Identification of Major Rhizobacterial Taxa Affected by a Glyphosate-Tolerant Soybean Line via Shotgun Metagenomic Approach

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Field Design and Sampling
2.2. DNA Extraction from Rhizospheric Soil Samples
2.3. Analyses of 16S rDNA via Deep Sequencing Amplicons
2.3.1. PCR Amplification of 16S rDNA (V5–V7) and Illumina MiSeq Sequencing
2.3.2. Operational Taxonomic Unit Selection and Analysis of Species Composition and Abundance
2.3.3. Alpha and Beta Diversity Analyses
2.4. Shotgun Metagenomic Approaches
2.4.1. Construction of Metagenomic DNA Library
2.4.2. Shotgun Metagenome Sequencing
2.4.3. Quality Control and De Novo Metagenome Assembly
2.4.4. Taxonomic Assignment
2.5. Statistical Analyses
3. Results
3.1. Composition and Structure of Rhizosphere Bacterial Community Revealed by 16S rDNA V5–V7 Amplicon Deep Sequencing
3.1.1. Overall Analysis of 16S rDNA (V5–V7 Hypervariable Region) Amplicon Sequencing Data-Based Illumina MiSeq
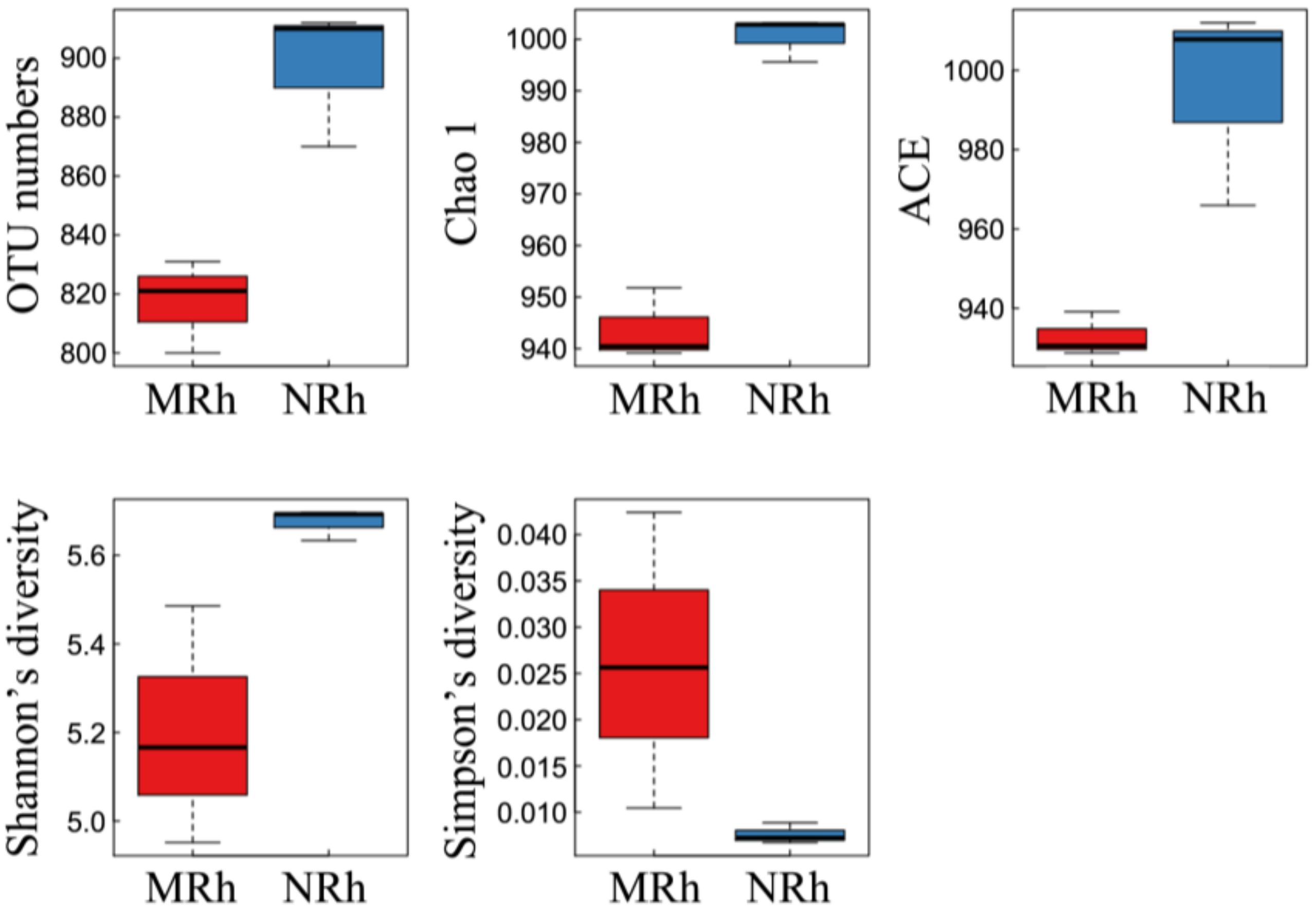
3.1.2. Alpha Diversity of Bacterial Communities in Rhizospheric Soil
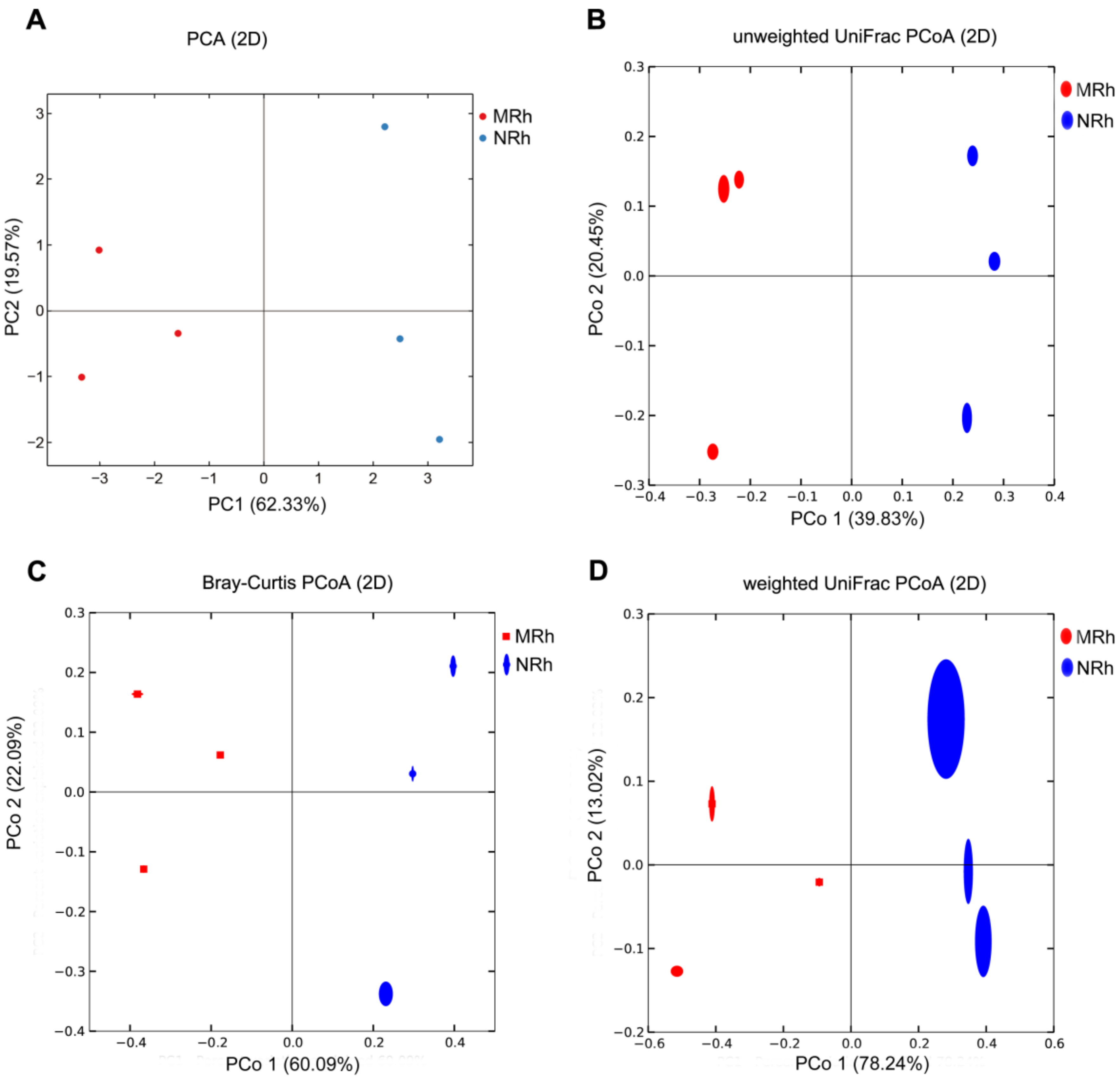
3.1.3. Beta Diversity of Bacterial Community in the Rhizosphere Soil
3.1.4. Comparison of Dominant Bacterial Phyla in the Rhizospheric Soil between N698 and MD12
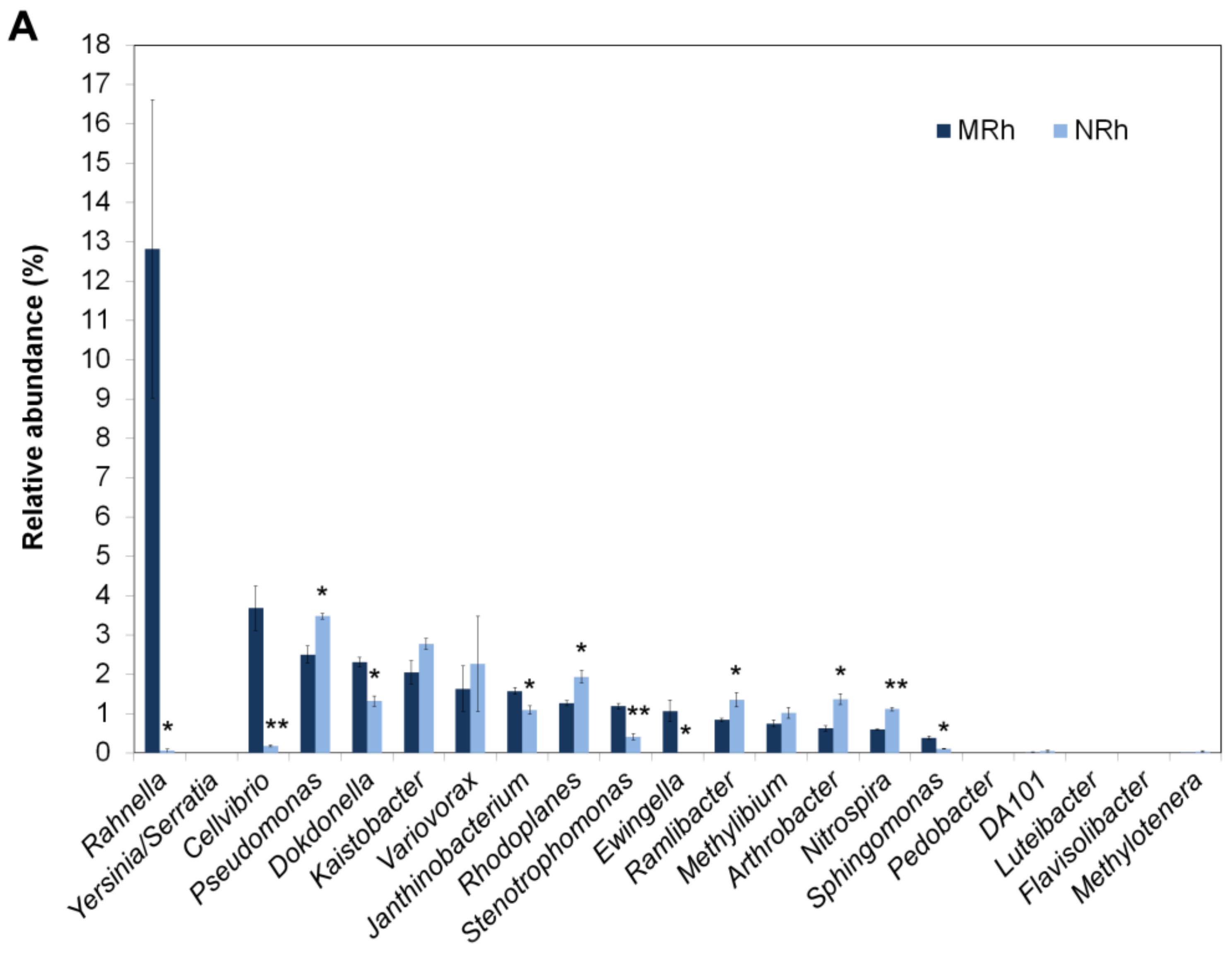
3.1.5. Comparison of Dominant Bacterial Genera in the Rhizospheric Soil between N698 and MD12
3.2. Composition of Rhizosphere Microbial Communities Revealed by Shotgun Metagenomic Approaches
3.2.1. Statistical Summary of Assembled Shotgun Metagenome Sequencing Data
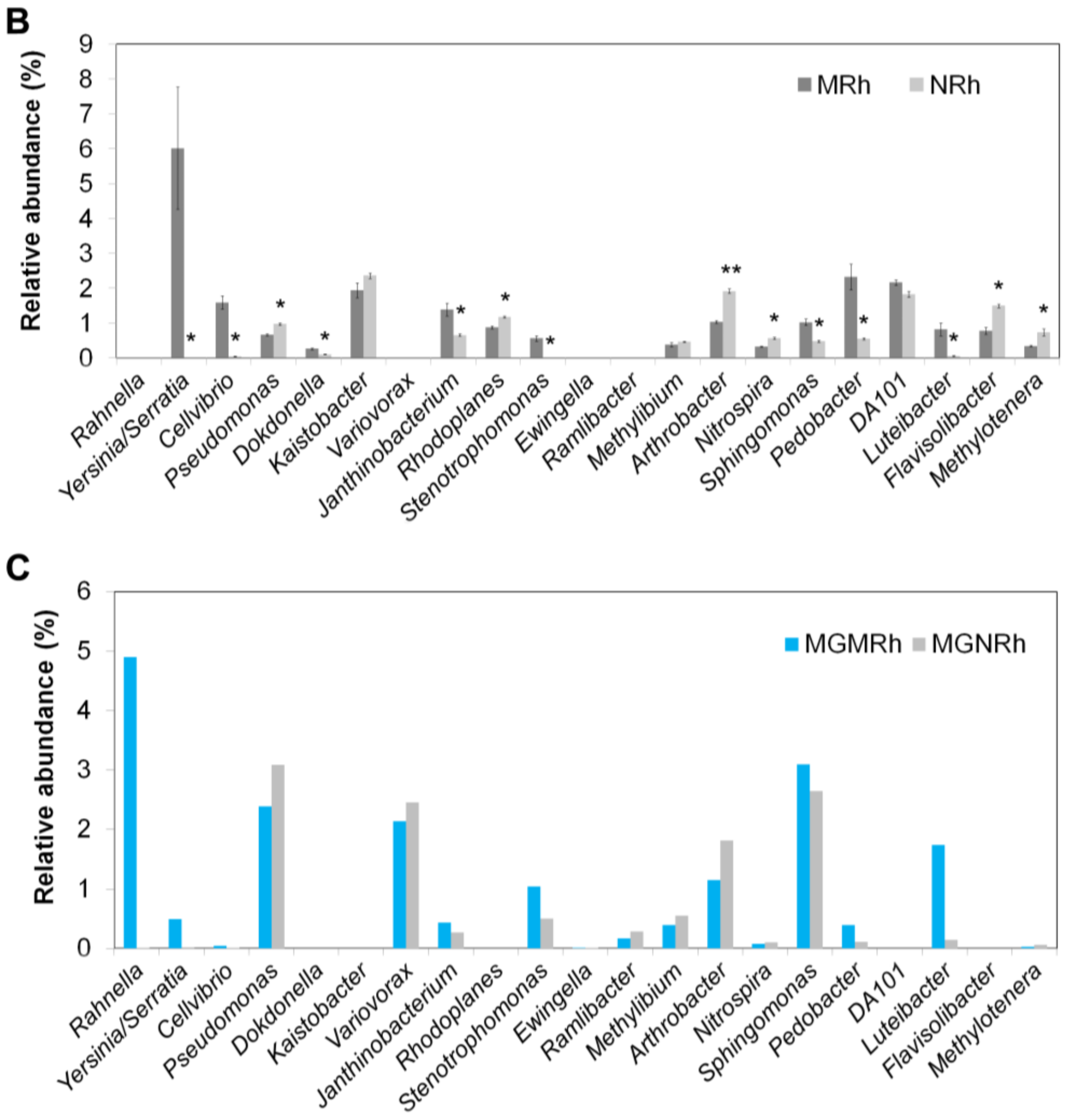
3.2.2. Computation and Comparison of Taxonomic Assignment between MGMRh and MGNRh by SOAPaligner Based on the NCBI Nucleotide Database
3.2.3. Computation and Comparison of Taxonomic Assignment between MGMRh and MGNRh by One Codex Based on One Codex DB
3.2.4. Computation and Comparison of Taxonomic Assignment between MGMRh and MGNRh by One Codex Based on RefSeq DB
3.2.5. Identification of Rhizobacterial Species of Rahnella and Serratia by Shotgun Metagenomic Approaches
4. Discussion
4.1. The Impact of the GT Soybean Line N698 on the Rhizobacteria Has Been Confirmed by 16S rDNA V5–V7 Amplicon Deep Sequencing
4.2. Shortcomings and Solutions of 16S rDNA Amplicon Deep Sequencing
4.3. Shotgun Metagenome Sequencing via NGS Technology and Bioinformatics Tool
4.4. Potential Reason and Consequences of Depletion of Rahnella Genus
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- James, C. Global status of commercialized biotech/GM Crops: 2015. China Biotechnol. 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Benbrook, C.M. Trends in glyphosate herbicide use in the United States and globally. Environ. Sci. Eur. 2016, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- ISAAA. Global status of commercialized biotech/GM Crops: 2016. China Biotechnol. 2017, 37, 1–8. [Google Scholar] [CrossRef]
