Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease

Institute of Biomedical and Clinical Sciences, University of Exeter Medical School, Exeter, EX1 2LU, UK
*
Authors to whom correspondence should be addressed.
Genes 2018, 9(2), 98; https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020098
Submission received: 2 December 2017
/
Revised: 8 February 2018
/
Accepted: 9 February 2018
/
Published: 15 February 2018
(This article belongs to the Special Issue Aberrant Pre-mRNA Splicing in Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Vascular endothelial growth factor A (VEGF-A) is a prominent pro-angiogenic and pro-permeability factor in the kidney. Alternative splicing of the terminal exon of VEGF-A through the use of an alternative 3′ splice site gives rise to a functionally different family of isoforms, termed VEGF-Axxxb, known to have anti-angiogenic and anti-permeability properties. Dysregulation of the VEGF-Axxx/VEGF-Axxxb isoform balance has recently been reported in several kidney pathologies, including diabetic nephropathy (DN) and Denys–Drash syndrome. Using mouse models of kidney disease where the VEGF-A isoform balance is disrupted, several reports have shown that VEGF-A165b treatment/over-expression in the kidney is therapeutically beneficial. Furthermore, inhibition of certain splice factor kinases involved in the regulation of VEGF-A terminal exon splicing has provided some mechanistic insight into how VEGF-A splicing could be regulated in the kidney. This review highlights the importance of further investigation into the novel area of VEGF-A splicing in chronic kidney disease pathogenesis and how future studies may allow for the development of splicing-modifying therapeutic drugs.
1. Introduction
The human genome is comprised of approximately 20,000 genes, but the human proteome is estimated to be formed of hundreds of thousands of proteins [1]. This diversity is mainly the result of a process known as alternative splicing (AS); a single gene transcript can give rise to multiple proteins depending on the way the gene is spliced [2]. In humans, more than 94% of genes can be alternatively spliced [3,4]. It is generally accepted that the more evolved a species is, the higher the percentage of genes that undergo AS [5].
It is becoming increasingly clear that changes to the tightly regulated process of AS in many genes can result in cellular dysfunction and disease. Cancer is the disease that is most commonly linked to AS dysregulation [6,7,8]; however, there are an increasing number of splice isoforms that have been recently implicated in kidney disease, including vascular endothelial growth factor A (VEGF-A) [9,10]. VEGF-A is key driver of angiogenesis, permeability, migration, and cell survival [11]. The abnormal expression of VEGF-A in the kidney has been widely reported in most types of kidney disease [12]. Alternative splicing of exon 8 of the VEGF-A gene results in the expression of an anti-angiogenic, anti-permeability, and cyto-protective family of isoforms, termed the VEGF-Axxxb family (the most common isoform being VEGF-A165b) [13], which has been shown to be protective in kidney disease [9,10].
This review discusses the mechanism of VEGF-A splicing, highlights the importance of a balance of the VEGF-A exon 8 splice variants in the kidney, and examines the ways in which VEGF-A splicing can be manipulated to obtain therapeutic benefit.
2. Alternative Splicing of VEGF-A
2.1. Modes of Alternative Splicing
The process of pre-mRNA AS involves the inclusion/exclusion of whole exons or parts of exons/introns in the final mRNA transcript. This process is achieved with a splicing reaction conducted by the spliceosome, a macromolecular assembly composed of small nuclear ribonucleoproteins and the associated accessory proteins [14]. In brief, the spliceosome assembles at a splice site (a conserved sequence in the pre-mRNA transcript at the exon-intron junction) and undergoes two transesterification reactions to achieve splicing. The main modes of AS are: cassette exon skipping or retention, intron retention, use of an alternative 3′ or 5′ splice sites in exons, and the inclusion/exclusion of two mutually exclusive exons.
2.2. Regulation of Alternative Splicing
AS is regulated by Cis-acting regulatory elements (auxiliary sequences), which can be divided into four subgroups; exonic and intronic sequence enhancers (ESEs and ISEs), and exonic and intronic sequence silencers (ESSs and ISSs). These Cis-acting regulatory elements recruit trans-acting splice factors to modulate the splicing reaction [2]. Splice factors are RNA-binding proteins, the most common being serine/arginine-rich (SR) proteins, that act with small nuclear ribonucleoproteins to form and mediate the action of the spliceosome [15]. Another key family of splice factors are the heterogeneous ribonucleoproteins (hnRNPs). In general, SR proteins bind to ESEs and recruit core splice factors to the polypyrimidine tract, activating splicing of a cassette exon, whereas hnRNPs classically block access of the spliceosome to the polypyrimidine tract, suppressing RNA splicing [16,17]. However, there are exceptions to these general rules.
