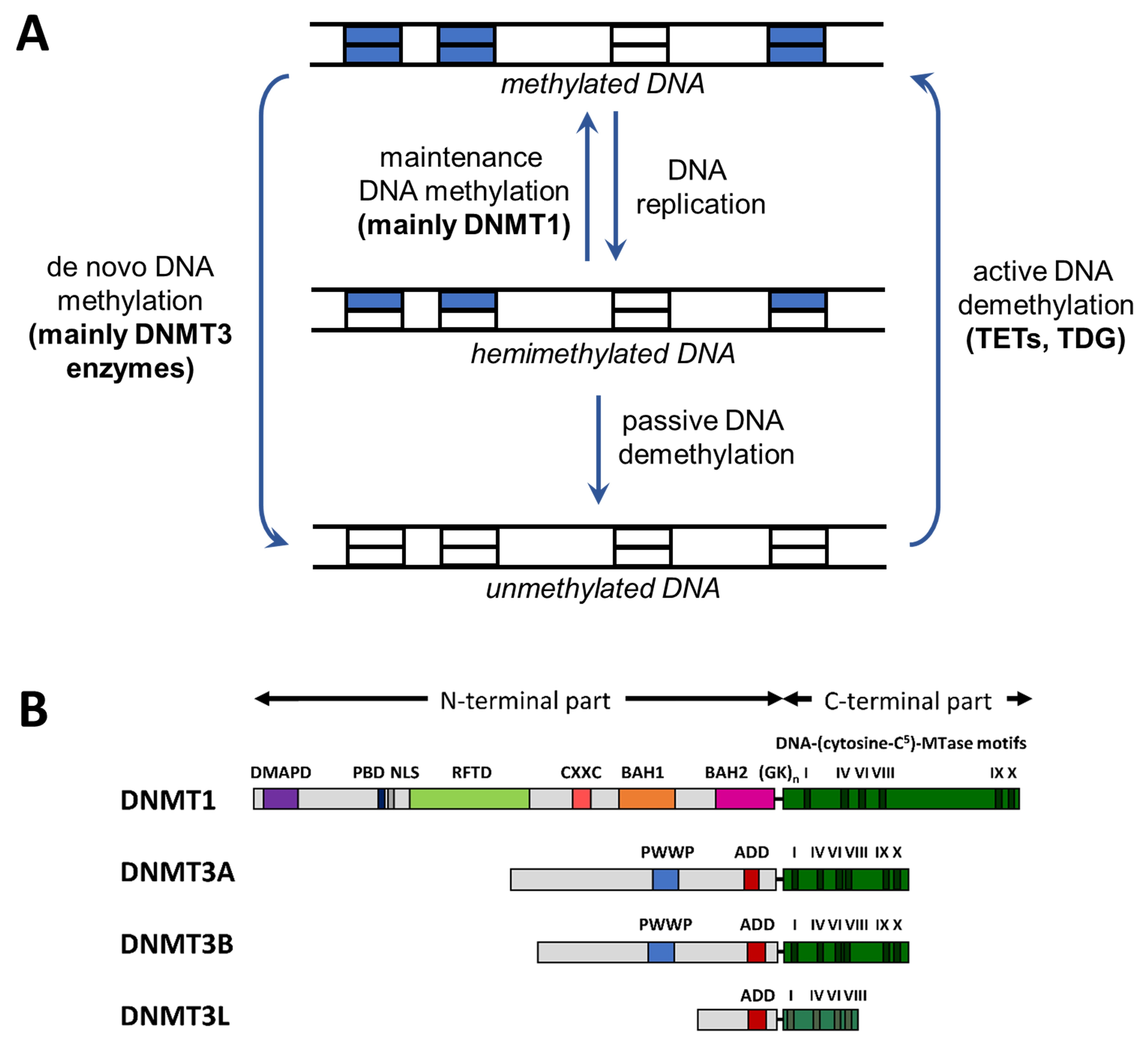
Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes



Abstract
:
1. Introduction
2. Evolutionary Impact of DNA Methylation
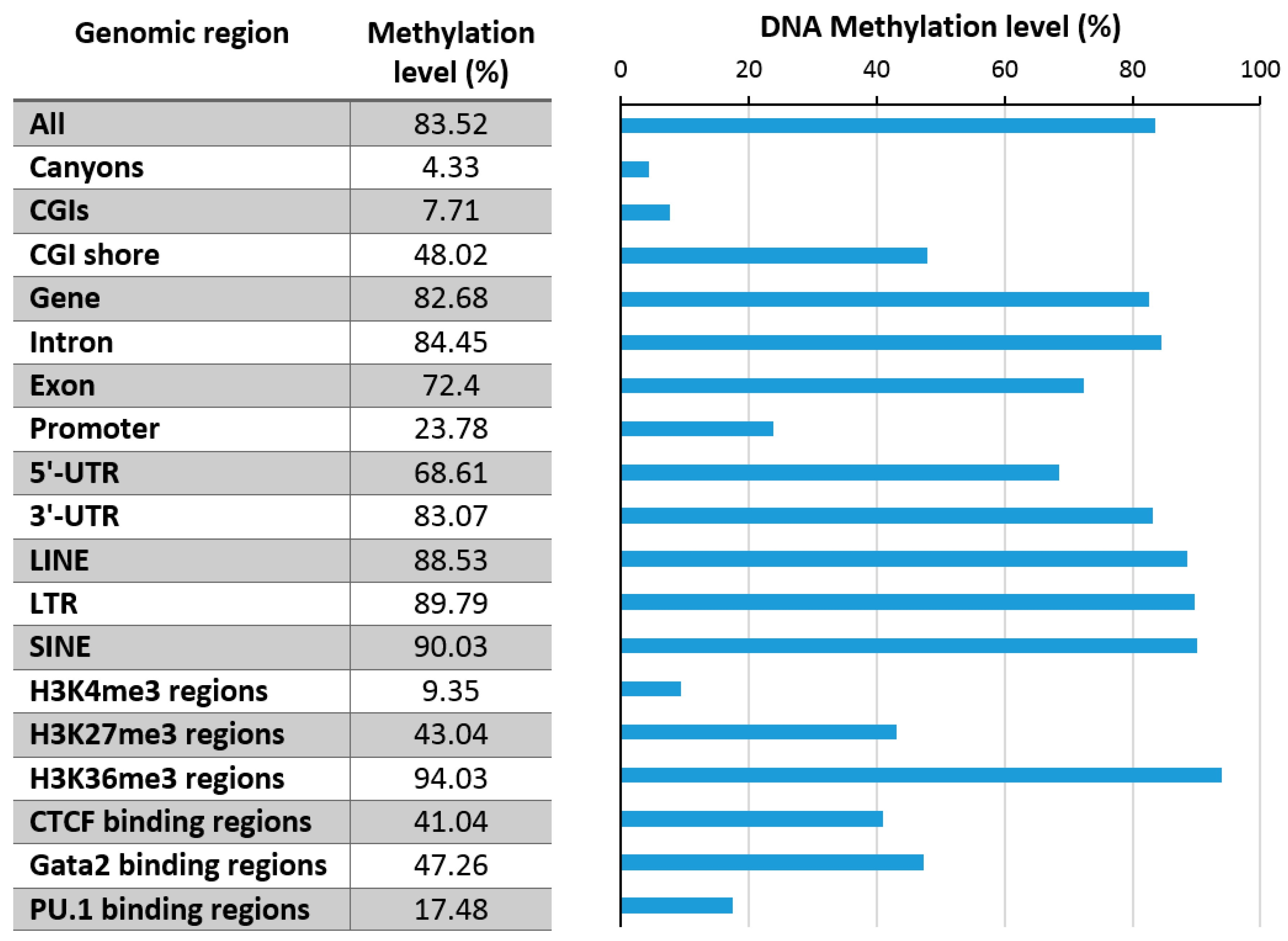
3. Genomic Distribution and Variability of DNA Methylation
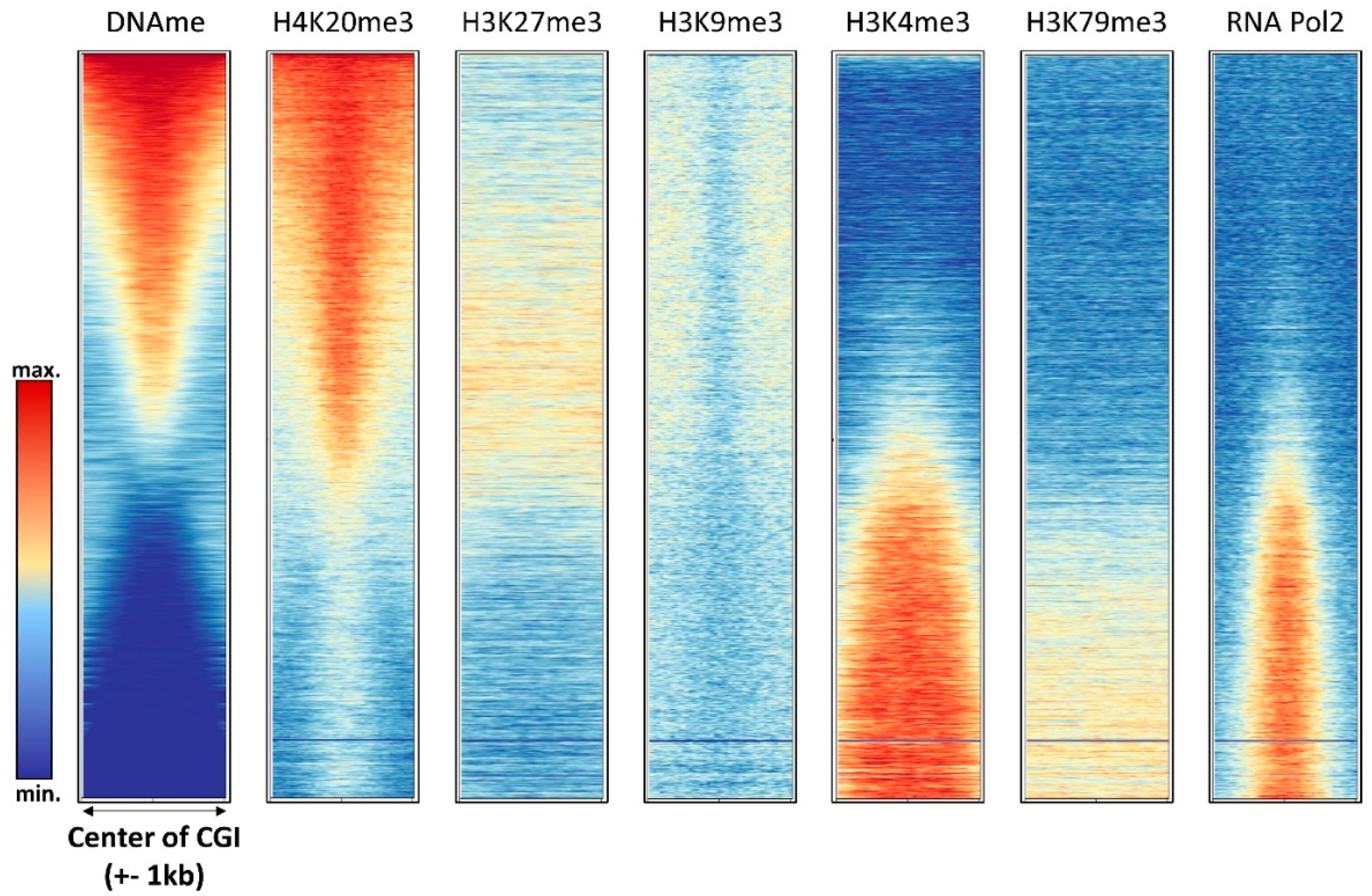
3.1. Promoter Methylation
3.2. Enhancer Methylation and Influence of DNA Methylation on TF Binding
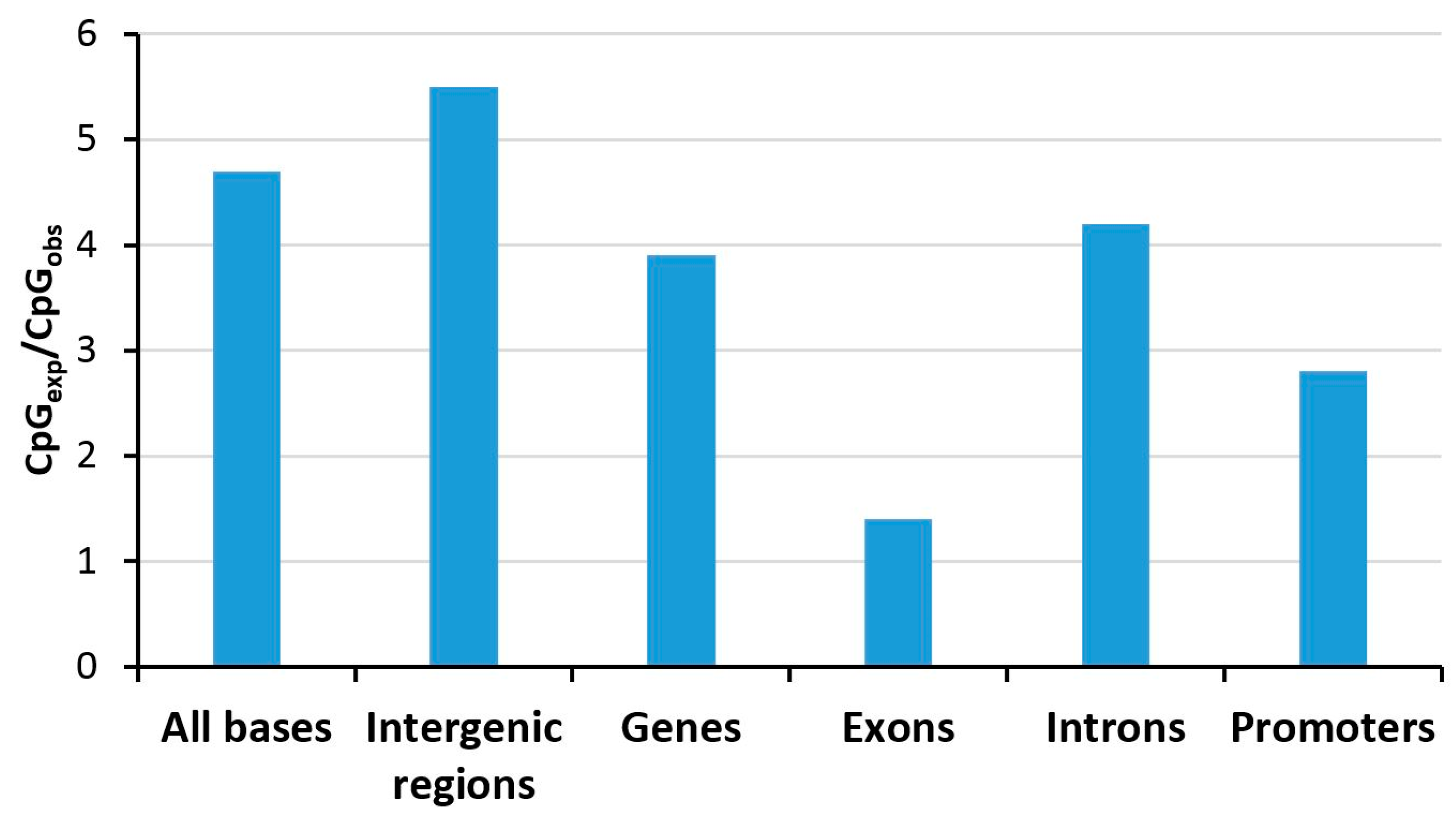
3.3. Repeat Methylation
3.4. DNA Methylation Canyons
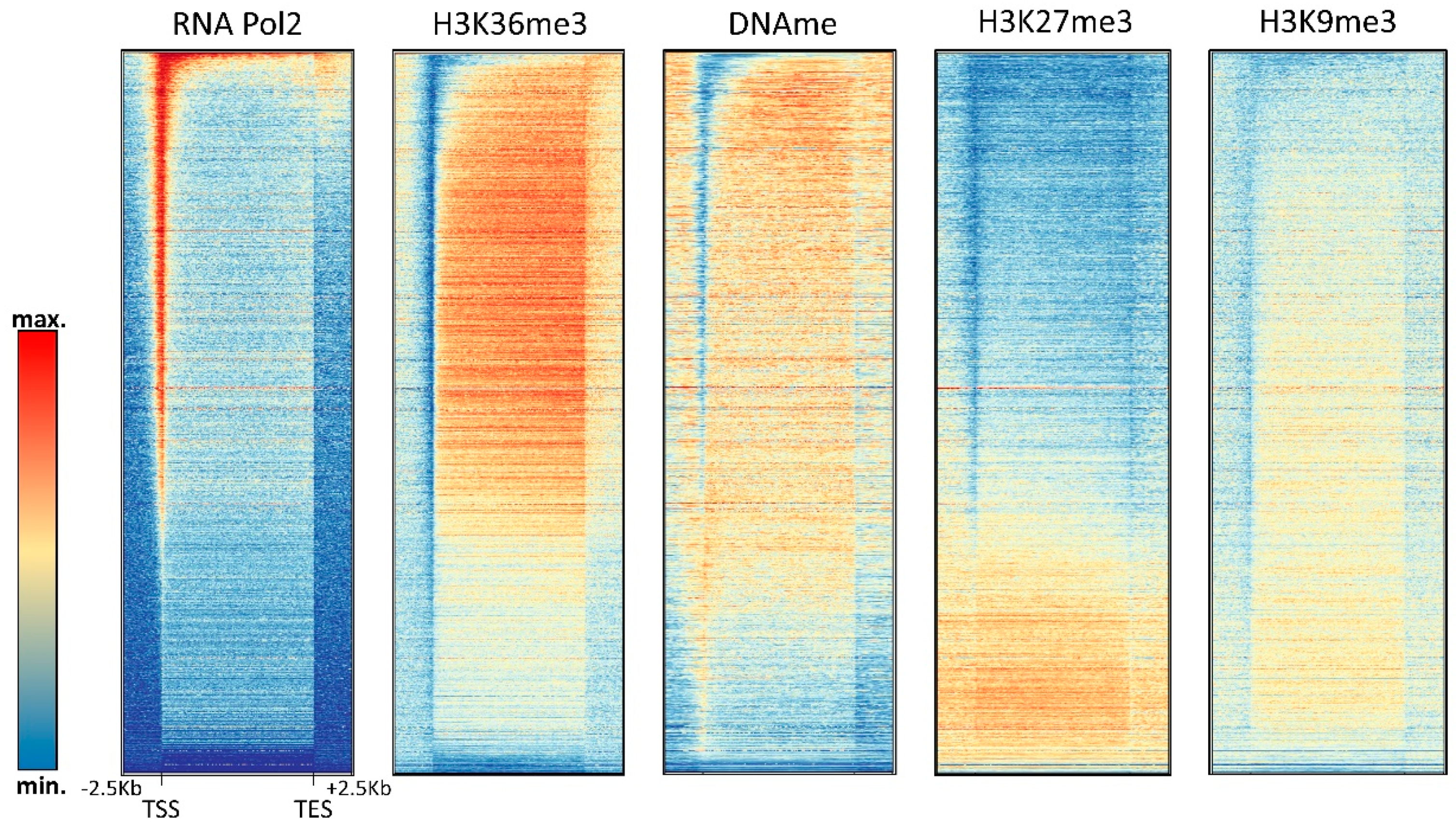
4. Relation of DNA Methylation and Chromatin Marks
4.1. H3K4me3
4.2. H3K36me3
4.3. H3K27me3
4.4. H3K9me3
4.5. Chromatin Remodeling and DNA Methylation
5. Additional DNA Methylation Events
5.1. Non-CpG Methylation: Enzymes, Patterns, and Role
5.2. Rare Methylation Events: Controversies about 6mA and Mitochondrial DNA Methylation
6. Outlook
Supplementary Materials
Funding
Conflicts of Interest
References
- Globisch, D.; Münzel, M.; Müller, M.; Michalakis, S.; Wagner, M.; Koch, S.; Brückl, T.; Biel, M.; Carell, T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010, 5, e15367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisanti, S.; Omar, W.A.; Tomaszewski, B.; De Prins, S.; Jacobs, G.; Koppen, G.; Mathers, J.C.; Langie, S.A. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS ONE 2013, 8, e79044. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Allis, C.D.; Li, H. Reading between the lines: “ADD”-ing histone and DNA methylation marks toward a new epigenetic “Sum”. ACS Chem. Biol. 2018, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: What’s next? J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Mirfattah, B.; Herring, J.; Tang, H.; Zhang, K. Probes and targets of DNA methylation and demethylation in drug development. Curr. Top. Med. Chem. 2017, 17, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Goodell, M.A. New answers to old questions from genome-wide maps of DNA methylation in hematopoietic cells. Exp. Hematol. 2014, 42, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Kong, X.; Zhang, H.; Liu, Y.; Shao, Z.; Jin, J.; Cai, Y.; Zhang, R.; Li, L.; Zhang, Y.W.; et al. Biochemical studies and molecular dynamic simulations reveal the molecular basis of conformational changes in DNA methyltransferase-1. ACS Chem. Biol. 2018, 13, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases—A new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Jeltsch, A. Mammalian DNA methyltransferases: New discoveries and open questions. Biochem. Soc. Trans. 2018, 46, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 2002, 3, 275–293. [Google Scholar] [CrossRef]