- Dunfield, K.E.; Germida, J.J. Impact of genetically modified crops on soil- and plant-associated microbial communities. J. Environ. Qual. 2004, 33, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zeng, Q.; Yan, F.M.; Xu, H.G.; Xu, C.R. Effects of transgenic plants on soil microorganisms. Plant Soil 2005, 271, 1–13. [Google Scholar] [CrossRef]
- Turrini, A.; Sbrana, C.; Giovannetti, M. Belowground environmental effects of transgenic crops: A soil microbial perspective. Res. Microbiol. 2015, 166, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.; Kamilova, F. Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 2009, 63, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.A.; Babujia, L.C.; Silva, A.P.; Guimaraes, M.d.F.; Arias, C.A.; Hungria, M. Impact of the ahas transgene and of herbicides associated with the soybean crop on soil microbial communities. Transgen. Res. 2013, 22, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.G.; Sun, S.; Ji, J.; Wu, H.Y.; Meng, F.; Zhang, M.R.; Zheng, X.B.; Wu, C.X.; Zhang, Z.G. Comparison of the rhizosphere bacterial communities of Zigongdongdou soybean and a high-methionine transgenic line of this cultivar. PLoS ONE 2014, 9, e103343. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.I.; Oh, Y.J.; Kim, B.Y.; Cho, H.S. Effects of CaMSRB2-Expressing transgenic rice cultivation on soil microbial communities. J. Microbiol. Biotechnol. 2016, 26, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.G.; Xu, L.P.; Gao, S.W.; Guo, J.L.; Luo, J.; You, Q.; Que, Y.X. Cry1Ac Transgenic sugarcane does not affect the diversity of microbial communities and has no significant effect on enzyme activities in rhizosphere soil within one crop season. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.J.; Lu, S.B.; Huo, Y.L.; Guan, Z.P.; Liu, B.A.; Wei, W. Do genetically modified plants affect adversely on soil microbial communities? Agric. Ecosyst. Environ. 2016, 235, 289–305. [Google Scholar] [CrossRef]
- Lee, Y.E.; Yang, S.H.; Bae, T.W.; Kang, H.G.; Lim, P.O.; Lee, H.Y. Effects of field-grown genetically modified zoysia grass on bacterial community structure. J. Microbiol. Biotechnol. 2011, 21, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Hungria, M.; Mendes, I.C.; Nakatani, A.S.; dos Reis-Junior, F.B.; Morais, J.Z.; Neves de Oliveira, M.C.; Fernandes, M.F. Effects of the glyphosate-resistance gene and herbicides on soybean: Field trials monitoring biological nitrogen fixation and yield. Field Crop Res. 2014, 158, 43–54. [Google Scholar] [CrossRef]