2.3. Alternative Splicing of VEGF-A
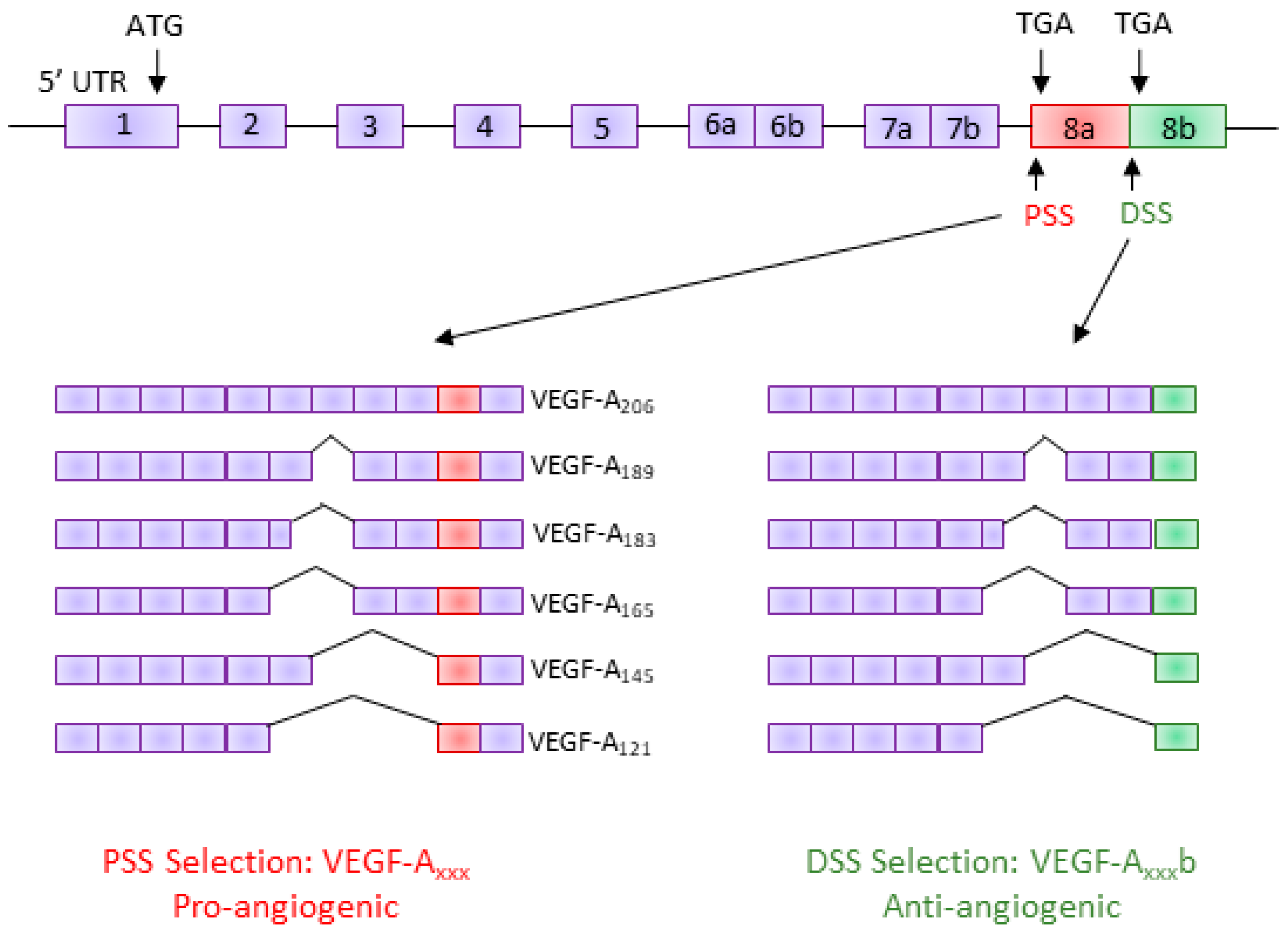
Human VEGF-A is a major regulator of angiogenesis and vessel permeability [11]. The VEGF-A gene consists of eight exons and seven introns [18]. AS of VEGF-A can occur through the inclusion/exclusion of various exons, giving rise to a family of pro-angiogenic isoforms generically known as VEGF-Axxx (VEGF-A121, VEGF-A145, VEGF-A165, VEGF-A189, and VEGF-A206—the number denotes the number of amino acids) [19]. It was not until 2002 that a novel alternative 3′ splice site (distal splice site; DSS) was discovered in exon 8 of the human VEGF-A gene, 66 base pairs downstream of the canonical proximal 3′ splice site (PSS); use of the DSS creates a new open reading frame and results in a functionally different family of isoforms, termed the VEGF-Axxxb family [13]. Although the VEGF-Axxxb isoforms have the same number of amino acids in total, they have an altered C-terminal sequence that differs by six amino acids (Figure 1). This small change in the terminal six amino acids results in VEGF-Axxxb having functionally opposite properties to VEGF-Axxx; VEGF-Axxxb has anti-angiogenic, anti-permeability, and anti-migratory properties [9,10,13,20]. However, like VEGF-Axxx, VEGF-Axxxb is a pro-survival factor [21].
