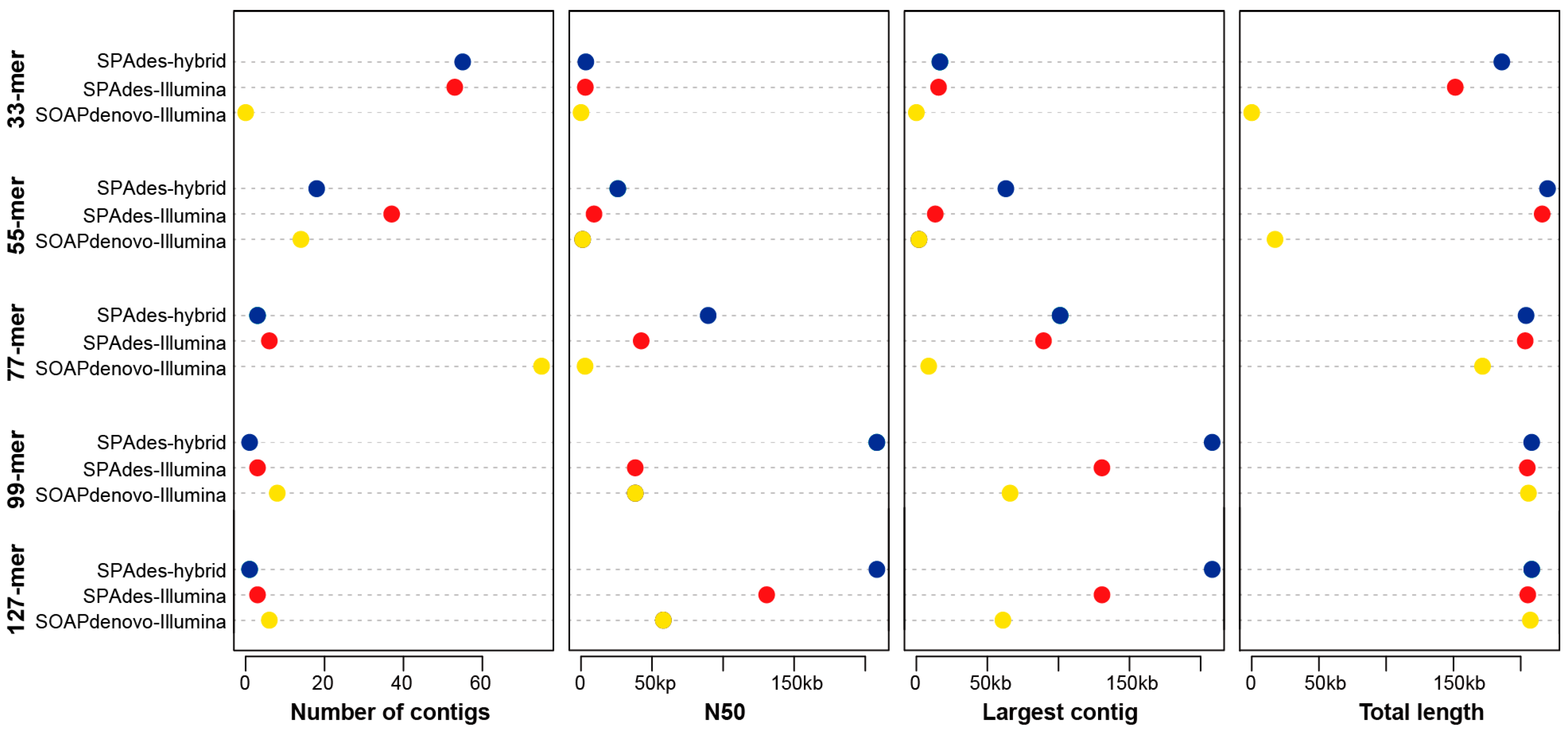
3.1. Comparison of Different Assembly Strategies
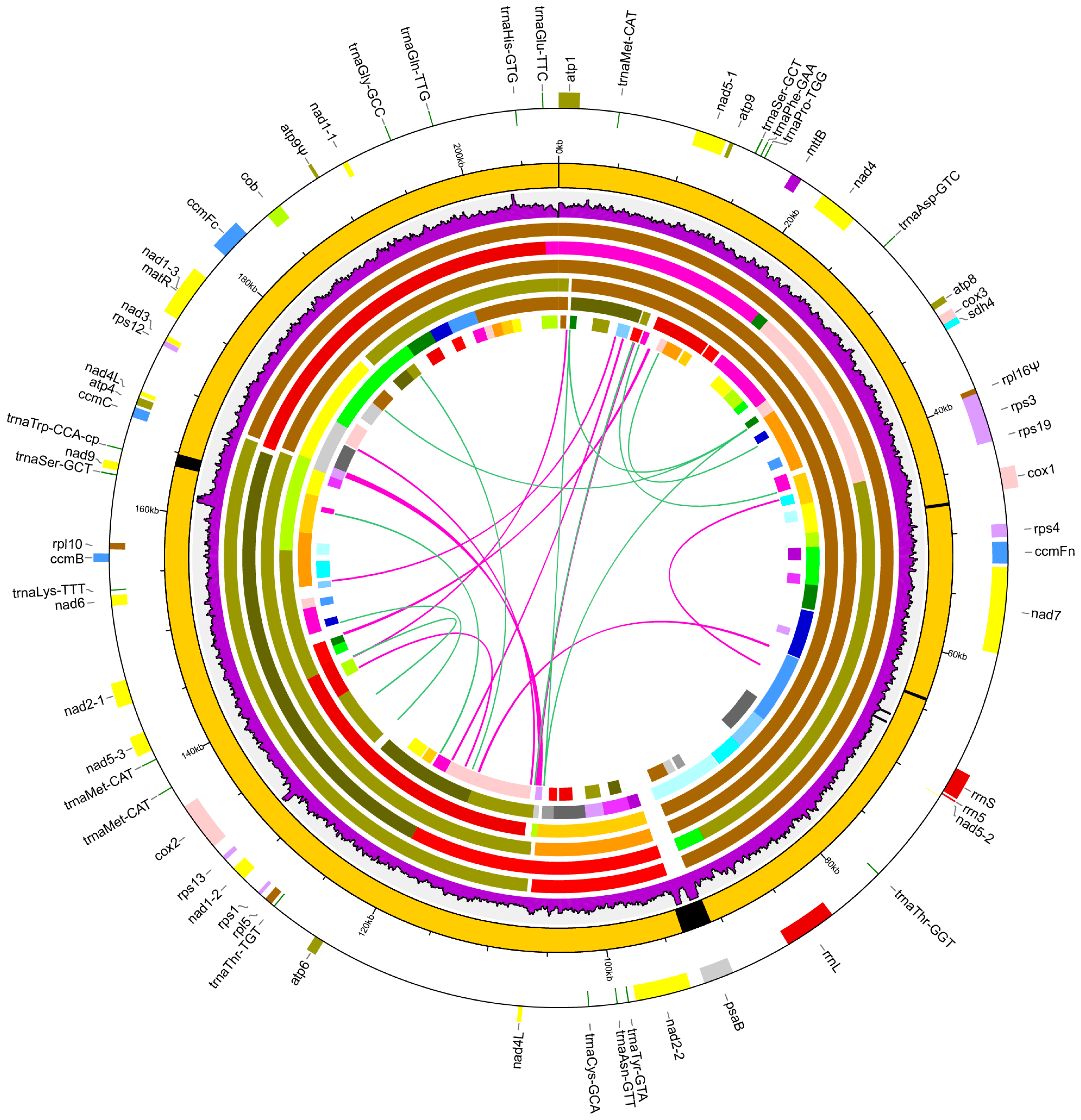
3.2. General Features of the Chrysanthemum nankingense Mitogenome
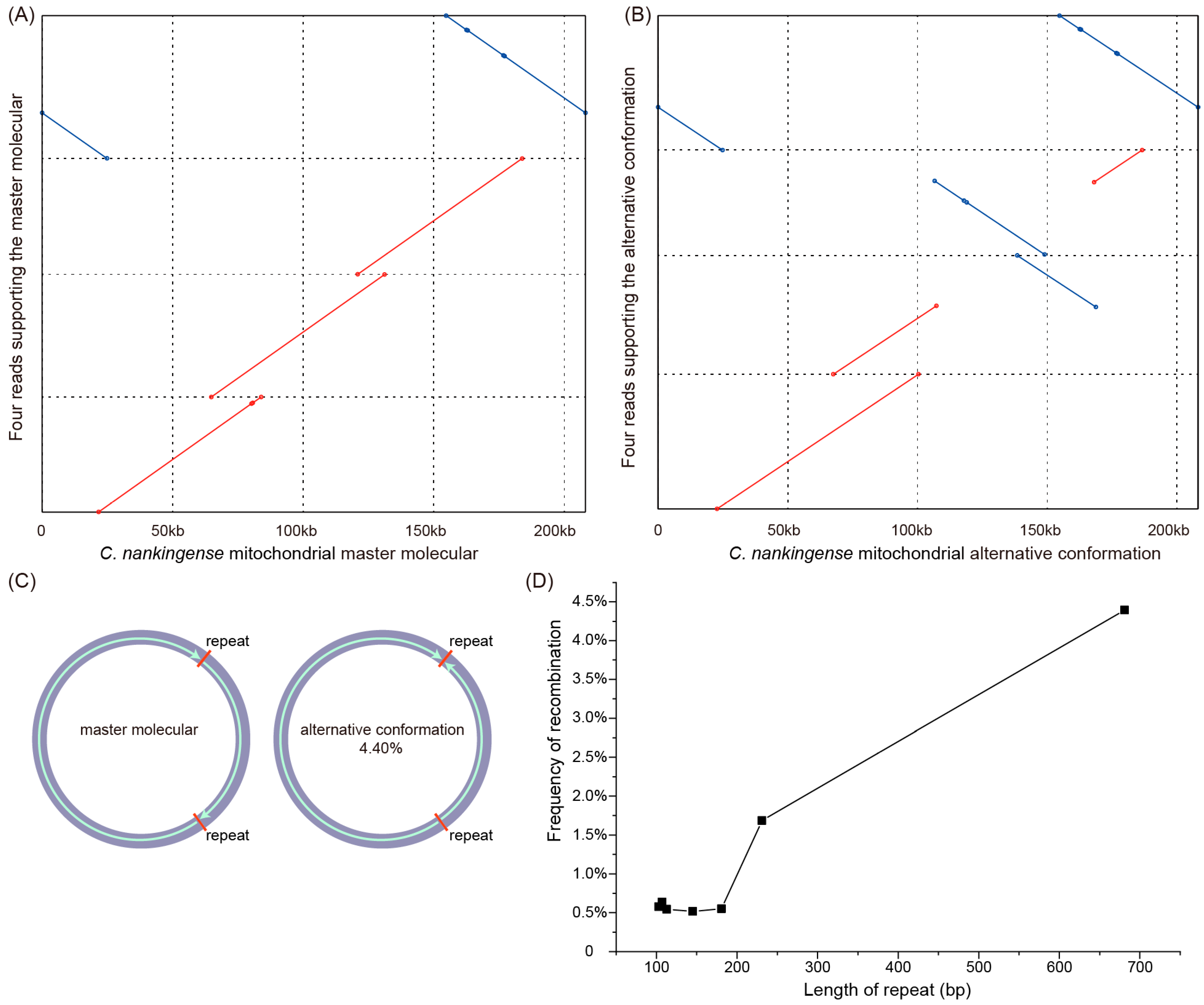
3.3. Repeats and Homologous Recombination
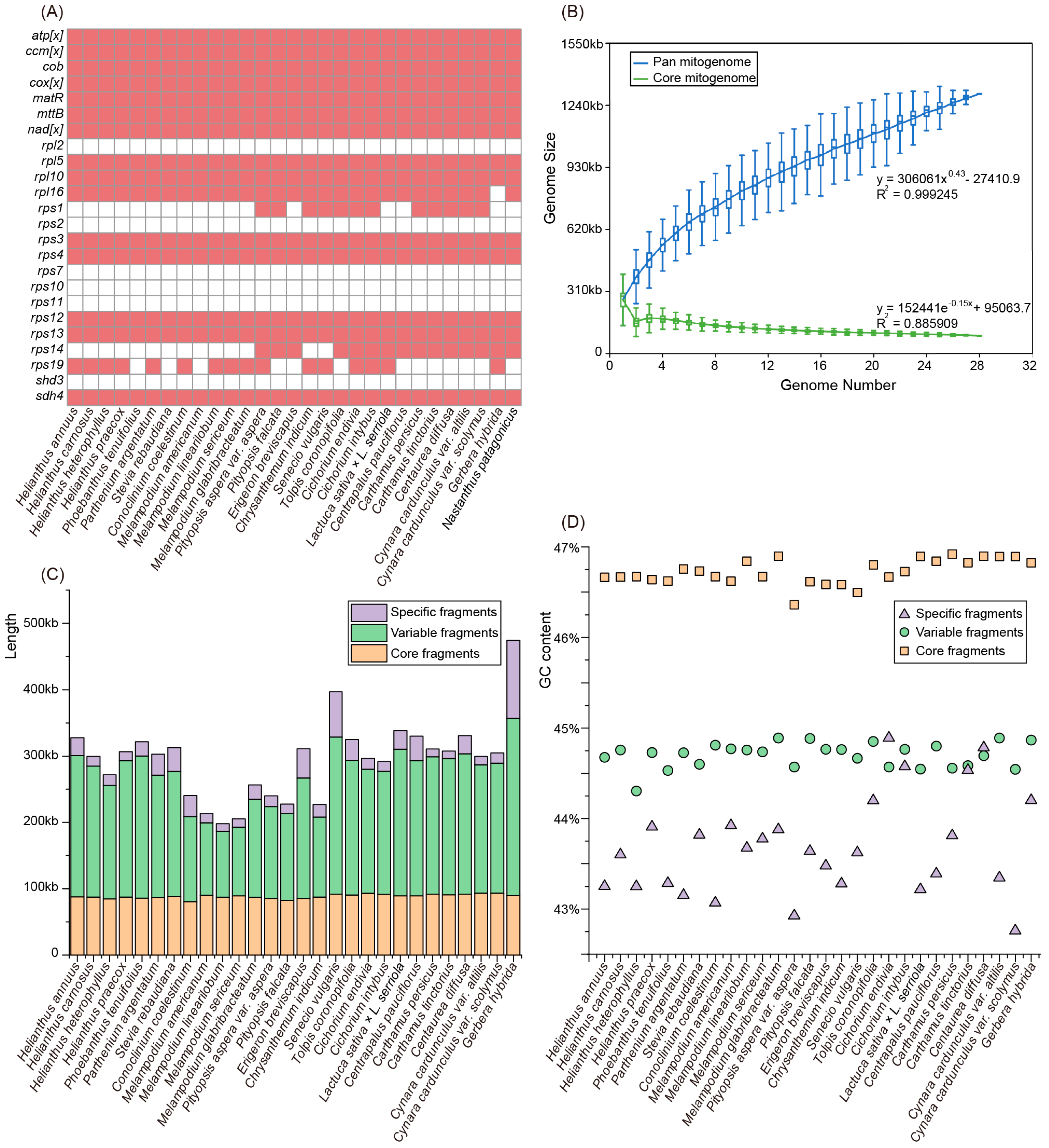
3.4. The Assemblies of the Asteraceae Mitogenomes
3.5. Variation in the Gene Content and Constitution in the Asteraceae Mitogenomes
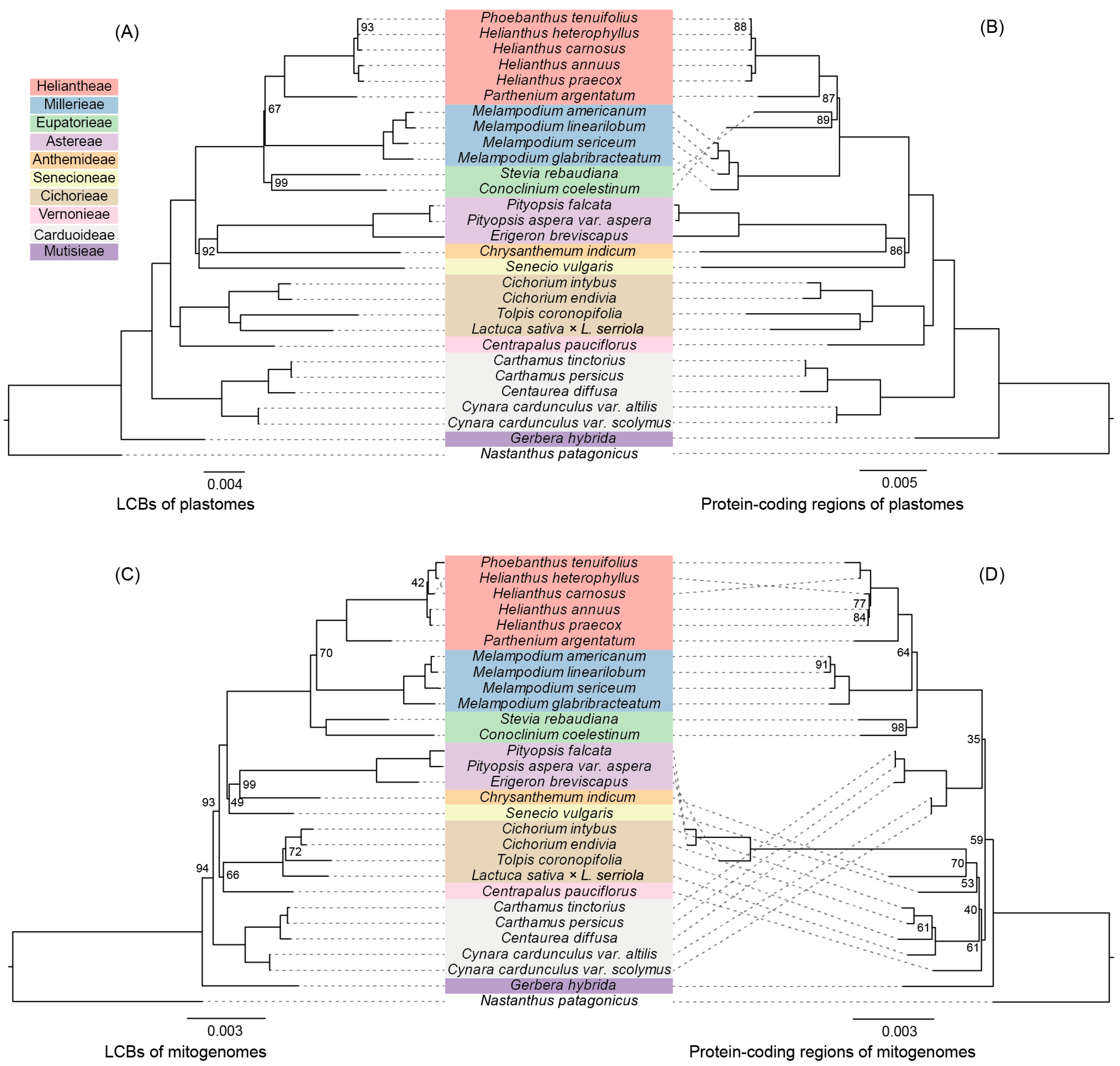
3.6. Phylogenetic Analysis Comparison
4. Discussion
4.1. The Effectiveness of Oxford Nanopore Technology in Mitogenome Research
4.2. The Diversity and Evolution of Asteraceae Mitogenomes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, B.L.; Anderson, R.S.; Bendich, A.J. The mitochondrial genome is large and variable in a family of plants (cucurbitaceae). Cell 1981, 25, 793–803. [Google Scholar] [CrossRef]
- Park, S.; Grewe, F.; Zhu, A.; Ruhlman, T.A.; Sabir, J.; Mower, J.P.; Jansen, R.K. Dynamic evolution of Geranium mitochondrial genomes through multiple horizontal and intracellular gene transfers. New Phytol. 2015, 208, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Paer, C.V.D.; Bouchez, O.; Besnard, G. Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 2017, 18, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. Using plants to elucidate the mechanisms of cytonuclear co-evolution. New Phytol. 2015, 205, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Horner, D.S.; Chiara, M.; Schiavon, R.; Valle, G.; Pesole, G. Large-scale detection and analysis of RNA editing in grape mtDNA by RNA deep-sequencing. Nucleic Acids Res. 2010, 38, 4755–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedge, V.; Arrieta-Montiel, M.; Christensen, A.C.; Mackenzie, S.A. Plant mitochondrial recombination surveillance requires unusual RecA and MutS homologs. Plant Cell 2007, 19, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, e3515–e3524. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Palmer, J.D. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenetics Evol. 2003, 29, 380–395. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Moreno, L.; Gonzalez, V.M.; Benjak, A.; Carmen Marti, M.; Puigdomenech, P.; Aranda, M.A.; Garcia-Mas, J. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genom. 2011, 12, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant mitochondrial genome diversity: The genomics revolution. In Plant Genome Diversity Volume 1: Plant Genomes, Their Residents, and Their Evolutionary Dynamics; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012; pp. 123–144. [Google Scholar]
- Sloan, D.B.; Muller, K.; McCauley, D.E.; Taylor, D.R.; Storchova, H. Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 2012, 196, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhao, Y.; Kong, X.; Khan, A.; Zhou, B.; Liu, D.; Kashif, M.H.; Chen, P.; Wang, H.; Zhou, R. Complete sequence of kenaf (Hibiscus cannabinus) mitochondrial genome and comparative analysis with the mitochondrial genomes of other plants. Sci. Rep. 2018, 8, 12714. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef] [PubMed]
- Funk, V.A.; Susanna, A.; Stuessy, T.F.; Robinson, H. Classification of compositae. In Systematics, Evolution, and Biogeography of Compositae; Funk, V.A., Susanna, A., Stuessy, T.F., Bayer, R.J., Eds.; International Association for Plant Taxonomy (IAPT): Vienna, Austria, 2009; pp. 171–192. [Google Scholar]
- Bock, D.G.; Kane, N.C.; Ebert, D.P.; Rieseberg, L.H. Genome skimming reveals the origin of the jerusalem artichoke tuber crop species: Neither from jerusalem nor an artichoke. New Phytol. 2014, 201, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, M.S.; Kornienko, I.V.; Azarin, K.V.; Usatov, A.V.; Logacheva, M.D.; Markin, N.V.; Gavrilova, V.A. Mitochondrial genomes organization in alloplasmic lines of sunflower (Helianthus annuus L.) with various types of cytoplasmic male sterility. PeerJ 2018, 6, e5266. [Google Scholar] [CrossRef] [PubMed]
