Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Material
2.2. DNA Isolation
2.3. Bacterial Artificial Chromosome Library Screening
2.4. Contig Assembly and Bacterial Artificial Chromosome Clone Sequencing
2.5. Functional Annotation of Bacterial Artificial Chromosome Sequences
2.6. Fluorescent In Situ Hybridization
2.7. Droplet Digital PCR
2.8. Genetic Mapping
2.9. Identification of Homolog Sequences in Legumes
2.10. Microsynteny Analysis
2.11. Phylogenetic Survey
2.12. Selection Pressure Analysis
2.13. In Silico Gene Expression Assay
3. Results
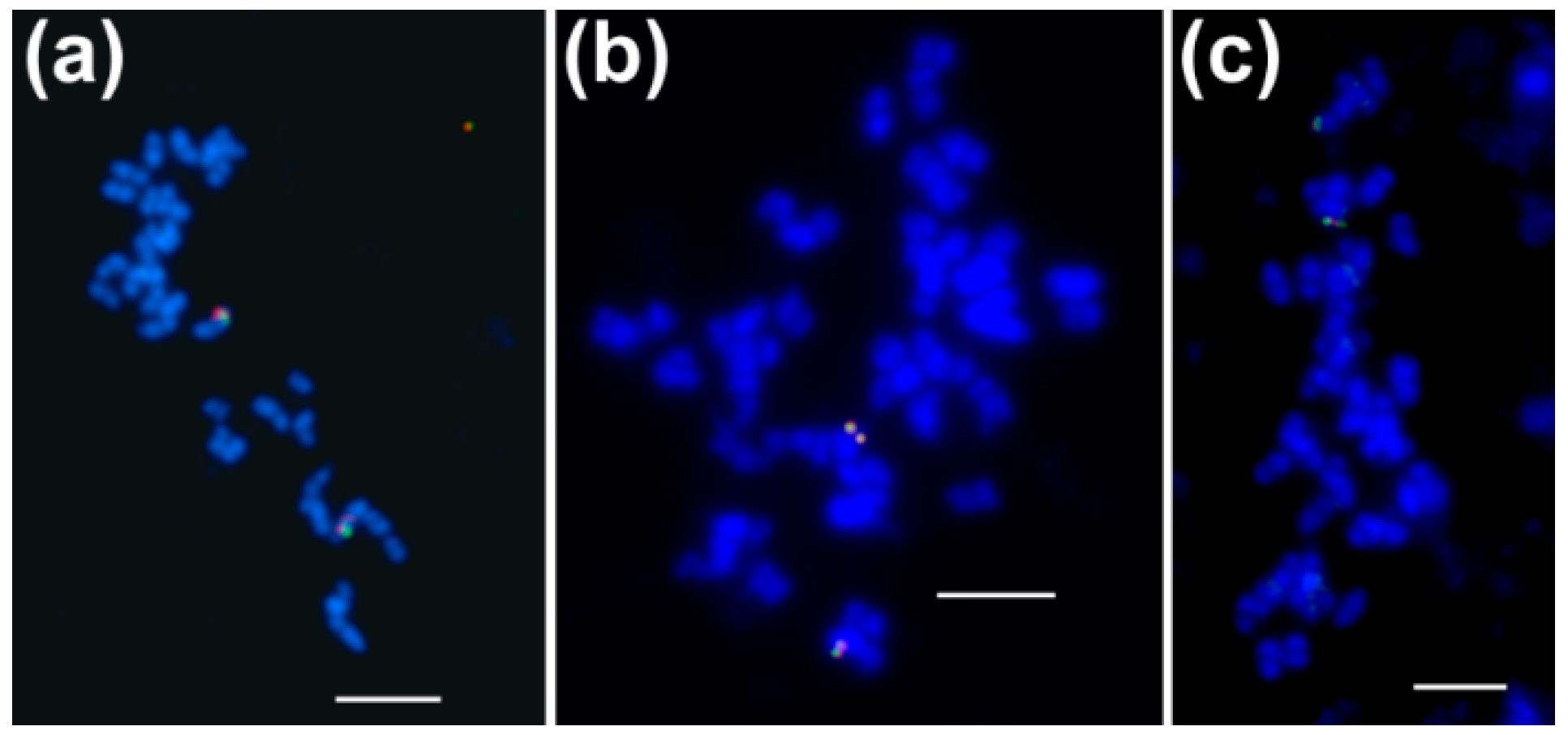
3.1. Both Homologous and Heterologous Probe(s) Were Applicable to Select BAC Clones Carrying Genes Encoding Cytosolic ACCase and Subunits of Plastid ACCase
3.2. Genes Encoding Cytosolic ACCase and Subunits of Plastid ACCase are Located in Different L. angustifolius Chromosomes
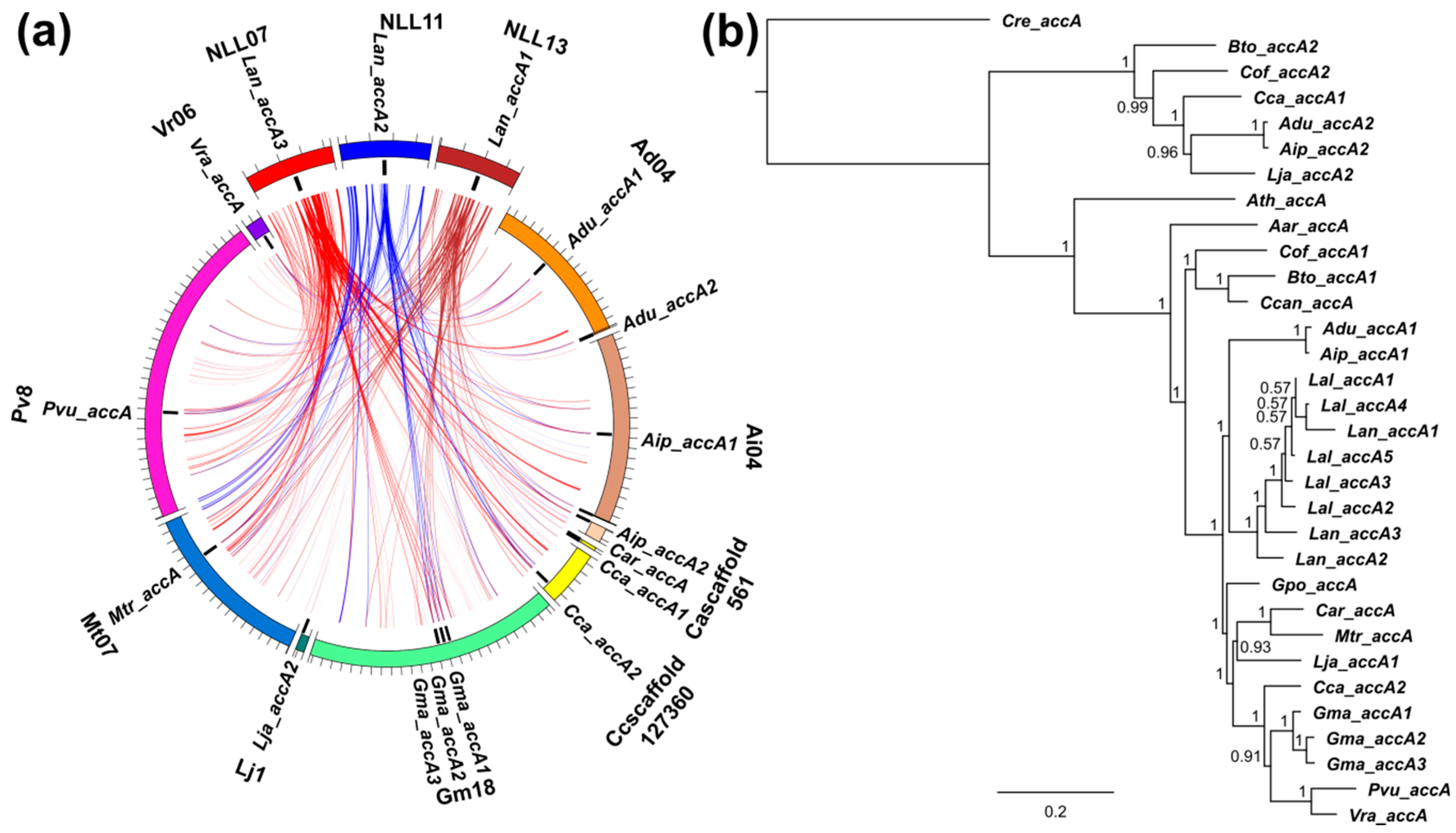
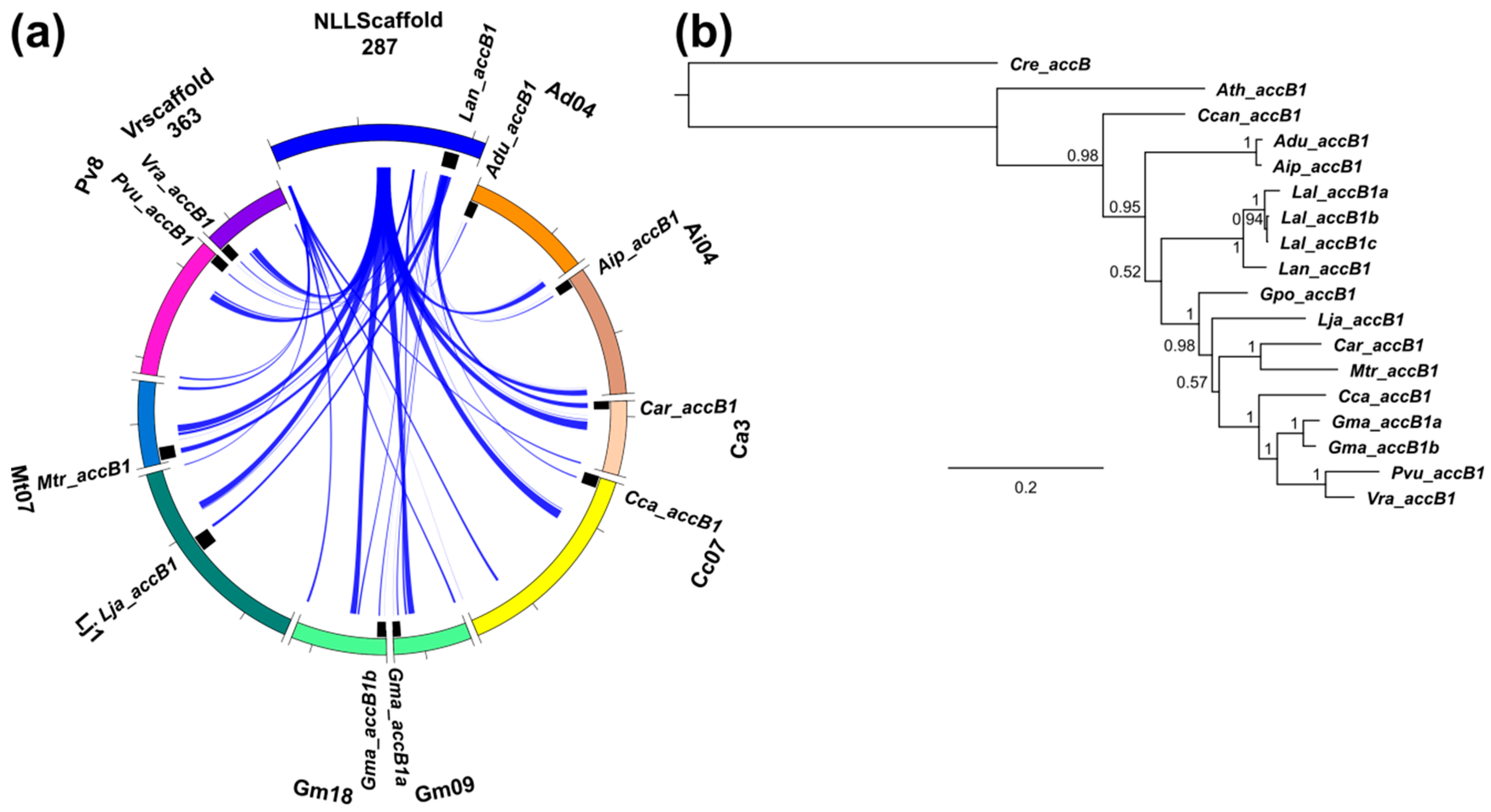
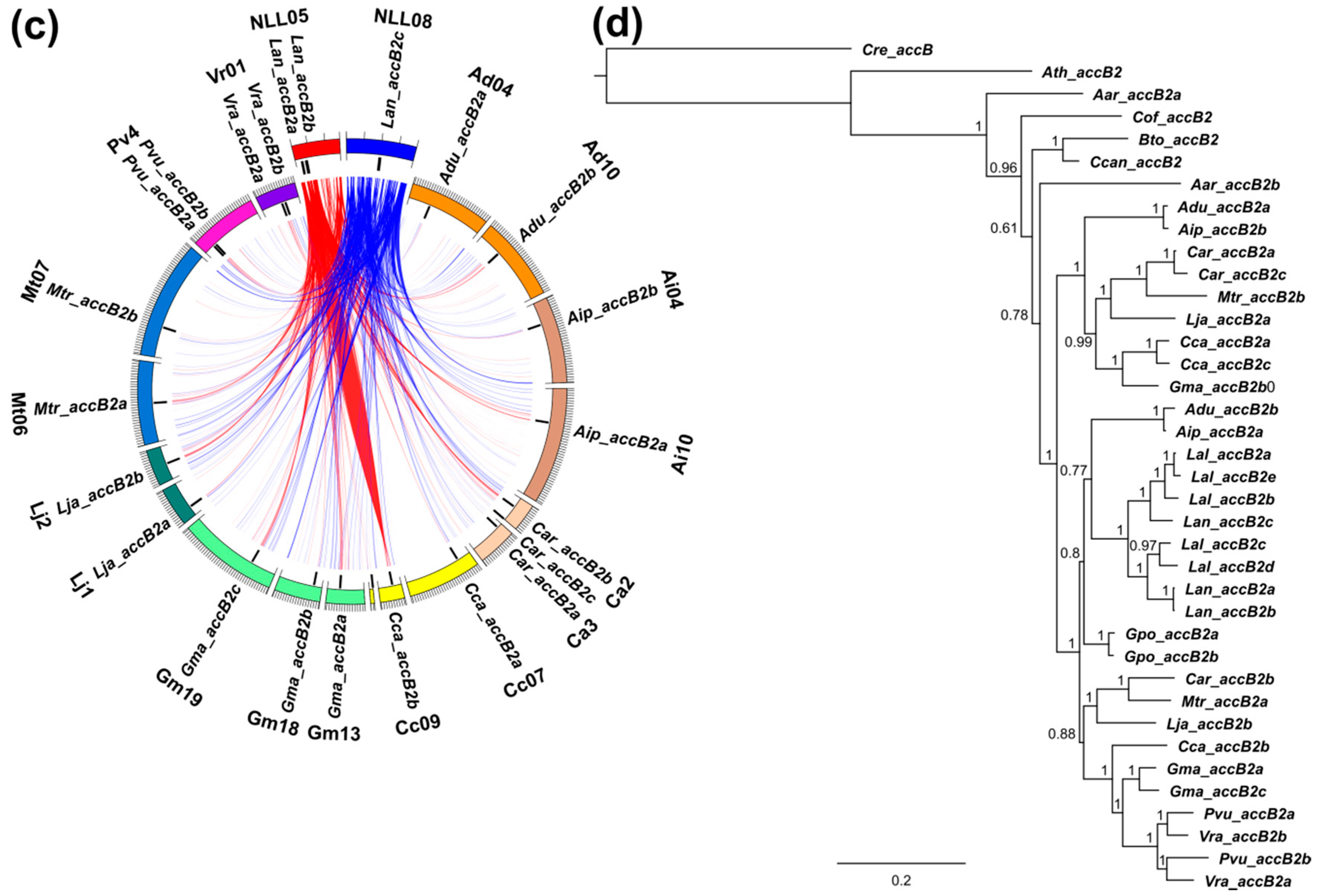
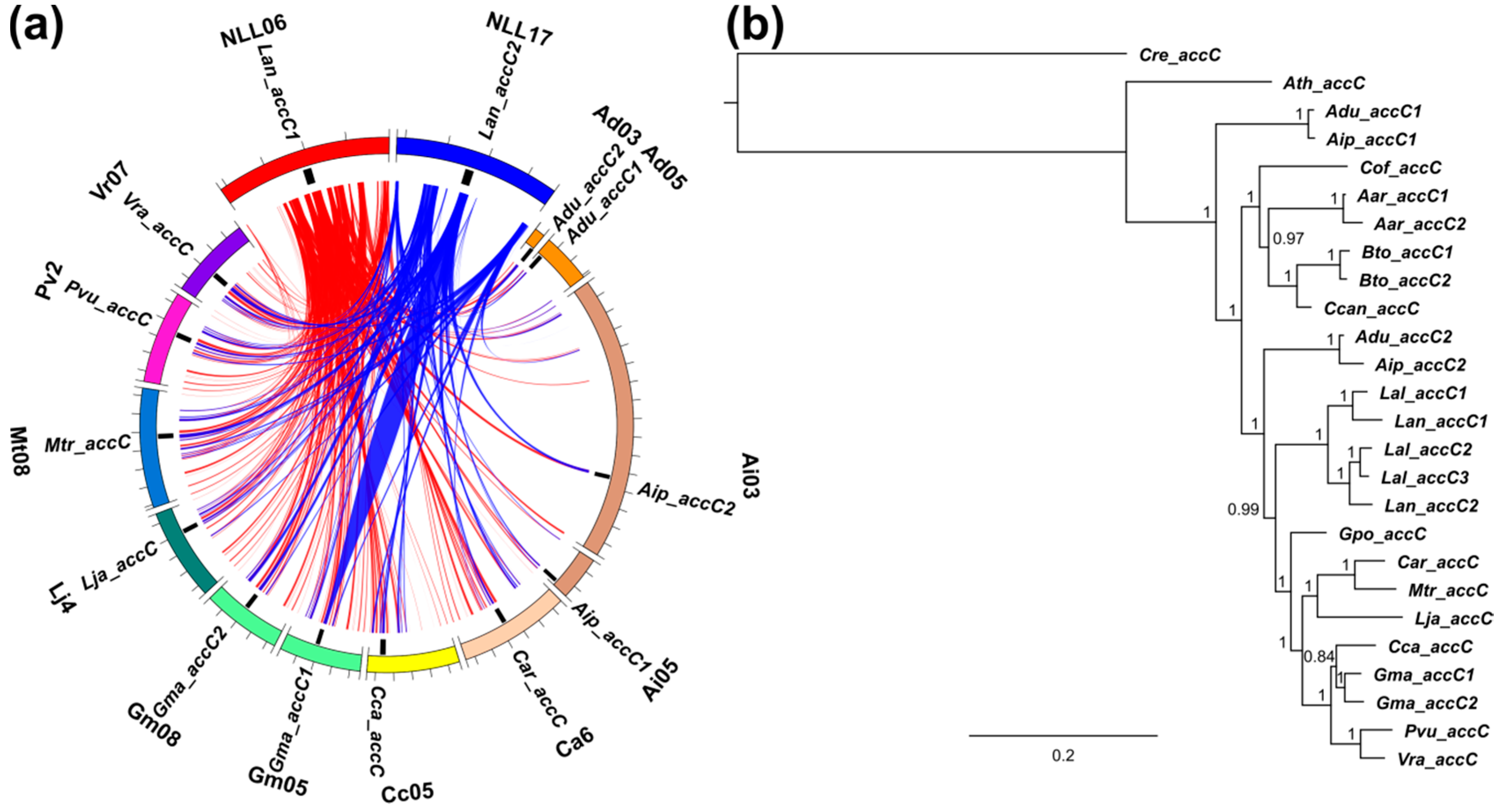
3.3. Lupins and Soybean Have Duplicates of All Nuclear ACCase Genes (ACC, accA, accB and accC)
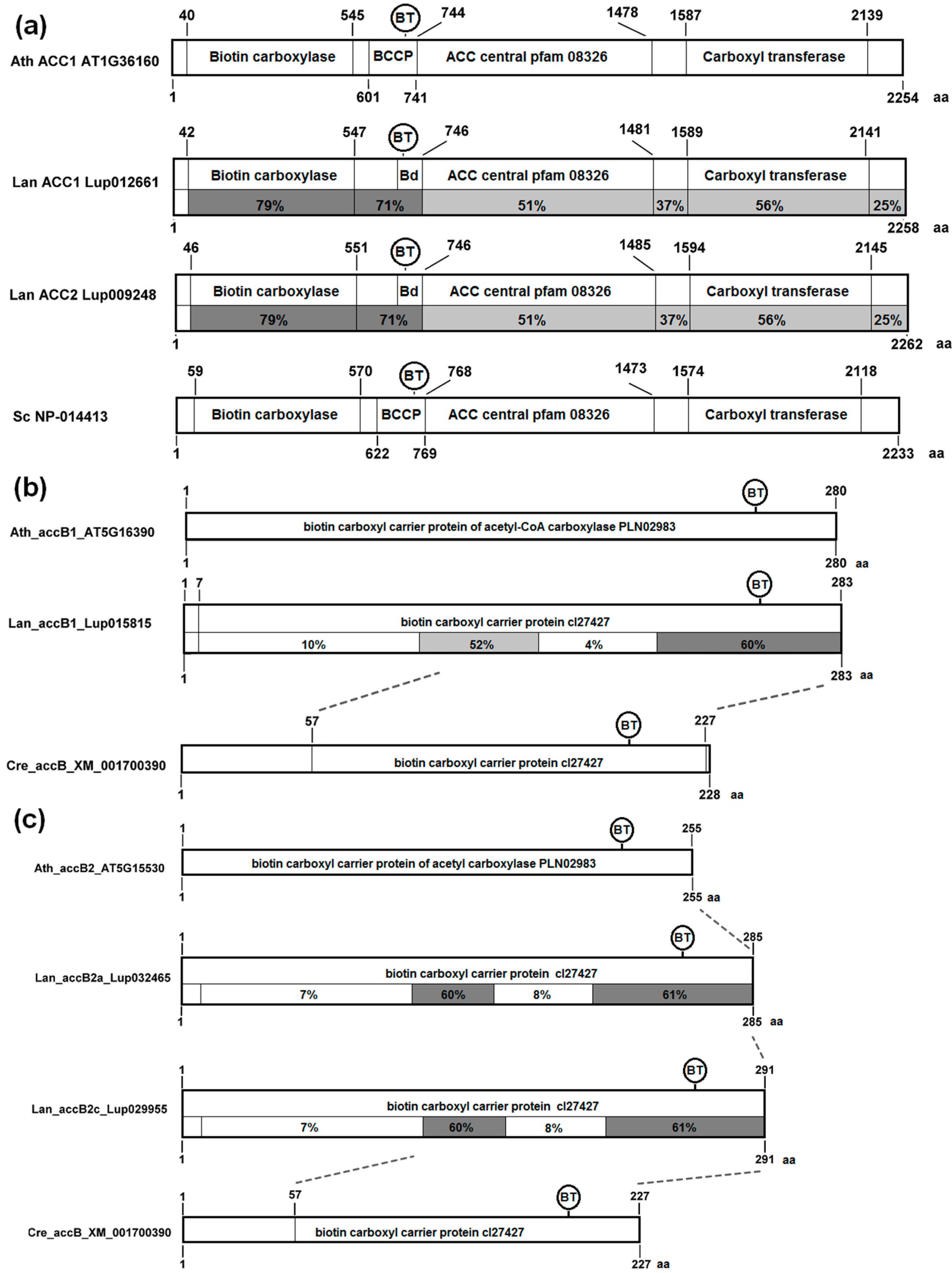
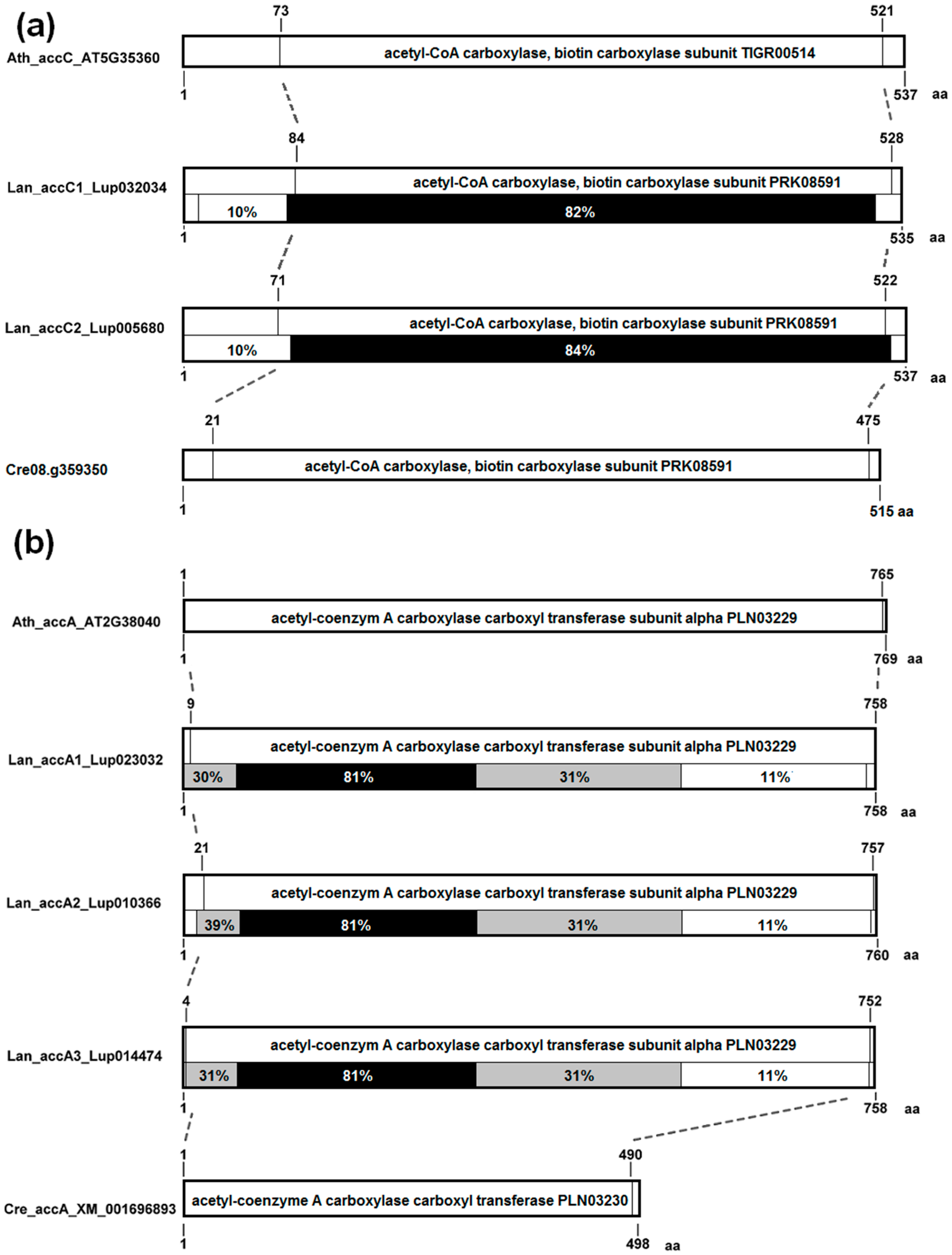
3.4. The Structure of Nuclear Genes Encoding Cytosolic ACCase and Subunits of Plastid ACCase Is Highly Conserved among the Legume Family
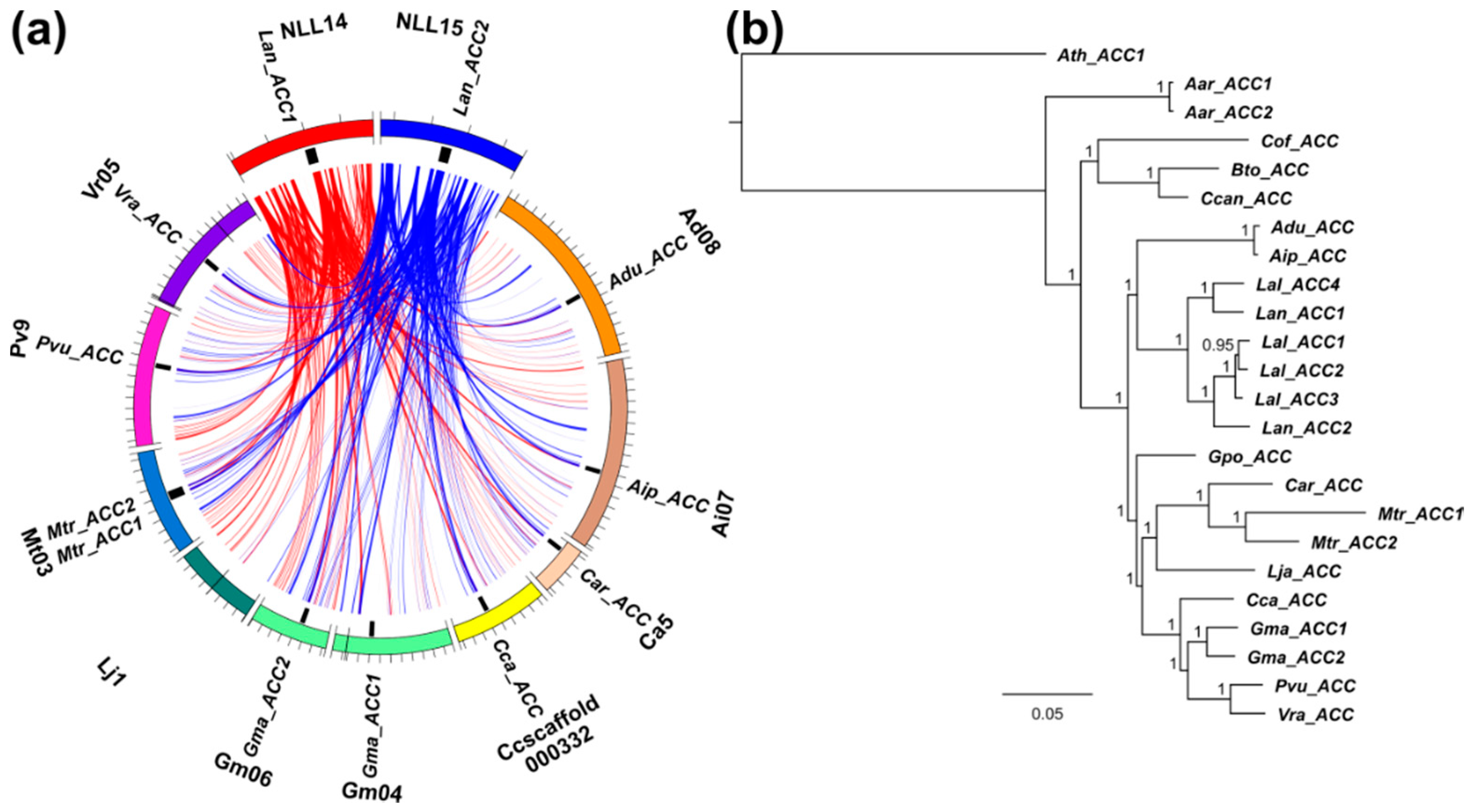
3.5. Whole-Genome Duplication Event(s) Shaped the Evolution of L. angustifolius Nuclear Genes for Cytosolic ACCase and Plastid ACCase Subunits
3.6. Legume ACC Genes Evolved by Lineage-Specific Duplications, Whereas accA, accB and accC Genes Both by Early and Lineage-Specific Duplications
3.7. Purifying Selection Shaped Evolution of Nuclear Genes Encoding Cytosolic ACCase and Subunits of Plastid ACCase
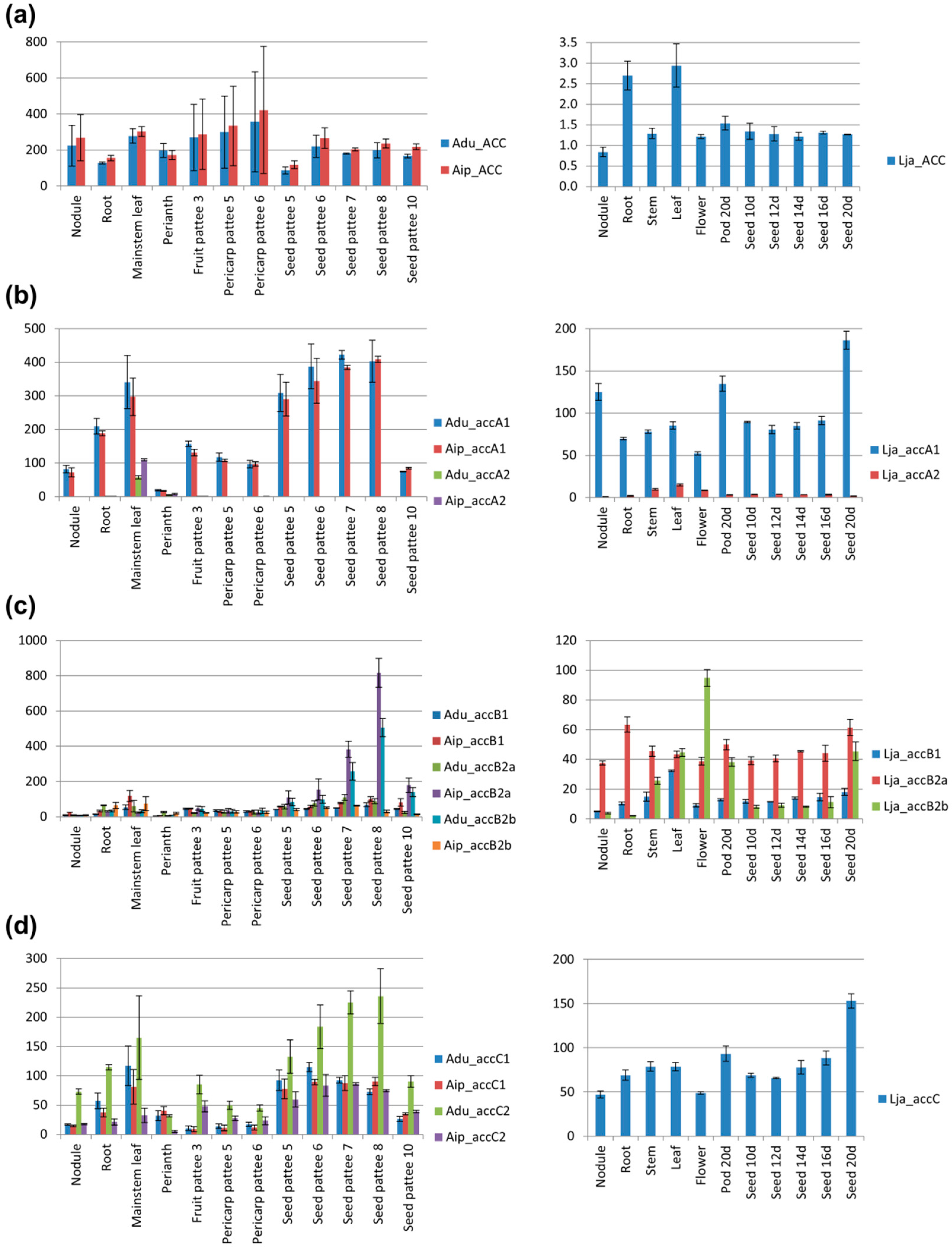
3.8. Transcription Profiles of accA, accB, and accC Duplicates are Different, Indicating the Possibility of Gene Sub-Functionalization
4. Discussion
4.1. Bacterial Artificial Chromosome-Based Approach Is Still Efficient in Current Genomic Analyses
4.2. Nuclear Genes Encoding Cytosolic ACCase and Plastid ACCase Subunits Evolved by Whole-Genome Duplication
4.3. Functional Differentiation of Duplicated ACCase Genes
4.4. Selection Constraints of Genes Encoding Cytosolic ACCase and Subunits of Plastid ACCase
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, P.H.; Vance, C.P. Legumes: importance and constraints to greater use. Plant Physiol. 2003, 131, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Anglade, J.; Billen, G.; Garnier, J. Relationships for estimating N2 fixation in legumes: incidence for N balance of legume-based cropping systems in Europe. Ecosphere 2015, 6, 1–24. [Google Scholar] [CrossRef]
- Hughes, C.; Eastwood, R. Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc. Natl. Acad. Sci. USA 2006, 103, 10334–10339. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.S.; Eastwood, R.J.; Miotto, S.T.S.; Hughes, C.E. Multiple continental radiations and correlates of diversification in Lupinus (Leguminosae): Testing for key innovation with incomplete taxon sampling. Syst. Biol. 2012, 61, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Mahe, F.; Markova, D.; Pasquet, R.; Misset, M.T.; Ainouche, A. Isolation, phylogeny and evolution of the SymRK gene in the legume genus Lupinus L. Mol. Phylogen. Evol. 2011, 60, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.S. Diversification of Lupinus (Leguminosae) in the western New World: Derived evolution of perennial life history and colonization of montane habitats. Mol. Phylogen. Evol. 2008, 48, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.S.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vega, J.J.; Ayling, S.; Hegarty, M.; Kudrna, D.; Goicoechea, J.L.; Ergon, Å.; Rognli, O.A.; Jones, C.; Swain, M.; Geurts, R.; et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci. Rep. 2015, 5, 17394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.-K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J.; et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Tian, Z.; Chen, C.; Luo, L.; Zhao, B.; Wang, Z.; Yu, L.; Li, Y.; Sun, Y.; Li, W.; et al. Genome sequencing of adzuki bean (Vigna angularis) provides insight into high starch and low fat accumulation and domestication. Proc. Natl. Acad. Sci. USA 2015, 112, 13213–13218. [Google Scholar] [CrossRef] [PubMed]