- Babujia, L.C.; Silva, A.P.; Nakatani, A.S.; Cantao, M.E.; Ribeiro Vasconcelos, A.T.; Visentainer, J.V.; Hungria, M. Impact of long-term cropping of glyphosate-resistant transgenic soybean Glycine max (L.) Merr. on soil microbiome. Transgenic Res. 2016, 25, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.-H.; Zhu, Y.-L.; Kong, L.-R.; Cheng, J.; Tang, C.-Y.; Hua, X.-M.; Meng, F.-F.; Pang, Y.-J.; Yang, R.-W.; Qi, J.-L.; et al. Impact of a glyphosate-tolerant soybean line on the rhizobacteria, revealed by illumina MiSeq. J. Microbiol. Biotechnol. 2017, 27, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Gans, J.; Wolinsky, M.; Dunbar, J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005, 309, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative metagenomics of microbial communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Inceoglu, O.; Abu Al-Soud, W.; Salles, J.F.; Semenov, A.V.; van Elsas, J.D. Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS ONE 2011, 6, e23321. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark Welch, D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Barriuso, J.; Valverde, J.R.; Mellado, R.P. Effect of Cry1Ab Protein on rhizobacterial communities of Bt-maize over a four-year cultivation period. PLoS ONE 2012, 7, e35481. [Google Scholar] [CrossRef] [PubMed]
- Verbruggen, E.; Kuramae, E.E.; Hillekens, R.; de Hollander, M.; Kiers, E.T.; Roling, W.F.M.; Kowalchuk, G.A.; van der Heijden, M.G.A. Testing potential effects of maize expressing the Bacillus thuringiensis Cry1Ab Endotoxin (BT Maize) on mycorrhizal fungal communities via DNA- and RNA-Based pyrosequencing and molecular fingerprinting. Appl. Environ. Microbiol. 2012, 78, 7384–7392. [Google Scholar] [CrossRef] [PubMed]
- Kuramae, E.E.; Verbruggen, E.; Hillekens, R.; de Hollander, M.; Roling, W.F.M.; van der Heijden, M.G.A.; Kowalchuk, G.A. Tracking Fungal Community Responses to Maize Plants by DNA- and RNA-Based Pyrosequencing. PLoS ONE 2013, 8, e69973. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.A.; Balint, M.; Greshake, B.; Bandow, C.; Rombke, J.; Schmitt, I. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 2013, 65, 128–132. [Google Scholar] [CrossRef]
- Xian, L.J.; Li, Y.; Jiang, Z.; Ma, J.D.; Jin, L.; Chen, L.; Zhou, C.W.; Zhang, J.; Liu, Y.K.; Zhu, L.; et al. Alterations in cecal microbiota of Jinhua piglets fostered by a Yorkshire sow. Chin. Sci. Bull. 2014, 59, 4304–4311. [Google Scholar] [CrossRef]
- Yang, C.Y.; Li, Y.; Zhou, B.; Zhou, Y.Y.; Zheng, W.; Tian, Y.; Van Nostrand, J.D.; Wu, L.Y.; He, Z.L.; Zhou, J.Z.; et al. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China. Sci. Rep. 2015, 5, 8476. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed]
- Mashiane, R.A.; Ezeokoli, O.T.; Adeleke, R.A.; Bezuidenhout, C.C. Metagenomic analyses of bacterial endophytes associated with the phyllosphere of a BT maize cultivar and its isogenic parental line from South Africa. World J. Microbiol. Biotechnol. 2017, 33, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.X.; Chu, Y.G.; Ding, C.J.; Huang, Q.J.; Zhang, B.Y.; Zhang, W.X.; Su, X.H. Assessing bacterial communities in the rhizosphere of 8-year-old genetically modified poplar (Populus spp.). J. For. Res. 2016, 27, 939–947. [Google Scholar] [CrossRef]