The most common VEGF-Axxxb isoform in the kidney is VEGF-A165b [22]. The differing properties of the anti-angiogenic VEGF-A165b are thought to be due to its inability to efficiently autophosphorylate VEGF receptor 2 (VEGFR-2), the key VEGF-A receptor for driving angiogenesis, permeability, and migration [20]. Several studies have shown that VEGF-A165b poorly activates the VEGFR-2 kinase domain, resulting in weak, transient phosphorylation of downstream targets including Akt and ERK1/2 [10,23].
2.4. Regulation of VEGF-A Exon 8 Splicing
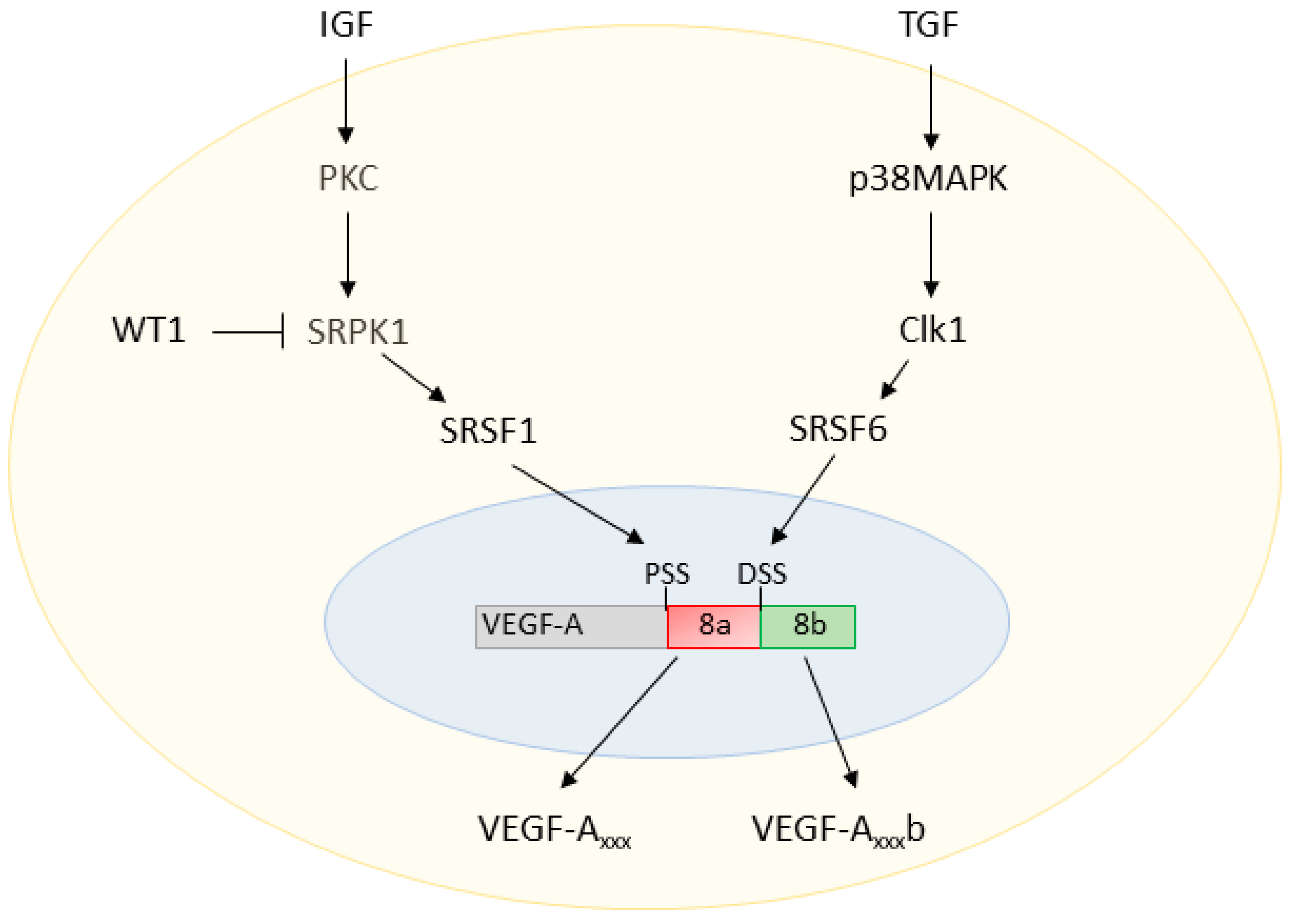
AS of exon 8 of the VEGF-A gene is known to be regulated by several SR proteins. Pre-mRNA sequence analysis revealed a predicted binding site for Serine/Arginine Rich Splicing Factor 1 (SRSF1) upstream of the DSS, and a predicted binding site for SRSF6 downstream of the DSS [24]. Binding of SRSF1 to the pre-mRNA promotes PSS selection, thus resulting in the expression of the VEGF-Axxx isoforms. On the other hand, binding of SRSF6 to the pre-mRNA promotes DSS selection and the expression of VEGF-Axxxb isoforms [25].