- Matasci, N.; Hung, L.H.; Yan, Z.; Carpenter, E.J.; Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Ayyampalayam, S.; Barker, M.; et al. Data access for the 1,000 Plants (1KP) project. Gigascience 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–4868. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; McKain, M.R.; Harkess, A.; Nelson, M.N.; Dash, S.; Deyholos, M.K.; Peng, Y.; Joyce, B.; Stewart, C.N.; Rolf, M.; et al. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 2015, 32, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.A.; Smith, P.M.C.; Gupta, S.; Jones, M.G.K.; Caligari, P.D.S. Genetics, Cytology and Biotechnology. In Lupins as Crop Plants: Biology, Production, and Utilization; Gladstones, J.S., Atkins, C.A., Hamblin, J., Eds.; CAB International: Oxfordshire, UK, 1998; pp. 67–92. [Google Scholar]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Biol. 2000, 42, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Naganowska, B.; Wolko, B.; Sliwińska, E.; Kaczmarek, Z. Nuclear DNA content variation and species relationships in the genus Lupinus (Fabaceae). Ann. Bot. 2003, 92, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, L.G.; Hane, J.K.; Nelson, M.N.; Gao, L.; Atkins, C.A.; Singh, K.B. Transcriptome sequencing of different narrow-leafed lupin tissue types provides a comprehensive uni-gene assembly and extensive gene-based molecular markers. Plant Biotechnol. J. 2015, 13, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Kroc, M.; Koczyk, G.; Święcicki, W.; Kilian, A.; Nelson, M.N. New evidence of ancestral polyploidy in the Genistoid legume Lupinus angustifolius L. (narrow-leafed lupin). Theor. Appl. Genet. 2014, 127, 1237–1249. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Moolhuijzen, P.M.; Boersma, J.G.; Chudy, M.; Lesniewska, K.; Bellgard, M.; Oliver, R.P.; Swiecicki, W.; Wolko, B.; Cowling, W.A.; et al. Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume, Lotus japonicus. DNA Res. 2010, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Phan, H.T.T.; Ellwood, S.R.; Moolhuijzen, P.M.; Hane, J.; Williams, A.; O’Lone, C.E.; Fosu-Nyarko, J.; Scobie, M.; Cakir, M.; et al. The first gene-based map of Lupinus angustifolius L.-location of domestication genes and conserved synteny with Medicago truncatula. Theor. Appl. Genet. 2006, 113, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Safár, J.; Janda, J.; Dolezel, J.; Wolko, B.; Naganowska, B. The bacterial artificial chromosome (BAC) library of the narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2006, 11, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-L.; Hane, J.K.; Kamphuis, L.G.; Foley, R.; Shi, B.-J.; Atkins, C.A.; Singh, K.B. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): Construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genom. 2011, 12, 521. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Majcherkiewicz, K.; Przysiecka, Ł.; Karlowski, W.; Wolko, B.; Naganowska, B. Comparative genomics of Lupinus angustifolius gene-rich regions: BAC library exploration, genetic mapping and cytogenetics. BMC Genom. 2013, 14, 79. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Zielezinski, A.; Wyrwa, K.; Szczepaniak, A.; Rychel, S.; Karlowski, W.; Wolko, B.; Naganowska, B. Remnants of the legume ancestral genome preserved in gene-rich regions: Insights from Lupinus angustifolius physical, genetic, and comparative mapping. Plant Mol. Biol. Rep. 2015, 33, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Wyrwa, K.; Książkiewicz, M.; Szczepaniak, A.; Susek, K.; Podkowiński, J.; Naganowska, B. Integration of Lupinus angustifolius L. (narrow-leafed lupin) genome maps and comparative mapping within legumes. Chromosome Res. 2016, 24, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Leśniewska, K.; Książkiewicz, M.; Nelson, M.N.; Mahé, F.; Aïnouche, A.; Wolko, B.; Naganowska, B. Assignment of 3 genetic linkage groups to 3 chromosomes of narrow-leafed lupin. J. Hered. 2011, 102, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Przysiecka, Ł.; Książkiewicz, M.; Wolko, B.; Naganowska, B. Structure, expression profile and phylogenetic inference of chalcone isomerase-like genes from the narrow-leafed lupin (Lupinus angustifolius L.) genome. Front. Plant Sci. 2015, 6, 268. [Google Scholar] [CrossRef] [PubMed]
- Książkiewicz, M.; Rychel, S.; Nelson, M.N.; Wyrwa, K.; Naganowska, B.; Wolko, B. Expansion of the phosphatidylethanolamine binding protein family in legumes: A case study of Lupinus angustifolius L. FLOWERING LOCUS T homologs, LanFTc1 and LanFTc2. BMC Genom. 2016, 17, 820. [Google Scholar] [CrossRef]
- Narożna, D.; Książkiewicz, M.; Przysiecka, Ł.; Króliczak, J.; Wolko, B.; Naganowska, B.; Mądrzak, C.J. Legume isoflavone synthase genes have evolved by whole-genome and local duplications yielding transcriptionally active paralogs. Plant Sci. 2017, 264, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Nikolau, B.J.; Ohlrogge, J.B.; Wurtele, E.S. Plant biotin-containing carboxylases. Arch. Biochem. Biophys. 2003, 414, 211–222. [Google Scholar] [CrossRef]
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 2013, 70, 863–891. [Google Scholar] [CrossRef] [PubMed]
- Huerlimann, R.; Heimann, K. Comprehensive guide to acetyl-carboxylases in algae. Crit. Rev. Biotechnol. 2013, 33, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Schwender, J.; Ohlrogge, J.B. Probing in vivo metabolism by stable isotope labeling of storage lipids and proteins in developing Brassica napus embryos. Plant Physiol. 2002, 130, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Konishi, T.; Nagano, Y. The compartmentation of acetyl-coenzyme A carboxylase in plants. Plant Physiol. 1995, 108, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Deusch, O.; Landan, G.; Roettger, M.; Gruenheit, N.; Kowallik, K.V.; Allen, J.F.; Martin, W.; Dagan, T. Genes of cyanobacterial origin in plant nuclear genomes point to a heterocyst-forming plastid ancestor. Mol. Biol. Evol. 2008, 25, 748–761. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.; Moreira, D. Early evolution of the biotin-dependent carboxylase family. BMC Evol. Biol. 2011, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Konishi, T.; Sasaki, Y. Compartmentalization of two forms of acetyl-CoA carboxylase in plants and the origin of their tolerance toward herbicides. Proc. Natl. Acad. Sci. USA 1994, 91, 3598–3601. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Nagano, Y. Plant acetyl-CoA carboxylase: structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci. Biotechnol. Biochem. 2004, 68, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Salie, M.J.; Thelen, J.J. Regulation and structure of the heteromeric acetyl-CoA carboxylase. Biochim. Biophys. Acta 2016, 1861, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Podkowinski, J.; Jelenska, J.; Sirikhachornkit, A.; Zuther, E.; Haselkorn, R.; Gornicki, P. Expression of cytosolic and plastid acetyl-coenzyme A carboxylase genes in young wheat plants. Plant Physiol. 2003, 131, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Shirley, B.W. Flavonoid biosynthesis: ‘New’ functions for an ‘old’ pathway. Trends Plant Sci. 1996, 1, 377–382. [Google Scholar] [CrossRef]
- Cronan, J.E., Jr.; Waldrop, G.L. Multi-subunit acetyl-CoA carboxylases. Prog. Lipid Res. 2002, 41, 407–435. [Google Scholar] [CrossRef]