- Debruyn, J.M.; Bevard, D.A.; Essington, M.E.; McKnight, J.Y.; Schaeffer, S.M.; Baxter, H.L.; Mazarei, M.; Mann, D.G.J.; Dixon, R.A.; Chen, F.; et al. Field-grown transgenic switchgrass (Panicum virgatum L.) with altered lignin does not affect soil chemistry, microbiology, and carbon storage potential. Glob. Chang. Biol. Bioenergy 2017, 9, 1100–1109. [Google Scholar] [CrossRef]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.W.; Chokhawala, H.; Schroth, G.; Luo, S.J.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zhang, T. Metagenomic and Metatranscriptomic analysis of microbial community structure and gene expression of activated sludge. PLoS ONE 2012, 7, e38183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Bremges, A.; Maus, I.; Belmann, P.; Eikmeyer, F.; Winkler, A.; Albersmeier, A.; Puhler, A.; Schluter, A.; Sczyrba, A. Deeply sequenced metagenome and metatranscriptome of a biogas-producing microbial community from an agricultural production-scale biogas plant. Gigascience 2015, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Mangrola, A.; Dudhagara, P.; Koringa, P.; Joshi, C.G.; Parmar, M.; Patel, R. Deciphering the microbiota of Tuwa hot spring, India using shotgun metagenomic sequencing approach. Genom. Data 2015, 4, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Mangrola, A.V.; Dudhagara, P.; Koringa, P.; Joshi, C.G.; Patel, R.K. Shotgun metagenomic sequencing based microbial diversity assessment of Lasundra hot spring, India. Genom. Data 2015, 4, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Tsurumaru, H.; Okubo, T.; Okazaki, K.; Hashimoto, M.; Kakizaki, K.; Hanzawa, E.; Takahashi, H.; Asanome, N.; Tanaka, F.; Sekiyama, Y.; et al. Metagenomic analysis of the bacterial community associated with the taproot of sugar beet. Microbes Environ. 2015, 30, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Nesme, J.; Cecillon, S.; Delmont, T.O.; Monier, J.M.; Vogel, T.M.; Simonet, P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr. Biol. 2014, 24, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.Y.; Jia, H.J.; Feng, Q.; Wang, D.H.; Liang, D.; Wu, X.N.; Li, J.H.; Tang, L.Q.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; Kohli, S.; Latka, C.; Bhatia, S.; Vellarikal, S.K.; Sivasubbu, S.; Scaria, V.; Ramachandran, S. Screening currency notes for microbial pathogens and antibiotic resistance genes using a shotgun metagenomic approach. PLoS ONE 2015, 10, e0128711. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Smartt, A.; Wang, J.; Utturkar, S.; Frank, A.; Bi, M.; Liu, J.; Williams, D.; Xu, T.; Eldridge, M.; et al. Integrated metagenomics and metatranscriptomics analyses of root-associated soil from transgenic switchgrass. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Hily, J.