The actions of SRSF1 and SRSF6 on the VEGF-A pre-mRNA are modulated by upstream regulators, as summarised in Figure 2. SRSF1 is a known target of the SR protein kinases 1 and 2 (SRPK1/2). Inhibition of SRPK1 activity with small molecule inhibitors or RNA interference (RNAi) knockdown of SRPK1 expression has been reported to block the phosphorylation and nuclear shuttling of SRSF1, switching the ratio of VEGF-A splicing to decrease the pro-angiogenic VEGF-Axxx and increase the anti-angiogenic VEGF-Axxxb isoforms [26,27].
AS of the VEGF-A terminal exon is understood to be modified by extracellular factors via signalling cascades, including various growth factors. Insulin-like growth factor 1 (IGF1) and tumour necrosis factor α (TNFα) have been shown to favour the use of the exon 8 PSS through protein kinase C (PKC)-induced activation of SRPK1; however, transforming growth factor β (TGFβ) promotes DSS use through cell division cycle (CDC)-like kinase 1 (Clk1) activation of p38 mitogen-activated kinases (p38MAPK), which phosphorylates SRSF6 [20,25].
3. VEGF-A Splice Variants in Chronic Kidney Disease
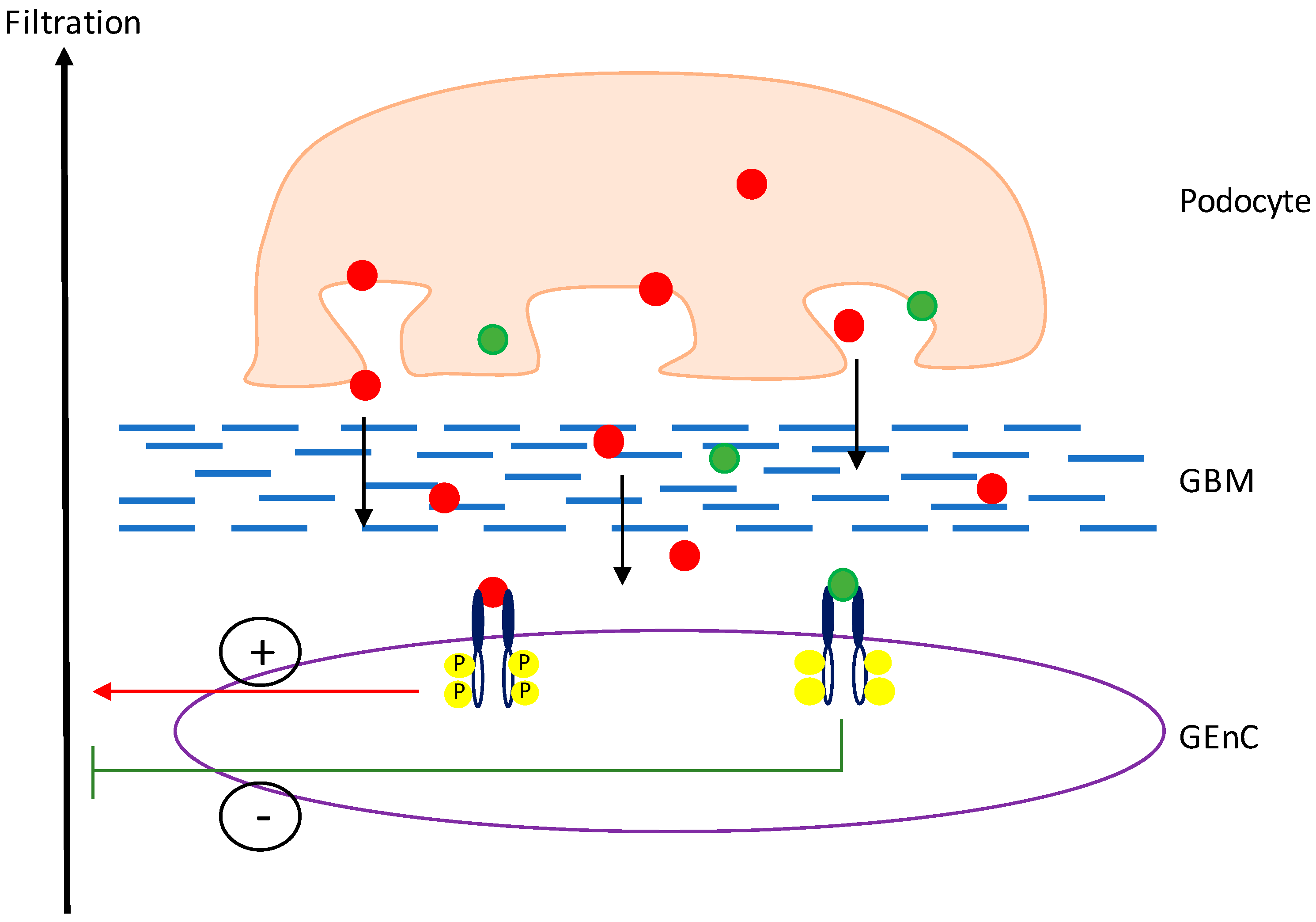
Within the glomerulus, VEGF-A is a key regulator of normal function and is predominately expressed in high levels by mature podocytes, which cross the glomerular basement membrane (GBM) to signal to VEGFR-2 on the glomerular endothelial cells (GEnCs) (Figure 3) [30]. In the physiologically normal kidney, despite high levels of podocyte VEGF-A expression, there is no angiogenesis. It is thought that the reason for this is that the podocytes express a balance of the pro- and anti-angiogenic isoforms of VEGF-A to maintain normal functioning of the glomerular filtration barrier (GFB) [21]. In culture, podocytes have been shown to express both VEGF-A165 and VEGF-A165b mRNA and protein when in a differentiated state [22]. Both isoforms have also been shown to be expressed at the mRNA level in the human kidney cortex [9].
Alterations in glomerular VEGF-A expression is implicated is almost every type of kidney disease; a report by Martini et al. [12] highlighted the strong correlation between VEGF-A expression in the kidney cortex and the estimated glomerular filtration rate (eGFR) in patients with a range of chronic kidney diseases (CKDs). However, it is only recently that the focus has shifted to assess the expression of the VEGF-Axxx/VEGF-Axxxb splice variant ratio in CKD. There is evidence to suggest that diabetic nephropathy (DN) is associated with a switch in VEGF-A splice isoform expression. Glomerular samples from DN patients have shown an increase in the expression of VEGF-Axxx mRNA relative to VEGF-Axxxb mRNA during the later stages of DN, when the kidney function has declined; however, during the early stages of DN, when the kidney is functioning relatively normally, there is an increase in the VEGF-Axxxb/VEGF-Axxx ratio [9]. It is suggested that during the early stages of DN, VEGF-Axxxb is acting to protect the GFB, preventing a decline in the eGFR.