- Reverdatto, S.; Beilinson, V.; Nielsen, N.C. A multisubunit acetyl coenzyme A carboxylase from soybean. Plant Physiol. 1999, 119, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Elizondo-Noriega, A.; Cantu, D.C.; Reilly, P.J. Structural classification of biotin carboxyl carrier proteins. Biotechnol. Lett. 2012, 34, 1869–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gornicki, P.; Podkowinski, J.; Scappino, L.A.; DiMaio, J.; Ward, E.; Haselkorn, R. Wheat acetyl-coenzyme A carboxylase: cDNA and protein structure. Proc. Natl. Acad. Sci. USA 1994, 91, 6860–6864. [Google Scholar] [CrossRef] [PubMed]
- Podkowinski, J.; Sroga, G.E.; Haselkorn, R.; Gornicki, P. Structure of a gene encoding a cytosolic acetyl-CoA carboxylase of hexaploid wheat. Proc. Natl. Acad. Sci. USA 1996, 93, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Roesler, K.R.; Savage, L.J.; Shintani, D.K.; Shorrosh, B.S.; Ohlrogge, J.B. Co-purification, co-immunoprecipitation, and coordinate expression of acetyl-coenzyme A carboxylase activity, biotin carboxylase, and biotin carboxyl carrier protein of higher plants. Planta 1996, 198, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Shorrosh, B.S.; Dixon, R.A.; Ohlrogge, J.B. Molecular cloning, characterization, and elicitation of acetyl-CoA carboxylase from alfalfa. Proc. Natl. Acad. Sci. USA 1994, 91, 4323–4327. [Google Scholar] [CrossRef] [PubMed]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Boersma, J.G.; Pallotta, M.; Li, C.; Buirchell, B.J.; Sivasithamparam, K.; Yang, H. Construction of a genetic linkage map using MFLP and identification of molecular markers linked to domestication genes in narrow-leafed lupin (Lupinus angustifolius L.). Cell. Mol. Biol. Lett. 2005, 10, 331–344. [Google Scholar] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Wolf, J.B.W. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 2014, 7, 1026–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V. Statistical approaches in eukaryotic gene prediction. In Handbook of Statistical Genetics; John Wiley & Sons, Ltd.: John Wiley & Sons, Ltd., 2004. [Google Scholar] [CrossRef]
- Neff, M.M.; Turk, E.; Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet. 2002, 18, 613–615. [Google Scholar] [CrossRef]
- Manly, K.F.; Robert, H.; Cudmore, J.; Meer, J.M. Map Manager QTX, cross-platform software for genetic mapping. Mamm. Genome 2001, 12, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed]
- Parra-González, L.B.; Aravena-Abarzúa, G.A.; Navarro-Navarro, C.S.; Udall, J.; Maughan, J.; Peterson, L.M.; Salvo-Garrido, H.E.; Maureira-Butler, I.J. Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC Genom. 2012, 13, 425. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Yang, S.S.; Miller, S.S.; Bucciarelli, B.; Liu, J.; Rydeen, A.; Bozsoki, Z.; Uhde-Stone, C.; Tu, Z.J.; Allan, D.; et al. An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 2013, 161, 705–724. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Revanna, K.V.; Chiu, C.-C.; Bierschank, E.; Dong, Q. GSV: A web-based genome synteny viewer for customized data. BMC Bioinform. 2011, 12, 316. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.-i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, J.; Chu, Y.; Scheffler, B.; Ozias-Akins, P. A developmental transcriptome map for allotetraploid Arachis hypogaea. Front. Plant Sci. 2016, 7, 1446. [Google Scholar] [CrossRef] [PubMed]
- Pazhamala, L.T.; Purohit, S.; Saxena, R.K.; Garg, V.; Krishnamurthy, L.; Verdier, J.; Varshney, R.K. Gene expression atlas of pigeonpea and its application to gain insights into genes associated with pollen fertility implicated in seed formation. J. Exp. Bot. 2017, 68, 2037–2054. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dai, X.; Liu, T.; Zhao, P.X. LegumeIP: An integrative database for comparative genomics and transcriptomics of model legumes. Nucleic Acids Res. 2012, 40, D1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.J.