-M.; Demaneche, S.; Poulicard, N.; Tannieres, M.; Djennane, S.; Beuve, M.; Vigne, E.; Demangeat, G.; Komar, V.; Gertz, C.; et al. Metagenomic-based impact study of transgenic grapevine rootstock on its associated virome and soil bacteriome. Plant Biotechnol. J. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Schlaeppi, K.; Dombrowski, N.; Oter, R.G.; van Themaat, E.V.L.; Schulze-Lefert, P. Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. USA 2014, 111, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Li, Y.R.; Cai, Z.M.; Li, S.H.; Zhu, J.F.; Zhang, F.; Liang, S.S.; Zhang, W.W.; Guan, Y.L.; Shen, D.Q.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.S.; Krumm, N.; Greenfield, N.B. One Codex: A Sensitive and accurate data platform for genomic microbial identification. bioRxiv 2015, 23. [Google Scholar] [CrossRef]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Yu, C.; Li, Y.R.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.-H.; Hua, X.-M.; Cheng, J.; Zhu, Y.-L.; Wang, G.-H.; Pang, Y.-J.; Yang, R.-W.; Zhang, L.; Shou, H.; Wang, X.-M.; et al. Impact of glyphosate on the rhizosphere microbial communities of an EPSPS-Transgenic Soybean Line ZUTS31 by metagenome sequencing. Curr. Genom. 2018, 19, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Zhou, J.Z.; He, Z.L.; Yang, Y.F.; Deng, Y.; Tringe, S.G.; Alvarez-Cohen, L. High-Throughput metagenomic technologies for complex microbial community analysis: Open and closed formats. mBio 2015, 6, e02288-14. [Google Scholar] [CrossRef] [PubMed]
- Lemos, L.N.; Fulthorpe, R.R.; Roesch, L.F.W. Low sequencing efforts bias analyses of shared taxa in microbial communities. Folia Microbiol. 2012, 57, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.J.; Raskin, L. PCR Biases Distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 2012, 7, e43093. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F.M.; Ferrera, I.; Sarmento, H.; Hingamp, P.; Ogata, H.; de Vargas, C.; Lima-Mendez, G.; et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, K.; Hall, M.W.; Lynch, M.D.J.; Moreno-Hagelsieb, G.; Neufeld, J.D. Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microbiol. 2014, 80, 5717–5722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, L.; Deng, Y.; Zhi, X.; Jiang, Y.H.; Tu, Q.; Xie, J.; Van Nostrand, J.D.; He, Z.; Yang, Y. Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 2011, 5, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, Y.H.; Deng, Y.; Shi, Z.; Zhou, B.Y.; Xue, K.; Wu, L.; He, Z.; Yang, Y. Random sampling process leads to overestimation of β-diversity of microbial communities. mBio 2013, 4, e00324-13. [Google Scholar] [CrossRef] [PubMed]