- Verma, N.; Pan, H.; Doré, L.C.; Shukla, A.; Li, Q.V.; Pelham-Webb, B.; Teijeiro, V.; González, F.; Krivtsov, A.; Chang, C.J.; et al. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet. 2018, 50, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Von Meyenn, F.; Iurlaro, M.; Habibi, E.; Liu, N.Q.; Salehzadeh-Yazdi, A.; Santos, F.; Petrini, E.; Milagre, I.; Yu, M.; Xie, Z.; et al. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol. Cell 2016, 62, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Gahurova, L.; Tomizawa, S.I.; Smallwood, S.A.; Stewart-Morgan, K.R.; Saadeh, H.; Kim, J.; Andrews, S.R.; Chen, T.; Kelsey, G.; et al. Transcription and chromatin determinants of de novo DNA methylation timing in oocytes. Epigenetics Chromatin 2017, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair (Amst) 2015, 32, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Rosic, S.; Amouroux, R.; Requena, C.E.; Gomes, A.; Emperle, M.; Beltran, T.; Rane, J.K.; Linnett, S.; Selkirk, M.E.; Schiffer, P.H.; et al. Evolutionary analysis indicates that DNA alkylation damage is a byproduct of cytosine DNA methyltransferase activity. Nat. Genet. 2018, 50, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Song, C.X.; He, C.; Zhang, Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef] [PubMed]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/α-ketoglutarate-dependent dioxygenases: Repairing nucleic acid alkylation damage and beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.; Czapinska, H.; Bochtler, M. CpG underrepresentation and the bacterial CpG-specific DNA methyltransferase M.MpeI. Proc. Natl. Acad. Sci. USA 2013, 110, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Rohde, C.; Tierling, S.; Jurkowski, T.P.; Bock, C.; Santacruz, D.; Ragozin, S.; Reinhardt, R.; Groth, M.; Walter, J.; et al. DNA methylation analysis of chromosome 21 gene promoters at single base pair and single allele resolution. PLoS Genet. 2009, 5, e1000438. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Downing, T.L.; Smith, Z.D.; Gu, H.; Clement, K.; Pop, R.; Akopian, V.; Klages, S.; Santos, D.P.; Tsankov, A.M.; et al. Global delay in nascent strand DNA methylation. Nat. Struct. Mol. Biol. 2018, 25, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, H.K.; King, H.W.; Patient, R.K.; Odom, D.T.; Klose, R.J. Protection of CpG islands from DNA methylation is DNA-encoded and evolutionarily conserved. Nucleic Acids Res. 2016, 44, 6693–6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, V.; Lurie, E.; Carrero, I.; Pawliczek, P.; Patel, R.Y.; Rozowsky, J.; Galeev, T.; Huang, Z.; Altshuler, R.C.; Zhang, Z.; et al. Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rohde, C.; Reinhardt, R.; Voelcker-Rehage, C.; Jeltsch, A. Non-imprinted allele-specific DNA methylation on human autosomes. Genome Biol. 2009, 10, R138. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, F.; Tan, L.; Kong, L.; Xiong, L.; Deng, J.; Barbera, A.J.; Zheng, L.; Zhang, H.; Huang, S.; et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell 2011, 42, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Pape, U.J.; Huang, Y.; Henderson, H.R.; Lister, R.; Ko, M.; McLoughlin, E.M.; Brudno, Y.; Mahapatra, S.; Kapranov, P.; et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 2011, 473, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Liu, K.; Lei, M.; Yang, A.; Li, Y.; Hughes, T.R.; Min, J. DNA Sequence recognition of human CXXC domains and their structural determinants. Structure 2018, 26, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Long, H.K.; Blackledge, N.P.; Klose, R.J. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013, 41, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Umata, T.; Okamoto, K.; Obuse, C.; Tsuneoka, M. CxxC-ZF domain is needed for KDM2A to demethylate histone in rDNA promoter in response to starvation. Cell Struct. Funct. 2014, 39, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Vertino, P.M.; Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010, 2, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. TET enzymes, variants, and differential effects on function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugino, K.; Hempel, C.M.; Okaty, B.W.; Arnson, H.A.; Kato, S.; Dani, V.S.; Nelson, S.B. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 2014, 34, 12877–12883. [Google Scholar] [CrossRef] [PubMed]
- Rajavelu, A.; Lungu, C.; Emperle, M.; Dukatz, M.; Bröhm, A.; Broche, J.; Hanelt, I.; Parsa, E.; Schiffers, S.; Karnik, R.; et al. Chromatin-dependent allosteric regulation of DNMT3A activity by MeCP2. Nucleic Acids Res. 2018, 46, 9044–9056. [Google Scholar] [CrossRef] [PubMed]