- Rothe, S.P.; Bokhad, M.N.; Kakpure, M.R. Medicinal significance of ornamental plants in human welfare from Akola district (M.S.) India. J. Exp. Sci. 2011, 2, 1807. [Google Scholar]
- Song, C.; Liu, Y.; Song, A.; Dong, G.; Zhao, H.; Sun, W.; Ramakrishnan, S.; Wang, Y.; Wang, S.; Li, T.; et al. The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant 2018. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. Nanopack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. HybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Team, R.D.C. R: A language and environment for statistical computing. Computing 2009, 14, 12–21. [Google Scholar]
- Kolde, R. Package ‘Pheatmap’. 2018. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 11 November 2018).
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cuthbert, J.M.; Taylor, D.R.; Sloan, D.B. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 10185–10191. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Virginia Sanchez-Puerta, M.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; dePamphilis, C.W.; et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-O.; Choi, J.-W.; Baek, J.-H.; Oh, J.-H.; Lee, S.-C.; Kim, C.-K. Assembly of the mitochondrial genome in the campanulaceae family using Illumina low-coverage sequencing. Genes 2018, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Van de Paer, C.; Hong-Wa, C.; Jeziorski, C.; Besnard, G. Mitogenomics of Hesperelaea, an extinct genus of Oleaceae. Gene 2016, 594, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.H.; Grewe, F.; Fan, W.S.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Arrieta-Montiel, M.P.; Shedge, V.; Davila, J.; Christensen, A.C.; Mackenzie, S.A. Diversity of the arabidopsis mitochondrial genome occurs via nuclear-controlled recombination activity. Genetics 2009, 183, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Fauron, C.M.R.; Moore, B.; Casper, M. Maize as a model of higher plant mitochondrial genome plasticity. Plant Sci. 1995, 112, 11–32. [Google Scholar] [CrossRef]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, O.M.; Ortiz, E.M.; Simpson, B.B. Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 2017, 214, 1736–1750. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Zhang, C.; Liu, M.; Hu, Y.; Gao, T.; Qi, J.; Ma, H. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 2016, 33, 2820–2835. [Google Scholar] [CrossRef] [PubMed]