- Pootakham, W.; Mhuantong, W.; Yoocha, T.; Putchim, L.; Sonthirod, C.; Naktang, C.; Thongtham, N.; Tangphatsornruang, S. High resolution profiling of coral-associated bacterial communities using full-length 16S rRNA sequence data from PacBio SMRT sequencing system. Sci. Rep. 2017, 7, 2774. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Freitas, T.A.K.; Li, P.E.; Scholz, M.B.; Chain, P.S.G. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Pan, Y.; Tooming-Klunderud, A.; Eijsink, V.G.; McHardy, A.C.; Nederbragt, A.J.; Pope, P.B. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep. 2016, 6, 25373. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.L.; Watson, M.; Minot, S.S.; Rivera, M.C.; Franklin, R.B. MinION (TM) nanopore sequencing of environmental metagenomes: A synthetic approach. Gigascience 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Kertesz, M.; Hug, L.A.; Pushkarev, D.; Blauwkamp, T.A.; Castelle, C.J.; Amirebrahimi, M.; Thomas, B.C.; Burstein, D.; Tringe, S.G.; et al. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res. 2015, 25, 534–543. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.; Herzfeld, T.; Wackernagel, W. Transfer of plastid DNA from tobacco to the soil bacterium Acinetobacter sp. by natural transformation. Mol. Microbiol. 2004, 53, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Fry, J.C.; Rogers, H.J.; Day, M.J. Transformation of Acinetobacter baylyi in non-sterile soil using recombinant plant nuclear DNA. Environ. Biosaf. Res. 2007, 6, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Pontiroli, A.; Brusetti, L.; Borin, S.; Sorlini, C.; Abruzzese, A.; Sacchi, G.A.; Vogel, T.M.; Simonet, P.; Bazzicalupo, M.; et al. Strategy for in situ detection of natural transformation-based horizontal gene transfer events. Appl. Environ. Microbiol. 2008, 74, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Pontiroli, A.; Rizzi, A.; Simonet, P.; Daffonchio, D.; Vogel, T.M.; Monier, J.M. Visual evidence of horizontal gene transfer between plants and bacteria in the phytosphere of transplastomic tobacco. Appl. Environ. Microbiol. 2009, 75, 3314–3322. [Google Scholar] [CrossRef] [PubMed]
- Ahrenholtz, I.; Harms, K.; de Vries, J.; Wackernagel, W. Increased killing of Bacillus subtilis on the hair roots of transgenic t4 lysozyme-producing potatoes. Appl. Environ. Microbiol. 2000, 66, 1862–1865. [Google Scholar] [CrossRef] [PubMed]
- Saxena, D.; Flores, S.; Stotzky, G. Bt toxin is released in root exudates from 12 transgenic corn hybrids representing three transformation events. Soil Biol. Biochem. 2002, 34, 133–137. [Google Scholar] [CrossRef]
- Tesfaye, M.; Dufault, N.S.; Dornbusch, M.R.; Allan, D.L.; Vance, C.P.; Samac, D.A. Influence of enhanced malate dehydrogenase expression by alfalfa on diversity of rhizobacteria and soil nutrient availability. Soil Biol. Biochem. 2003, 35, 1103–1113. [Google Scholar] [CrossRef]
- Poerschmann, J.; Gathmann, A.; Augustin, J.; Langer, U.; Gorecki, T. Molecular composition of leaves and stems of genetically modified BT and near-isogenic non-BT maize–Characterization of lignin patterns. J. Environ. Qual. 2005, 34, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Flores, S.; Saxena, D.; Stotzky, G. Transgenic BT plants decompose less in soil than non-BT plants. Soil Biol. Biochem. 2005, 37, 1073–1082. [Google Scholar] [CrossRef]