- Reizel, Y.; Sabag, O.; Skversky, Y.; Spiro, A.; Steinberg, B.; Bernstein, D.; Wang, A.; Kieckhaefer, J.; Li, C.; Pikarsky, E.; et al. Postnatal DNA demethylation and its role in tissue maturation. Nat. Commun. 2018, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Kribelbauer, J.F.; Laptenko, O.; Chen, S.; Martini, G.D.; Freed-Pastor, W.A.; Prives, C.; Mann, R.S.; Bussemaker, H.J. Quantitative analysis of the dna methylation sensitivity of transcription factor complexes. Cell Rep. 2017, 19, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Wang, H.; John, S.; Shafer, A.; Canfield, T.; Lee, K.; Stamatoyannopoulos, J.A. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015, 12, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Schubeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Shimbo, T.; Wade, P.A. Proteins that read DNA methylation. Adv. Exp. Med. Biol. 2016, 945, 303–320. [Google Scholar] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Mayran, A.; Khetchoumian, K.; Hariri, F.; Pastinen, T.; Gauthier, Y.; Balsalobre, A.; Drouin, J. Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat. Genet. 2018, 50, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Su, Y.; Song, Q.; Tung, B.; Oyinlade, O.; Liu, S.; Ying, M.; Ming, G.L.; Song, H.; Qian, J.; et al. Methylated cis-regulatory elements mediate KLF4-dependent gene transactivation and cell migration. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlet, J.; Duymich, C.E.; Lay, F.D.; Mundbjerg, K.; Dalsgaard Sørensen, K.; Liang, G.; Jones, P.A. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol. Cell 2016, 62, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; Von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Shao, X.; Liu, H.; Liu, S.; Wu, Q.; Zhang, Y. Genome-wide dynamic changes of DNA methylation of repetitive elements in human embryonic stem cells and fetal fibroblasts. Genomics 2012, 99, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 2017, 8, 2057. [Google Scholar] [CrossRef] [PubMed]
- Lupo, A.; Cesaro, E.; Montano, G.; Zurlo, D.; Izzo, P.; Costanzo, P. KRAB-Zinc finger proteins: A repressor family displaying multiple biological functions. Curr. Genomics 2013, 14, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jönsson, M.E.; Männe, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Jeong, S.; Escobar, S.G.; Cortez, C.C.; Li, T.W.; Saito, Y.; Yoo, C.B.; Jones, P.A.; Liang, G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc. Natl. Acad. Sci. USA 2006, 103, 14080–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, K.M.; Wang, H.; Kim, H.R.; Wenderski, W.; Fang, F.; Li, C.H.; Dewell, S.; Hughes, S.H.; Melnick, A.M.; Patel, D.J.; et al. Engineering of a histone-recognition domain in Dnmt3a alters the epigenetic landscape and phenotypic features of mouse ESCs. Mol. Cell 2015, 59, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Petell, C.J.; Alabdi, L.; He, M.; San Miguel, P.; Rose, R.; Gowher, H. An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 2016, 44, 7605–7617. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaniel, S.L.; Strahl, B.D. Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci. 2017, 74, 3317–3334. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forné, I.; Pichler, G.; Hörl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galupa, R.; Heard, E. X-chromosome inactivation: New insights into cis and trans regulation. Curr. Opin. Genet. Dev. 2015, 31, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Paucek, R.D.; Gooding, A.R.; Brown, Z.Z.; Ge, E.J.; Muir, T.W.; Cech, T.R. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol. 2017, 24, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holoch, D.; Margueron, R. Mechanisms Regulating PRC2 Recruitment and Enzymatic Activity. Trends Biochem. Sci. 2017, 42, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaru, H.; Selker, E.U. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 2001, 414, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Krajewski, K.; Nady, N.; Tempel, W.; Xue, S.; Badeaux, A.I.; Barsyte-Lovejoy, D.; Martinez, J.Y.; Bedford, M.T.; Fuchs, S.M.; et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012, 19, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Bostick, M.; Kim, J.K.; Estève, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Berkyurek, A.C.; Suetake, I.; Arita, K.; Takeshita, K.; Nakagawa, A.; Shirakawa, M.; Tajima, S. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J. Biol. Chem. 2014, 289, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Bashtrykov, P.; Rajavelu, A.; Hackner, B.; Ragozin, S.; Carell, T.; Jeltsch, A. Targeted mutagenesis results in an activation of DNA methyltransferase 1 and confirms an autoinhibitory role of its RFTS domain. Chembiochem 2014, 15, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Jakoncic, J.; Qian, C. UHRF1 double Tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J. Mol. Biol. 2012, 415, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Dickson, B.M.; Ong, M.S.; Krajewski, K.; Houliston, S.; Kireev, D.B.; Arrowsmith, C.H.; Strahl, B.D. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013, 27, 1288–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nady, N.; Lemak, A.; Walker, J.R.; Avvakumov, G.V.; Kareta, M.S.; Achour, M.; Xue, S.; Duan, S.; Allali-Hassani, A.; Zuo, X.; et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011, 286, 24300–24311. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, J.; Chen, R.; Wang, L.; Li, B.; Cheng, H.; Duan, X.; Zhu, H.; Wei, W.; Li, J.; et al. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat. Commun. 2016, 7, 12464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veland, N.; Hardikar, S.; Zhong, Y.; Gayatri, S.; Dan, J.; Strahl, B.D.; Rothbart, S.B.; Bedford, M.T.; Chen, T. The arginine methyltransferase PRMT6 regulates DNA methylation and contributes to global DNA hypomethylation in cancer. Cell Rep. 2017, 21, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.; Schotta, G.; Sorensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Geiman, T.M.; Xi, S.; Jiang, Q.; Schmidtmann, A.; Chen, T.; Li, E.; Muegge, K. Lsh is involved in de novo methylation of DNA. EMBO J. 2006, 25, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; McIntosh, C.; Lister, R.; Zhu, I.; Han, Y.; Ren, J.; Landsman, D.; Lee, E.; Briones, V.; Terashima, M.; et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014, 24, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lungu, C.; Muegge, K.; Jeltsch, A.; Jurkowska, R.Z. An ATPase-deficient variant of the SNF2 family member HELLS shows altered dynamics at pericentromeric heterochromatin. J. Mol. Biol. 2015, 427, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Briones, V.; Barbour, S.; Yu, W.; Han, Y.; Terashima, M.; Muegge, K. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015, 43, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Teplova, M.; Ishibe-Murakami, S.; Patel, D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012, 335, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Verreault, A. Maintenance DNA methylation of nucleosome core particles. J. Biol. Chem. 2004, 279, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Stockdale, C.J.; Goyal, R.; Ferreira, H.; Owen-Hughes, T.; Jeltsch, A. De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry 2005, 44, 9899–9904. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Suetake, I.; Shimahara, H.; Ura, K.; Tate, S.; Tajima, S. Distinct DNA methylation activity of Dnmt3a and Dnmt3b towards naked and nucleosomal DNA. J. Biochem. 2006, 139, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Felle, M.; Hoffmeister, H.; Rothammer, J.; Fuchs, A.; Exler, J.H.; Längst, G. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011, 39, 6956–6969. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.; et al. Characterization of Dnmt1 binding and DNA methylation on nucleosomes and nucleosomal arrays. PLoS ONE 2015, 10, e0140076. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- De Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6, 7870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Jenness, C.; Giunta, S.; Müller, M.M.; Kimura, H.; Muir, T.W.; Funabiki, H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc. Natl. Acad. Sci. USA 2018, 115, E876–E885. [Google Scholar] [CrossRef] [PubMed]
- Bashtrykov, P.; Ragozin, S.; Jeltsch, A. Mechanistic details of the DNA recognition by the Dnmt1 DNA methyltransferase. FEBS Lett. 2012, 586, 1821–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ecker, J.R. Non-CG methylation in the human genome. Annu. Rev. Genomics Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.J.; Nakai, K. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef] [PubMed]
- Kinde, B.; Wu, D.Y.; Greenberg, M.E.; Gabel, H.W. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc. Natl. Acad. Sci. USA 2016, 113, 15114–15119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yang, H.; Fang, J.; Ma, L.; Gong, R.; Wang, P.; Li, Z.; Xu, Y. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 2015, 6, 7023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keown, C.L.; Berletch, J.B.; Castanon, R.; Nery, J.R.; Disteche, C.M.; Ecker, J.R.; Mukamel, E.A. Allele-specific non-CG DNA methylation marks domains of active chromatin in female mouse brain. Proc. Natl. Acad. Sci. USA 2017, 114, E2882–E2890. [Google Scholar] [CrossRef] [PubMed]
- O’Brown, Z.K.; Greer, E.L. N6-Methyladenine: A conserved and dynamic DNA mark. Adv. Exp. Med. Biol. 2016, 945, 213–246. [Google Scholar] [PubMed]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizábal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Huang, H.; Liu, D.; Cheng, Y.; Liu, X.; Zhang, W.; Yin, R.; Zhang, D.; Zhang, P.; Liu, J.; et al. N6-methyladenine DNA modification in Drosophila. Cell 2015, 161, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Koziol, M.J.; Bradshaw, C.R.; Allen, G.E.; Costa, A.S.H.; Frezza, C.; Gurdon, J.B. Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat. Struct. Mol. Biol. 2016, 23, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 2018, 71, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Schiffers, S.; Ebert, C.; Rahimoff, R.; Kosmatchev, O.; Steinbacher, J.; Bohne, A.V.; Spada, F.; Michalakis, S.; Nickelsen, J.; Müller, M.; et al. Quantitative LC-MS provides no evidence for m6 dA or m4 dC in the genome of mouse embryonic stem cells and tissues. Angew. Chem. Int. Ed. Engl. 2017, 56, 11268–11271. [Google Scholar] [CrossRef] [PubMed]