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Verdier, J.; Torres-Jerez, I.; Wang, M.; Andriankaja, A.; Allen, S.N.; He, J.; Tang, Y.; Murray, J.D.; Udvardi, M.K. Establishment of the Lotus japonicus Gene Expression Atlas (LjGEA) and its use to explore legume seed maturation. Plant J. 2013, 74, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Benedito, V.A.; Torres-Jerez, I.; Murray, J.D.; Andriankaja, A.; Allen, S.; Kakar, K.; Wandrey, M.; Verdier, J.; Zuber, H.; Ott, T.; et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008, 55, 504–513. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Benedito, V.A.; Wang, M.; Murray, J.D.; Zhao, P.X.; Tang, Y.; Udvardi, M.K. The Medicago truncatula gene expression atlas web server. BMC Bioinform. 2009, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Iniguez, L.P.; Fu, F.; Bucciarelli, B.; Miller, S.S.; Jackson, S.A.; McClean, P.E.; Li, J.; Dai, X.; Zhao, P.X.; et al. An RNA-Seq based gene expression atlas of the common bean. BMC Genom. 2014, 15, 866. [Google Scholar] [CrossRef]
- Yao, S.; Jiang, C.; Huang, Z.; Torres-Jerez, I.; Chang, J.; Zhang, H.; Udvardi, M.; Liu, R.; Verdier, J. The Vigna unguiculata Gene Expression Atlas (VuGEA) from de novo assembly and quantification of RNA-seq data provides insights into seed maturation mechanisms. Plant J. 2016, 88, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.N.; Książkiewicz, M.; Rychel, S.; Besharat, N.; Taylor, C.M.; Wyrwa, K.; Jost, R.; Erskine, W.; Cowling, W.A.; Berger, J.D.; et al. The loss of vernalization requirement in narrow-leafed lupin is associated with a deletion in the promoter and de-repressed expression of a Flowering Locus T (FT) homologue. New Phytol. 2017, 213, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Lehti-Shiu, M.D.; Thibaud-Nissen, F.; Prakash, T.; Buell, C.R.; Shiu, S.-H. Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol. 2009, 151, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Visendi, P.; Berkman, P.J.; Hayashi, S.; Golicz, A.A.; Bayer, P.E.; Ruperao, P.; Hurgobin, B.; Montenegro, J.; Chan, C.-K.K.; Staňková, H.; et al. An efficient approach to BAC based assembly of complex genomes. Plant Methods 2016, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Shi, C.; Thudi, M.; Mariac, C.; Wallace, J.; Qi, P.; Zhang, H.; Zhao, Y.; Wang, X.; Rathore, A.; et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. 2017, 35, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, Q.; Hu, W.; Zhang, X.; Wang, L.; Hua, X.; Yu, Q.; Ming, R.; Zhang, J. Evolution and expression of the fructokinase gene family in Saccharum. BMC Genom. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Rispail, N.; Rubiales, D. Genome-wide identification and comparison of legume MLO gene family. Sci. Rep. 2016, 6, 32673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Wu, H.; Zhang, R.; Li, S.; He, W.; Wong, F.-L.; Li, G.; Zhao, S.; Lam, H.-M. Molecular phylogeny and dynamic evolution of disease resistance genes in the legume family. BMC Genom. 2016, 17, 402. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Wall, P.K.; Leebens-Mack, J.H.; Lindsay, B.G.; Soltis, D.E.; Doyle, J.J.; Soltis, P.S.; Carlson, J.E.; Arumuganathan, K.; Barakat, A.; et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006, 16, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Peer, Y. A mystery unveiled. Genome Biol. 2011, 12, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlueter, J.A.; Dixon, P.; Granger, C.; Grant, D.; Clark, L.; Doyle, J.J.; Shoemaker, R.C. Mining EST databases to resolve evolutionary events in major crop species. Genome 2004, 47, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, S.B.; Ilut, D.; Farmer, A.D.; Maki, S.L.; May, G.D.; Singer, S.R.; Doyle, J.J. Polyploidy did not predate the evolution of nodulation in all legumes. PLoS ONE 2010, 5, e11630. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.; Herendeen, P.S.; Wojciechowski, M.F. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Pfeil, B.E.; Schlueter, J.A.; Shoemaker, R.C.; Doyle, J.J. Placing paleopolyploidy in relation to taxon divergence: a phylogenetic analysis in legumes using 39 gene families. Syst. Biol. 2005, 54, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Moretzsohn, M.C.; Madsen, L.H.; Sandal, N.; Leal-Bertioli, S.C.M.; Guimarães, P.M.; Hougaard, B.K.; Fredslund, J.; Schauser, L.; Nielsen, A.M.; et al. An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure, stability and evolution of legume genomes. BMC Genom. 2009, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Z.; Liu, Y.; Liu, T.; Li, Q.; Ji, Y.; Li, C.; Fang, C.; Wang, M.; Wu, M.; et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and Arabidopsis. Plant Cell 2015, 27, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhao, H.; Parsons, E.P.; Xu, C.; Kosma, D.K.; Xu, X.; Chao, D.; Lohrey, G.; Bangarusamy, D.K.; Wang, G.; et al. The glossyhead1 allele of ACC1 reveals a principal role for multidomain acetyl-coenzyme A carboxylase in the biosynthesis of cuticular waxes by Arabidopsis. Plant Physiol. 2011, 157, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Post-Beittenmiller, D. Biochemistry and molecular biology of wax production in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, J.J.; Choi, J.-K.; Wurtele, E.S.; Nikolau, B.J. Tissue distribution of acetyl-CoA carboxylase in leaves of leek (Allium porrum L.). J. Plant Physiol. 1998, 153, 265–269. [Google Scholar] [CrossRef]
- Garcia-Ponce, B.; Rocha-Sosa, M. The octadecanoid pathway is required for pathogen-induced multi-functional acetyl-CoA carboxylase accumulation in common bean (Phaseolus vulgaris L.). Plant Sci. 2000, 157, 181–190. [Google Scholar] [CrossRef]
- Baud, S.; Guyon, V.; Kronenberger, J.; Wuilleme, S.; Miquel, M.; Caboche, M.; Lepiniec, L.; Rochat, C. Multifunctional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 2003, 33, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Wen, T.N.; Nikolau, B.J.; Wurtele, E.S. Coordinate regulation of the nuclear and plastidic genes coding for the subunits of the heteromeric acetyl-coenzyme A carboxylase. Plant Physiol. 2000, 122, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Cui, Y.; Ding, Y.; Mei, J.; Dong, H.; Zhang, W.; Wu, S.; Liang, Y.; Zhang, C.; Li, J.; et al. Time-series analyses of transcriptomes and proteomes reveal molecular networks underlying oil accumulation in canola. Front. Plant Sci. 2016, 7, 2007. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Singh, S.K.; Du, C.; Li, C.; Fan, J.; Pattanaik, S.; Yuan, L. Comparative transcriptomic analysis of two Brassica napus near-isogenic lines reveals a network of genes that influences seed oil accumulation. Front. Plant Sci. 2016, 7, 1498. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mooney, B.P.; Hajduch, M.; Joshi, T.; Zhou, M.; Xu, D.; Thelen, J.J. System analysis of an Arabidopsis mutant altered in de novo fatty acid synthesis reveals diverse changes in seed composition and metabolism. Plant Physiol. 2009, 150, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Salie, M.J.; Zhang, N.; Lancikova, V.; Xu, D.; Thelen, J.J. A family of negative regulators targets the committed step of de novo fatty acid biosynthesis. Plant Cell 2016, 28, 2312–2325. [Google Scholar] [CrossRef] [PubMed]
- Feria Bourrellier, A.B.; Valot, B.; Guillot, A.; Ambard-Bretteville, F.; Vidal, J.; Hodges, M. Chloroplast acetyl-CoA carboxylase activity is 2-oxoglutarate-regulated by interaction of PII with the biotin carboxyl carrier subunit. Proc. Natl. Acad. Sci. USA 2010, 107, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Focks, N.; Benning, C. wrinkled1: A novel, low-seed-oil mutant of Arabidopsis with a deficiency in the seed-specific regulation of carbohydrate metabolism. Plant Physiol. 1998, 118, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, V.S.; Zaytseva, O.O.; Mglinets, A.V.; Shatskaya, N.V.; Kosterin, O.E.; Vasiliev, G.V. Nuclear-cytoplasmic conflict in pea (Pisum sativum L.) is associated with nuclear and plastidic candidate genes encoding acetyl-CoA carboxylase subunits. PLoS ONE 2015, 10, e0119835. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ilarslan, H.; Brachova, L.; Qian, H.R.; Li, L.; Che, P.; Wurtele, E.S.; Nikolau, B.J. Reverse-genetic analysis of the two biotin-containing subunit genes of the heteromeric acetyl-coenzyme A carboxylase in Arabidopsis indicates a unidirectional functional redundancy. Plant Physiol. 2011, 155, 293–314. [Google Scholar] [CrossRef] [PubMed]