- Berge, O.; Heulin, T.; Achouak, W.; Richard, C.; Bally, R.; Balandreau, J. Rahnella aquatilis—A nitrogen-fixing enteric bacterium associated with the rhizosphere of wheat and maize. Can. J. Microbiol. 1991, 37, 195–203. [Google Scholar] [CrossRef]
- Heulin, T.; Berge, O.; Mavingui, P.; Gouzou, L.; Hebbar, K.P.; Balandreau, J. Bacillus polymyxa and Rahnella aquatilis, the dominant N2-fixing bacteria associated with wheat rhizosphere in French soils. Eur. J. Soil Biol. 1994, 30, 35–42. [Google Scholar]
- Kim, K.Y.; Jordan, D.; Krishnan, H.B. Rahnella aquatilis, a bacterium isolated from soybean rhizosphere, can solubilize hydroxyapatite. FEMS Microbiol. Lett. 1997, 153, 273–277. [Google Scholar] [CrossRef]
- Vyas, P.; Joshi, R.; Sharma, K.C.; Rahi, P.; Gulati, A.; Gulati, A. Cold-adapted and rhizosphere-competent strain of Rahnella sp. with broad-spectrum plant growth-promotion potential. 2010, 20, 1724–1734. [Google Scholar] [CrossRef]
- Li, G.E.; Wu, X.Q.; Ye, J.R.; Hou, L.; Zhou, A.D.; Zhao, L. Isolation and identification of phytate-degrading rhizobacteria with activity of improving growth of poplar and masson pine. World J. Microbiol. Biotechnol. 2013, 29, 2181–2193. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Guo, Y.B.; Wang, J.H.; Li, J.Y.; Wang, H.M. Biological control of grape crown gall by Rahnella aquatilis HX2. Plant Dis. 2007, 91, 957–963. [Google Scholar] [CrossRef]
- Palmieri, D.; Vitullo, D.; De Curtis, F.; Lima, G. A microbial consortium in the rhizosphere as a new biocontrol approach against Fusarium decline of chickpea. Plant Soil 2017, 412, 425–439. [Google Scholar] [CrossRef]
- He, H.D.; Ye, Z.H.; Yang, D.J.; Yan, J.L.; Xiao, L.; Zhong, T.; Yuan, M.; Cai, X.D.; Fang, Z.Q.; Jing, Y.X. Characterization of endophytic Rahnella sp JN6 from Polygonum pubescens and its potential in promoting growth and Cd, Pb, Zn uptake by Brassica napus. Chemosphere 2013, 90, 1960–1965. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioinformatics Tool | SOAPaligner | One Codex Data Platform | ||||
|---|---|---|---|---|---|---|
| Database | NCBI nucleotide database | One Codex DB | RefSeq DB | |||
| Sample name | MGMRh | MGNRh | MGMRh | MGNRh | MGMRh | MGNRh |
| Total mapped reads | 790,310 | 318,187 | 7,413,129 | 6,118,123 | 4,878,689 | 4,058,863 |
| Genus name | Mapped reads (proportions of total mapped reads) | |||||
| Rahnella 1 | 316,533 (40.052%) | 385 (0.1210%) | 363,110 (4.898%) | 510 (0.00834%) | 216,768 (4.443%) | 364 (0.00897%) |
| Serratia | 28,900 (3.657%) | 38 (0.0120%) | 36,565 (0.4932%) | 1416 (0.02314%) | 20,940 (0.4293%) | 1959 (0.04826%) |
| Yersinia 2 | 381 (0.0482%) | 6 (0.0019%) | 337 (0.0045%) | 37 (0.00060%) | 1144 (0.0234%) | 111 (0.00273%) |
| Variovorax 1 | 27,824 (3.521%) | 21,698 (6.819%) | 158,661 (2.1403%) | 150,119 (2.4537%) | 123,709 (2.536%) | 109,798 (2.7051%) |