Another example of the dysregulation of VEGF-A exon 8 splicing in CKD is in Denys–Drash syndrome, where a mutation occurs in the Wilm’s tumour suppression gene, WT1 [31]. In healthy podocytes, WT1 binds to the SRPK1 promoter and represses the expression of SRPK1; however, in WT1 mutant podocytes, a reduction in VEGF-A165b is observed due to the mutated WT1 not transcriptionally repressing SRPK1 [26]. This dysregulation of the VEGF-A splicing balance plays a role in the development of CKD in these patients.
There are several types of ischaemic kidney disease where low VEGF-A expression levels have been shown to contribute to the pathology, which can be rescued by VEGF administration, such as in acute kidney injury [32] and renovascular disease [33]. However, there is no reported evidence of the VEGF-Axxx/VEGF-Axxxb splicing balance/expression levels in these diseases.
4. Manipulation of VEGF-A Splicing as a Potential Therapeutic Avenue in Kidney Disease
As the number of disease entities known to be associated with AS increases, we are beginning to explore whether manipulation of AS could be used for therapeutic benefit. In terms of VEGF-A splicing in the kidney, the general aim is to try to switch the splicing ratio to increase the anti-angiogenic VEGF-Axxxb isoforms, and decrease the pro-angiogenic VEGF-Axxx isoforms (summarised in Figure 4). There has been promising evidence from both in vitro assays and in vivo models to suggest that VEGF-A165b is therapeutic in CKD.
In mice, podocyte-specific over-expression of the pro-angiogenic VEGF-A164 has been reported to be detrimental to kidney function. Constitutive over-expression of VEGF-A164 results in albuminuria at birth and a lack in formation of a fully functional GFB, consistent with the phenotype of congenital nephrosis [34,35]. Similarly, inducible over-expression of VEGF-A164 in adult mice results in glomerular disease characterised by proteinuria, increased glomerular water permeability, and GFB abnormalities [36,37], although the severity of the disease phenotype remains inconsistent between different models. When inducible podocyte-specific VEGF-A164 over-expressing mice also over-expressed VEGF-A165b in a constitutive manner, the functional phenotype was rescued, indicating that the balance of VEGF-A isoforms plays a critical role in the regulation of glomerular permeability [37]. This was also true with the dual insult of inducible podocyte VEGF-A164 over-expression and Streptozotocin (STZ)-induced DN; constitutive over-expression of VEGF-A165b prevented the onset of albuminuria in this model [9].