- Lee-Yaw, J.A.; Grassa, C.J.; Joly, S.; Andrew, R.L.; Rieseberg, L.H. An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). New Phytol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rydin, C.; Wikstrom, N.; Bremer, B. Conflicting results from mitochondrial genomic data challenge current views of Rubiaceae phylogeny. Am. J. Bot. 2017, 104, 1522–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Song, Q.; Li, S.; Hu, Z.; Dong, G.; Song, C.; Huang, H.; Liu, Y. Assembly of a Complete Mitogenome of Chrysanthemum nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae Mitogenomes. Genes 2018, 9, 547. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110547
Wang S, Song Q, Li S, Hu Z, Dong G, Song C, Huang H, Liu Y. Assembly of a Complete Mitogenome of Chrysanthemum nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae Mitogenomes. Genes. 2018; 9(11):547. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110547
Chicago/Turabian StyleWang, Shuaibin, Qingwei Song, Shanshan Li, Zhigang Hu, Gangqiang Dong, Chi Song, Hongwen Huang, and Yifei Liu. 2018. "Assembly of a Complete Mitogenome of Chrysanthemum nankingense Using Oxford Nanopore Long Reads and the Diversity and Evolution of Asteraceae Mitogenomes" Genes 9, no. 11: 547. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110547