| Ewingella 1 | ND | ND | 1077 (0.01453%) | 67 (0.00110%) | ND | ND |
| Ramlibacter 1 | 1846 (02336%) | 2518 (0.7913%) | 12,642 (0.1705%) | 17,414 (0.28463%) | 17,680 (0.3624%) | 23,159 (0.57058%) |
| Luteibacter 2 | 24 (0.0030%) | 2 (0.0006%) | 129,081 (1.7413%) | 9029 (0.14758%) | ND | ND |
| Pedobacter 2 | 638 (0.0807%) | 166 (0.0522%) | 29,399 (0.3966%) | 6812 (0.11134%) | 5210 (0.1068%) | 2026 (0.04991%) |
| Flavisolibacter 2 | 20 (0.0025%) | 19 (0.0060%) | ND | ND | ND | ND |
| Bioinformatics Tool | SOAPaligner | One Codex Data Platform | ||||
|---|---|---|---|---|---|---|
| Database | NCBI nucleotide database | One Codex DB | RefSeq DB | |||
| Sample name | MGMRh | MGNRh | MGMRh | MGNRh | MGMRh | MGNRh |
| Total mapped reads | 790,310 | 318,187 | 7,413,129 | 6,118,123 | 4,878,689 | 4,058,863 |
| Species name 1,2 | ||||||
| Rahnella aquatilis | 8260 | 9 | 26,556 | 46 | 216,768 | 364 |
| Rahnella sp. WP5 | ND | ND | 89,230 | 86 | ND | ND |
| Rahnella sp. Y9602 | 37,445 | 35 | 12,208 | 25 | ND | ND |
| SUM | 45,705 | 44 | 127,994 | 157 | 216,768 | 364 |
| Serratia liquefaciens | 24,395 | 25 | 32,253 | 91 | ND | ND |
| Serratia proteamaculans | 267 | 0 | ND | ND | 7693 | 184 |
| Serratia plymuthica | 258 | 1 | 304 | 37 | 6042 | 289 |
| Serratia marcescens | 41 | 4 | 452 | 149 | 4184 | 1223 |
| Serratia rubidaea | ND | ND | 142 | 134 | ND | ND |
| Serratia sp. S4 | ND | ND | 229 | 0 | ND | ND |
| Serratia quinivorans | ND | ND | 227 | 0 | ND | ND |
| Serratia fonticola | 8 | 0 | 114 | 33 | 1047 | 134 |
| Serratia entomophila | 88 | 0 | ND | ND | ND | ND |
| Serratia odorifera | 65 | 0 | 83 | 54 | ND | ND |
| Serratia sp. 506_PEND | ND | ND | 102 | 0 | ND | ND |
| Serratia sp. SCBI | 52 | 2 | 15 | 0 | ND | ND |
| Serratia grimesii | 1 | 0 | 72 | 0 | ND | ND |
| SUM | 25,175 | 32 | 33,993 | 498 | 18,966 | 1830 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, G.-H.; Hua, X.-M.; Liang, L.; Wen, Z.-L.; Du, M.-H.; Meng, F.-F.; Pang, Y.-J.; Qi, J.-L.; Tang, C.-Y.; Yang, Y.-H. Identification of Major Rhizobacterial Taxa Affected by a Glyphosate-Tolerant Soybean Line via Shotgun Metagenomic Approach. Genes 2018, 9, 214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040214
Lu G-H, Hua X-M, Liang L, Wen Z-L, Du M-H, Meng F-F, Pang Y-J, Qi J-L, Tang C-Y, Yang Y-H. Identification of Major Rhizobacterial Taxa Affected by a Glyphosate-Tolerant Soybean Line via Shotgun Metagenomic Approach. Genes. 2018; 9(4):214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040214
Chicago/Turabian StyleLu, Gui-Hua, Xiao-Mei Hua, Li Liang, Zhong-Ling Wen, Mei-Hang Du, Fan-Fan Meng, Yan-Jun Pang, Jin-Liang Qi, Cheng-Yi Tang, and Yong-Hua Yang. 2018. "Identification of Major Rhizobacterial Taxa Affected by a Glyphosate-Tolerant Soybean Line via Shotgun Metagenomic Approach" Genes 9, no. 4: 214. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040214