On the other hand, low renal expression of VEGF-A has been shown to contribute to disease pathologies, which can be rescued by VEGF-A administration [32,33]. Although the VEGF-A isoform expression levels have not been characterised in specific disease types, a mouse model with podocyte-specific knockdown of VEGF-A has been reported to be detrimental to kidney function. Heterozygous constitutive knockdown of podocyte VEGF-A resulted in nephrotic syndrome and end-stage renal disease (ESRD) by 9 weeks of age, whereas homozygous knockdown resulted in perinatal death [35]. Knockdown of VEGF-A expression in the mature glomerulus has been reported to result in a range of phenotypes, ranging from profound thrombotic glomerular injury [38], to mild albuminuria and ultra-structural changes to the GFB [10]. These differences are likely to result from differing genetic backgrounds and degrees of VEGF-A knockdown. When inducible podocyte-specific VEGF-A knockdown mice over-expressed the VEGF-A165b isoform in the podocytes, the kidney injury phenotype was rescued [10]. Our group has recently shown in a quadruple transgenic mouse model that podocyte-specific overexpression of VEGF-A165b only, while all other VEGF isoforms are deleted, rescues albuminuria and the increase in water permeability [10].
As described above, DN is associated with dysregulation of VEGF-A splicing. When type I and type II diabetic mice were treated with intraperitoneal injections of VEGF-A165b recombinant protein, they did not develop the DN phenotype observed in the diabetic controls [9]. Therefore, VEGF-A165b appears to play a therapeutic role in mouse models of CKD, indicating that manipulation of VEGF-A splice isoforms could be a novel therapeutic avenue in CKD. It is important to note that podocyte-specific VEGF-A165b over-expression alone resulted in no detrimental changes to kidney function in the long term [39].
A key study examined how the splicing of VEGF-A can be manipulated in cultured WT1 mutant podocytes. As described above, WT1 mutant podocytes are unable to transcriptionally repress SRPK1 expression, resulting in increased activation of SRSF1 and high expression of the pro-angiogenic VEGF-A165 with reduced expression of the anti-angiogenic VEGF-A165b. This alteration in VEGF-A splicing could be reversed by wild-type WT1, a knockdown of SRSF1 or SRPK1, and inhibition of SRPK1, thus increasing the expression of the anti-angiogenic VEGF-A165b isoform [26].
Podocyte expressed VEGF-A signals to VEGFR-2 located on the GEnCs. Mechanistically, mice over-expressing podocyte-specific VEGF-A165b have been shown to have an increase in glomerular VEGFR-2 expression [9]. This is also true for cultured GEnCs treated with VEGF-A165b; however, unlike VEGF-A165, VEGF-A165b is unable to fully phosphorylate VEGFR2 at the tyrosine residues required for full activation, resulting in an inhibition in some of the downstream signalling pathways involved in migration [10]. Interestingly, VEGF-A165b can activate pro-survival factors downstream of VEGFR2, Akt and ERK1/2 [10], suggesting that VEGF-A165b may either weakly phosphorylate some tyrosine residues of VEGFR2 in a transient manner, or could be acting through other receptors such as VEGFR1. Further research is required to gain more information on the mechanistic action of VEGF-A165b in the kidney.
5. Discussion
The balance of the VEGF-Axxx/VEGF-Axxxb appears to be essential in the normal functioning of the GFB. This ratio of splice isoforms has been found to be altered in certain types of CKD, including DN and Denys–Drash syndrome [9,31]. However, manipulation of the pathological balance of isoforms to increase levels of VEGF-A165b has been shown to have therapeutic benefit via inhibition of increased angiogenesis and permeability, and through the activation of pro-survival factors [9,10]. This evidence highlights the importance for further investigation into the novel area of the contribution of VEGF-A splicing towards CKD pathogenesis and how we can manipulate the isoform balance for therapeutic benefit. We envision that future studies will provide more insights into the mechanistic aspects of VEGF-A splicing dysregulation in CKD, allowing for the development of a novel class of splicing-modifying therapeutic drugs.