- Roesler, K.; Shintani, D.; Savage, L.; Boddupalli, S.; Ohlrogge, J. Targeting of the Arabidopsis homomeric acetyl-coenzyme A carboxylase to plastids of rapeseeds. Plant Physiol. 1997, 113, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Gu, K.; Chiam, H.; Tian, D.; Yin, Z. Molecular cloning and expression of heteromeric ACCase subunit genes from Jatropha curcas. Plant Sci. 2011, 180, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Harwood, J.L. Fatty acid metabolism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 101–138. [Google Scholar] [CrossRef]
- Cork, J.M.; Purugganan, M.D. The evolution of molecular genetic pathways and networks. Bioessays 2004, 26, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, H.; Rieseberg, L.H.; Ritland, K. The correlation of evolutionary rate with pathway position in plant terpenoid biosynthesis. Mol. Biol. Evol. 2009, 26, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Clotault, J.; Peltier, D.; Soufflet-Freslon, V.; Briard, M.; Geoffriau, E. Differential selection on carotenoid biosynthesis genes as a function of gene position in the metabolic pathway: A study on the carrot and dicots. PLoS ONE 2012, 7, e38724. [Google Scholar] [CrossRef]
- Olson-Manning, C.F.; Lee, C.R.; Rausher, M.D.; Mitchell-Olds, T. Evolution of flux control in the glucosinolate pathway in Arabidopsis thaliana. Mol. Biol. Evol. 2013, 30, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Rausher, M.D.; Miller, R.E.; Tiffin, P. Patterns of evolutionary rate variation among genes of the anthocyanin biosynthetic pathway. Mol. Biol. Evol. 1999, 16, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Accession Number of Sequence Used for Primer Design | Accession Number of Probe Sequence | Tm 1 | Probe Size (bp) | Primer Pairs |
|---|---|---|---|---|---|
| La ACC | XM_003638746 | MF378602 | 58 °C | 404 | Pri-129 + Pri-131 |
| La BCCP | XM_003534455 | MF378603 | 56 °C | 251 | Pri-139 + Pri-140 |
| La CT-α | NM_001249264 | MF378604 | 58 °C | 437 | Pri-142 + Pri-145 |
| Mt BC | XM_003630560 | MF378605 | 60 °C | 1141 | J08-35 + J08-36 |
| 009k09 | 073e17 | 002f03 | 005g02 | 009a01 | 049f04 | 089l06 | 096g16 | 126d16 | 011g20 | 016j11 | 046i04 | 048n08 | 051f15 | 060f02 | |
| 009k06 | - | ||||||||||||||
| 073e17 | N * | - | |||||||||||||
| 002f03 | N | N | - | ||||||||||||
| 005g02 | N/A | N/A | N | - | |||||||||||
| 009a01 | N | N | N | N | - | ||||||||||
| 049f04 | N | N/A | N/A | N/A | N/A | - | |||||||||
| 089l06 | N/A | N | N | N | N | N | - | ||||||||
| 096g16 | N/A | N | N | N/A | N | N/A | N | - | |||||||
| 126d14 | N | N | N | N | Y | N | N | N | - | ||||||
| 011g20 | N | N | N | N/A | N | N/A | N/A | N/A | N | - | |||||
| 016j11 | N | N | N | N/A | N | N/A | N | N | N | N | - | ||||
| 046i04 | N | N | N | N/A | N | N/A | N | N | N | N | Y | - | |||
| 048n08 | N | N | N | N/A | N | N/A | N | N | N | N | Y | Y | - | ||
| 051f15 | N | N | N | N/A | N | N/A | N/A | N | N | N/A | N | N | N | - | |
| 060f02 | N | N | N | N/A | N | N/A | N | N | N | N | Y | Y | Y | N | - |
| Marker | Marker Type | Enzyme | Products 83A:476 (bp) | Products P27255 (bp) | Chi-Square p-Value | NLL | Distance to Other Markers (cM) | LOD Values |
|---|---|---|---|---|---|---|---|---|
| 002F03_5 | CAPS | MnlI | 315, 168, 59, 58 | 209, 168, 106, 59, 58 | 0.526 | NLL-10 | 1.3, 3.0 | 20.3, 14.7 |
| 005G02_3 | CAPS | TaqI | 352, 268, 109 | 292, 267, 109, 60 | 0.332 | NLL-03 | 0.6, 1.8 | 22.3, 14.0 |