- Van der Wijst, M.G.; Rots, M.G. Mitochondrial epigenetics: An overlooked layer of regulation? Trends Genet. 2015, 31, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Mangalhara, K.C.; Prakasam, G.; Bamezai, R.N.K. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci. Rep. 2017, 7, 1525. [Google Scholar] [CrossRef] [PubMed]
- Van der Wijst, M.G.; van Tilburg, A.Y.; Ruiters, M.H.; Rots, M.G. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Du, Q.; Chen, L.; Fu, G.; Li, S.; Fu, L.; Zhang, X.; Ma, C.; Bin, C. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 2016, 6, 23421. [Google Scholar] [CrossRef] [PubMed]
- Mechta, M.; Ingerslev, L.R.; Fabre, O.; Picard, M.; Barrès, R. Evidence suggesting absence of mitochondrial DNA methylation. Front. Genet. 2017, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Kungulovski, G.; Jeltsch, A. Epigenome editing: State of the art, concepts, and perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Rots, M.G.; Jeltsch, A. Editing the epigenome: Overview, open questions, and directions of future development. Methods Mol. Biol. 2018, 1767, 3–18. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat Type | Total Number | Mean Length (bps) | Mean GC Content | Mean CpGexp/obs | DNA Methylation |
|---|---|---|---|---|---|
| SINE | 1,426,563 | 244 | 0.50 | 2.98 | H1: 81.4–90.2 |
| IMR90: 62.8–90.9 | |||||
| mHSC: 90.0 | |||||
| LINE | 947,779 | 578 | 0.38 | 4.01 | H1: 82.6–90.7 |
| IMR90: 41.9–88.4 | |||||
| mHSC: 88.53 | |||||
| LTR | 530,763 | 443 | 0.44 | 4.84 | H1: 81.4–90.7 |
| IMR90: 37.2–83.7 | |||||
| mHSC: 89.79 | |||||
| DNA Transposon | 273,586 | 272 | 0.40 | 3.63 | H1: 83.7–91.9 |
| IMR90: 45.4–90.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeltsch, A.; Broche, J.; Bashtrykov, P. Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes. Genes 2018, 9, 566. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110566
Jeltsch A, Broche J, Bashtrykov P. Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes. Genes. 2018; 9(11):566. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110566
Chicago/Turabian StyleJeltsch, Albert, Julian Broche, and Pavel Bashtrykov. 2018. "Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes" Genes 9, no. 11: 566. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9110566