Acknowledgments
This work was supported by grants to Seb Oltean from British Heart Foundation (PG/15/53/31371), Richard Bright VEGF Research Trust and Diabetes UK (17/000568). These grants include funds for Open Access publishing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Klerk, E.; t Hoen, P.A. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evoluntionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Weiss, W.A. Alternative splicing in cancer: Implications for biology and therapy. Oncogene 2015, 34, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Qiu, Y.; Ferguson, J.K.; Stevens, M.; Neal, C.; Russell, A.; Kaura, A.; Arkill, K.P.; Harris, K.; Symonds, C.; et al. Vascular endothelial growth factor-A165b is protective and restores endothelial glycocalyx in diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Neal, C.R.; Salmon, A.H.J.; Bates, D.O.; Harper, S.J.; Oltean, S. VEGF-A165 b protects against proteinuria in a mouse model with progressive depletion of all endogenous VEGF-A splice isoforms from the kidney. J. Physiol. 2017, 595, 6281–6298. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Claesson-Welsh, L. VEGF receptor signal transduction. Sci. STKE 2001, 11, re21. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.; Nair, V.; Keller, B.J.; Eichinger, F.; Hawkins, J.J.; Randolph, A.; Boger, C.A.; Gadegbeku, C.A.; Fox, C.S.; Cohen, C.D.; et al. Integrative biology identifies shared transcriptional networks in CKD. J. Am. Soc. Nephrol. 2014, 25, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.C.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Bevan, H.S.; van den Akker, N.M.; Qiu, Y.; Polman, J.A.; Foster, R.R.; Yem, J.; Nishikawa, A.; Satchell, S.C.; Harper, S.J.; Gittenberger-de Groot, A.C.; et al. The alternatively spliced anti-angiogenic family of VEGF isoforms VEGFxxxb in human kidney development. Nephron Physiol. 2008, 110, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.G.; Foster, R.R.; Saleem, M.; Mathieson, P.W.; Gillatt, D.A.; Bates, D.O.; Harper, S.J. Differentiated human podocytes endogenously express an inhibitory isoform of vascular endothelial growth factor (VEGF165b) mRNA and protein. Am. J. Physiol. Renal Physiol. 2004, 286, F767–F773. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.O.; Claesson-Welsh, L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M. The role of VEGF 165b in pathophysiology. Cell Adh. Migr. 2012, 6, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.V.; Dick, A.D.; Harper, S.J.; Bates, D.O. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Invest. Opthalmol. Vis. Sci. 2013, 54, 5797–5806. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.F.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Baelde, H.J.; Quaggin, S.E. Role of the VEGF-a signaling pathway in the glomerulus: Evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007, 106, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, V.A.; Jeruschke, S.; Eitner, F.; Becker, J.U.; Pitschke, G.; Ince, Y.; Miner, J.H.; Leuschner, I.; Engers, R.; Everding, A.S.; et al. Impaired glomerular maturation and lack of VEGF165b in Denys-Drash syndrome. J. Am. Soc. Nephrol. 2007, 18, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Y.; Yang, S.; Wu, M.; Fang, Y.; Feng, J.; Liu, B. Protective effect of vascular endothelial growth factor against cardiopulmonary bypass-associated acute kidney injury in beagles. Exp. Ther. Med. 2018, 15, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Lgue, O.C.; McGowan, J.W.; George, E.M.; Bidwell, G.L., 3rd. Therapeutic angiogenesis by vascular endothelial growth factor supplementation for treatment of renal disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Reidy, K.; Marlier, A.; Bertuccio, C.; Villegas, G.; Jimenez, J.; Kashgarian, M.; Tufro, A. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid-resistant nephrotic syndrome. Am. J. Pathol. 2010, 177, 2225–2233. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Neal, C.R.; Mavrou, A.; Patel, P.; Ahad, T.; Alsop, C.; Lee, T.; Sison, K.; Qiu, Y.; Harper, S.J.; et al. VEGF165b overexpression restores normal glomerular water permeability in VEGF164-overexpressing adult mice. Am. J. Physiol. Renal Physiol. 2012, 303, F1026–F1036. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Ferguson, J.; Oltean, S.; Neal, C.R.; Kaura, A.; Bevan, H.; Wood, E.; Sage, L.M.; Lanati, S.; Nowak, D.G.; et al. Overexpression of VEGF165b in podocytes reduces glomerular permeability. J. Am. Soc. Nephrol. 2010, 21, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vascular endothelial growth factor A (VEGF-A) splice variants. The VEGF-A pre-mRNA is comprised of eight exons; inclusion/exclusion of these exons gives rise to several isoforms with differing amino acid lengths. In the terminal exon (exon 8), an alternative 3′ splice site results in a new family of isoforms, the VEGF-Axxxb family. These isoforms have the same number of amino acids but a different C-terminus sequence, resulting in them being functionally different (anti-angiogenic). PSS: proximal splice site; DSS: distal splice site; UTR: untranslated region.
Figure 1.
Vascular endothelial growth factor A (VEGF-A) splice variants. The VEGF-A pre-mRNA is comprised of eight exons; inclusion/exclusion of these exons gives rise to several isoforms with differing amino acid lengths. In the terminal exon (exon 8), an alternative 3′ splice site results in a new family of isoforms, the VEGF-Axxxb family. These isoforms have the same number of amino acids but a different C-terminus sequence, resulting in them being functionally different (anti-angiogenic). PSS: proximal splice site; DSS: distal splice site; UTR: untranslated region.