| 009K06_5 | CAPS | MjaIV | 289, 196, 84 | 485, 84 | 0.272 | NLL-14 | 0.0, 0.8 | 16.9, 17.9 |
| 011G20_5 | PCR | - | 624 | - | 0.546 | NLL-13 | 0.7, 2.8 | 18.8, 16.1 |
| 016J11_5 | CAPS | MaeIII | 353 | 254, 99 | 0.019 | NLL-11 | 1.4, 0.7 | 17.4, 20.9 |
| 040M06_3 | CAPS | DdeI | 350, 303 | 350, 207, 96 | 0.174 | NLL-14 | 0.8, 0.0 | 16.7, 16.9 |
| 042C13_3 | CAPS | HphI | 280, 223, 162 | 385, 280 | 0.669 | NLL-08 | 0.0, 1.4 | 22.6, 18.6 |
| 051F15_5 | CAPS | MfeI | 342, 273 | 615 | 0.652 | NLL-13 | 3.6, 8.5 | 14.6, 9.2 |
| 060F02_3 | CAPS | AciI | 158, 67 | 225 | 0.003 | NLL-11 | 0.7, 1.3 | 20.9, 19.7 |
| 069L17_3 | CAPS | Hpy188I | 467 | 353, 112 | 0.583 | NLL-13 | 0.7, 0.7 | 19.7, 18.8 |
| 073E17_5 | CAPS | MnlI | 186, 131, 84, 82, 82, 52, 3 | 219, 211, 84, 82, 3 | 0.113 | NLL-15 | 0.5, 1.6 | 16.0, 17.6 |
| 077J19_5 | PCR | - | 352 | - | 0.599 | NLL-07 | 0.6, 1.9 | 22.0, 18.8 |
| 112B24_5 | CAPS | MnlI | 255, 135, 70, 48 | 255, 183, 70 | 0.669 | NLL-05 | 1.4, 4.0 | 17.7, 12.7 |
| 126D14_5 | CAPS | BclI | 628 | 354, 275 | 0.654 | NLL-06 | 1.3, 1.3 | 20.0, 19.2 |
| BAC | Length (bp) | GC (%) | Repetitive Elements (%) | Major Fractions of Repetitive Elements | BAC-FISH Signals | No. of Predicted Genes |
|---|---|---|---|---|---|---|
| 009A01 | 45,596 | 34.3 | 16.4 | LTR/Gypsy, NonLTR/RTE, Simple repeats | S | 5 |
| 009K06 | 54,264 | 32.4 | 2.2 | Simple repeats | S | 7 |
| 042E20 | 35,419 | 33.7 | 25.6 | LTR/Copia, DNA/Helitron, DNA/EnSpm/CACT | R | 1 |
| 051F15 | 57,486 | 32.0 | 9.3 | LTR/Copia, LTR/Gypsy, DNA/hAT | S | 6 |
| 060F02 | 79,573 | 31.8 | 23.8 | LTR/Gypsy, LTR/Copia, DNA/Helitron | S | 3 |
| 070D20 | 46,840 | 33.4 | 7.1 | LTR/Copia, Simple repeats | R | 2 |
| 073E17 | 42,375 | 33.8 | 11.1 | DNA/MuDR, LTR/Copia | S | 2 |
| 075C05 | 94,179 | 35.7 | 33.2 | LTR/Copia, LTR/Gypsy, Simple repeats | R | 3 |
| 089L06 1 | 23,289 | 32.3 | 37.3 | LTR/Copia, NonLTR/L1, Simple repeats | S | 0 |
| 092K09 | 162,642 | 33.4 | 36.2 | LTR/Copia, LTR/Gypsy, DNA/EnSpm/CACTA | R | 3 |
| 096G16 1 | 4599 | 31.2 | 83.2 | NonLTR/L1 | S | 1 |
| Species | Sequence Type | ACC | accA | accB | accC |
|---|---|---|---|---|---|
| Acacia argyrophylla | transcriptome | 2 | 1 | 2 | 2 |
| Arachis duranensis | genome | 1 | 2 | 3 | 2 |
| Arachis ipaensis | genome | 1 | 2 | 3 | 2 |
| Arabidopsis thaliana1 | genome | 2 | 1 | 2 | 1 |
| Bauhinia tomentosa | transcriptome | 1 | 2 | 1 | 2 |
| Cicer arietinum | genome | 1 | 1 | 4 | 1 |
| Cajanus cajan | genome | 1 | 2 | 4 | 1 |
| Cercis canadensis | transcriptome | 1 | 1 | 2 | 1 |
| Copaifera officinalis | transcriptome | 1 | 2 | 1 | 1 |
| Chlamydomonas reinhardtii2 | genome | 0 | 1 | 1 | 1 |
| Glycine max | genome | 2 | 3 | 5 | 2 |
| Gompholobium polymorphum | transcriptome | 1 | 1 | 3 | 1 |
| Lupinus albus | transcriptome | 4 | 5 | 8 | 3 |
| Lupinus angustifolius | genome | 2 | 3 | 4 | 2 |
| Lotus japonicus | genome | 1 | 2 | 3 | 1 |
| Medicago truncatula | genome | 2 | 1 | 3 | 1 |
| Phaseolus vulgaris | genome | 1 | 1 | 3 | 1 |
| Vigna radiata | genome | 1 | 1 | 3 | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczepaniak, A.; Książkiewicz, M.; Podkowiński, J.; Czyż, K.B.; Figlerowicz, M.; Naganowska, B. Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications. Genes 2018, 9, 563. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110563
Szczepaniak A, Książkiewicz M, Podkowiński J, Czyż KB, Figlerowicz M, Naganowska B. Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications. Genes. 2018; 9(11):563. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110563
Chicago/Turabian StyleSzczepaniak, Anna, Michał Książkiewicz, Jan Podkowiński, Katarzyna B. Czyż, Marek Figlerowicz, and Barbara Naganowska. 2018. "Legume Cytosolic and Plastid Acetyl-Coenzyme—A Carboxylase Genes Differ by Evolutionary Patterns and Selection Pressure Schemes Acting before and after Whole-Genome Duplications" Genes 9, no. 11: 563. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110563