Figure 2.
VEGF-A splicing regulation. Insulin-like growth factor (IGF) activates protein kinase C (PKC), which in turn phosphorylates SR protein kinase 1 (SRPK1). Activated SRPK1 can then shuttle the splice factor Serine/Arginine Rich Splicing Factor 1 (SRSF1) to the nucleus, resulting in proximal splice site (PSS) selection in exon 8 of VEGF-A. The transcription factor Wilms Tumor 1 (WT1) inhibits the synthesis of SRPK1, downregulating PSS selection. On the other hand, transforming growth factor (TGF) signalling activates p38 mitogen-activated protein kinases (p38MAPK), which then activate cell division cycle (CDC)-like kinase 1 (Clk1). Activates Clk1 causes shuttling of the splice factor SRSF6 to the nucleus, resulting in distal splice site (DSS) selection.
Figure 2.
VEGF-A splicing regulation. Insulin-like growth factor (IGF) activates protein kinase C (PKC), which in turn phosphorylates SR protein kinase 1 (SRPK1). Activated SRPK1 can then shuttle the splice factor Serine/Arginine Rich Splicing Factor 1 (SRSF1) to the nucleus, resulting in proximal splice site (PSS) selection in exon 8 of VEGF-A. The transcription factor Wilms Tumor 1 (WT1) inhibits the synthesis of SRPK1, downregulating PSS selection. On the other hand, transforming growth factor (TGF) signalling activates p38 mitogen-activated protein kinases (p38MAPK), which then activate cell division cycle (CDC)-like kinase 1 (Clk1). Activates Clk1 causes shuttling of the splice factor SRSF6 to the nucleus, resulting in distal splice site (DSS) selection.

Figure 3.
VEGF-A signalling in the glomerulus. Both VEGF-Axxx (red dots) and VEGF-Axxxb (green dots) are expressed by the podocytes and diffuse through the glomerular basement membrane (GBM) to bind to VEGFR2 on the glomerular endothelial cells (GEnCs), against the flow of filtrate. Upon binding of VEGF-Axxx to VEGFR2, the receptor is phosphorylated, initiating a pro-permeability response. However, upon binding of VEGF-Axxxb to VEGFR2, the receptor is not activated and an anti-permeability response is initiated.
Figure 3.
VEGF-A signalling in the glomerulus. Both VEGF-Axxx (red dots) and VEGF-Axxxb (green dots) are expressed by the podocytes and diffuse through the glomerular basement membrane (GBM) to bind to VEGFR2 on the glomerular endothelial cells (GEnCs), against the flow of filtrate. Upon binding of VEGF-Axxx to VEGFR2, the receptor is phosphorylated, initiating a pro-permeability response. However, upon binding of VEGF-Axxxb to VEGFR2, the receptor is not activated and an anti-permeability response is initiated.

Figure 4.
Switching VEGF-A splicing with splicing-modifying drugs for therapeutic benefit. SRPK1 is a key regulator of VEGF-A exon 8 splicing; therefore, it is an ideal target for therapeutic modulation of VEGF-A splicing to increase the VEGF-Axxxb isoform expression. Splicing-modifying drugs designed to up-regulate WT1 (thus initiating SRPK1 transcriptional repression), inhibit SRPK1 activation or increase the activation/expression of SRSF6, resulting in a splicing switch to increase the VEGF-Axxxb/VEGF-Axxx ratio. As such, they provide a novel therapeutic avenue in the treatment/prevention of chronic kidney disease. CKD: chronic kidney disease; GFR: glomerular filtration rate.
Figure 4.
Switching VEGF-A splicing with splicing-modifying drugs for therapeutic benefit. SRPK1 is a key regulator of VEGF-A exon 8 splicing; therefore, it is an ideal target for therapeutic modulation of VEGF-A splicing to increase the VEGF-Axxxb isoform expression. Splicing-modifying drugs designed to up-regulate WT1 (thus initiating SRPK1 transcriptional repression), inhibit SRPK1 activation or increase the activation/expression of SRSF6, resulting in a splicing switch to increase the VEGF-Axxxb/VEGF-Axxx ratio. As such, they provide a novel therapeutic avenue in the treatment/prevention of chronic kidney disease. CKD: chronic kidney disease; GFR: glomerular filtration rate.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stevens, M.; Oltean, S. Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease. Genes 2018, 9, 98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020098
AMA Style
Stevens M, Oltean S. Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease. Genes. 2018; 9(2):98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020098
Chicago/Turabian StyleStevens, Megan, and Sebastian Oltean. 2018. "Modulation of VEGF-A Alternative Splicing as a Novel Treatment in Chronic Kidney Disease" Genes 9, no. 2: 98. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020098
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.