Full Mitogenomes in the Critically Endangered Kākāpō Reveal Major Post-Glacial and Anthropogenic Effects on Neutral Genetic Diversity


Abstract
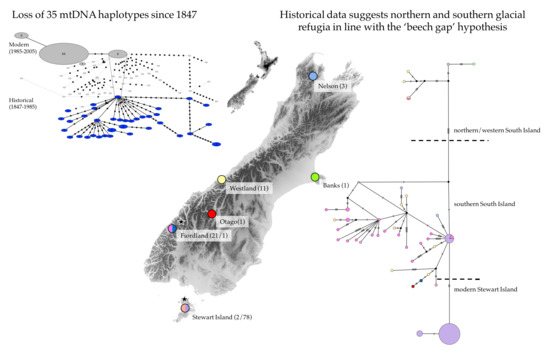
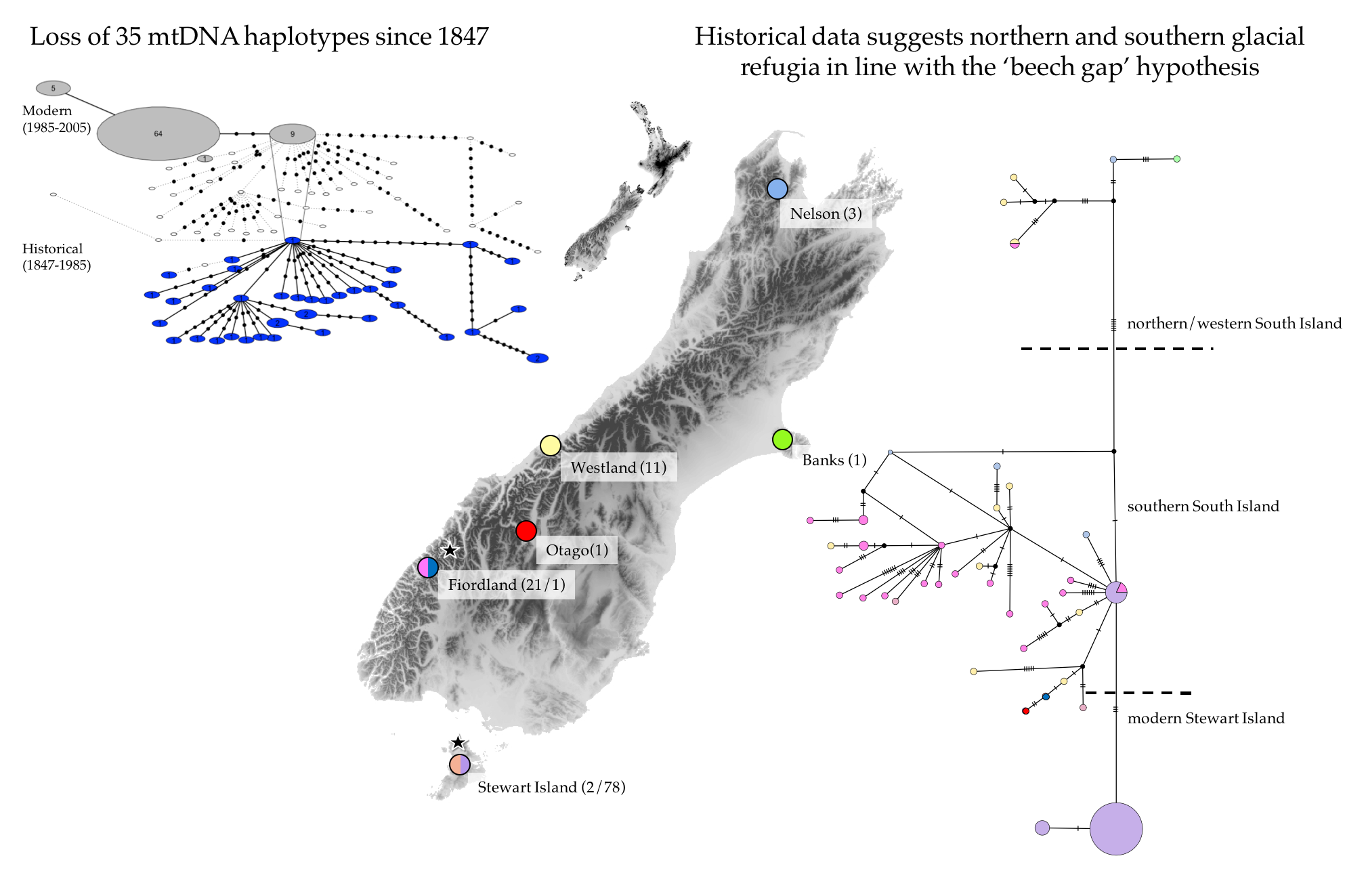
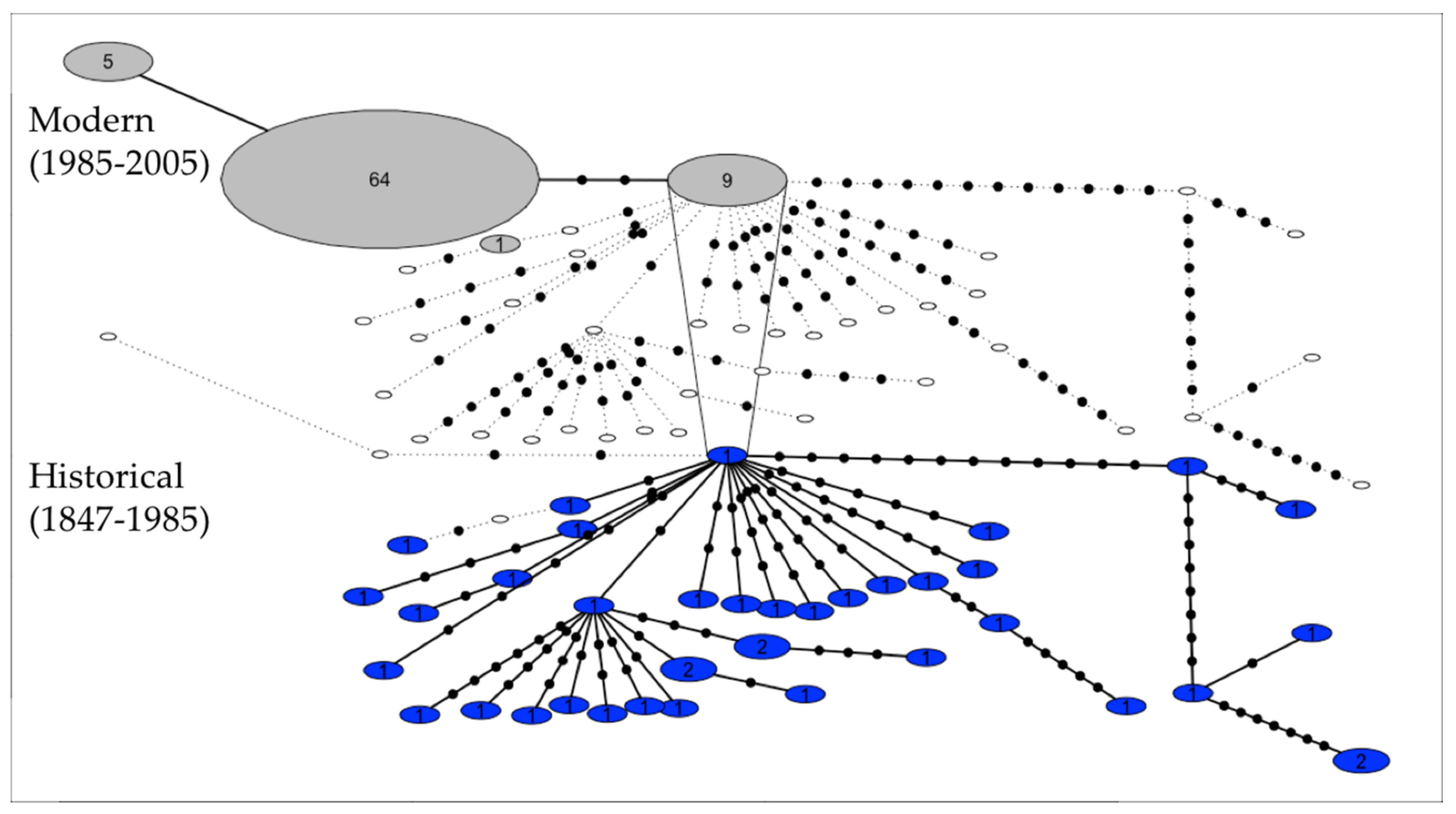
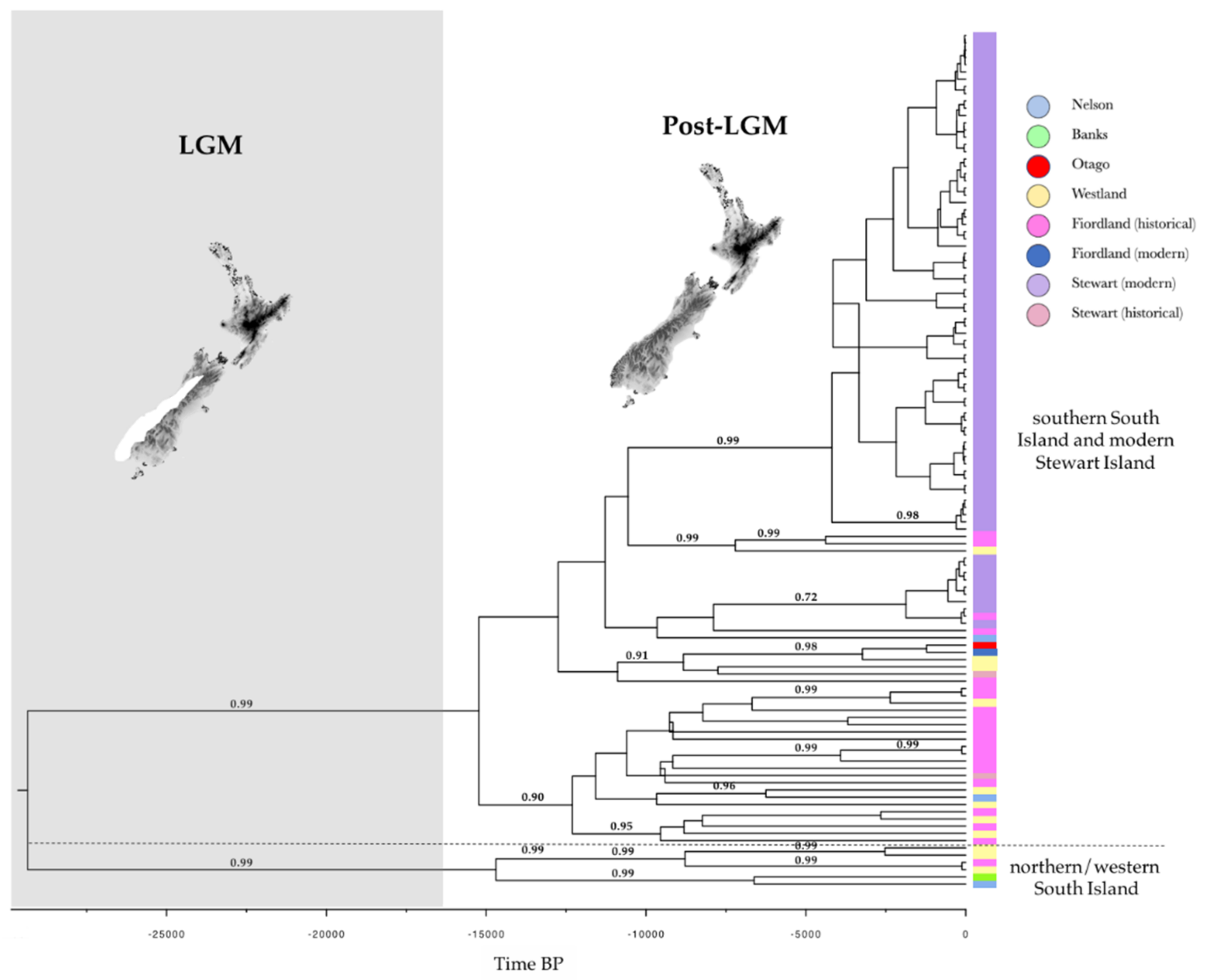
:
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extractions and DNA Library Preparation
2.3. Data Processing
2.4. Changes in Genetic Diversity
2.5. Phylogenetic and Demographic Analyses
2.6. Data Availability
3. Results
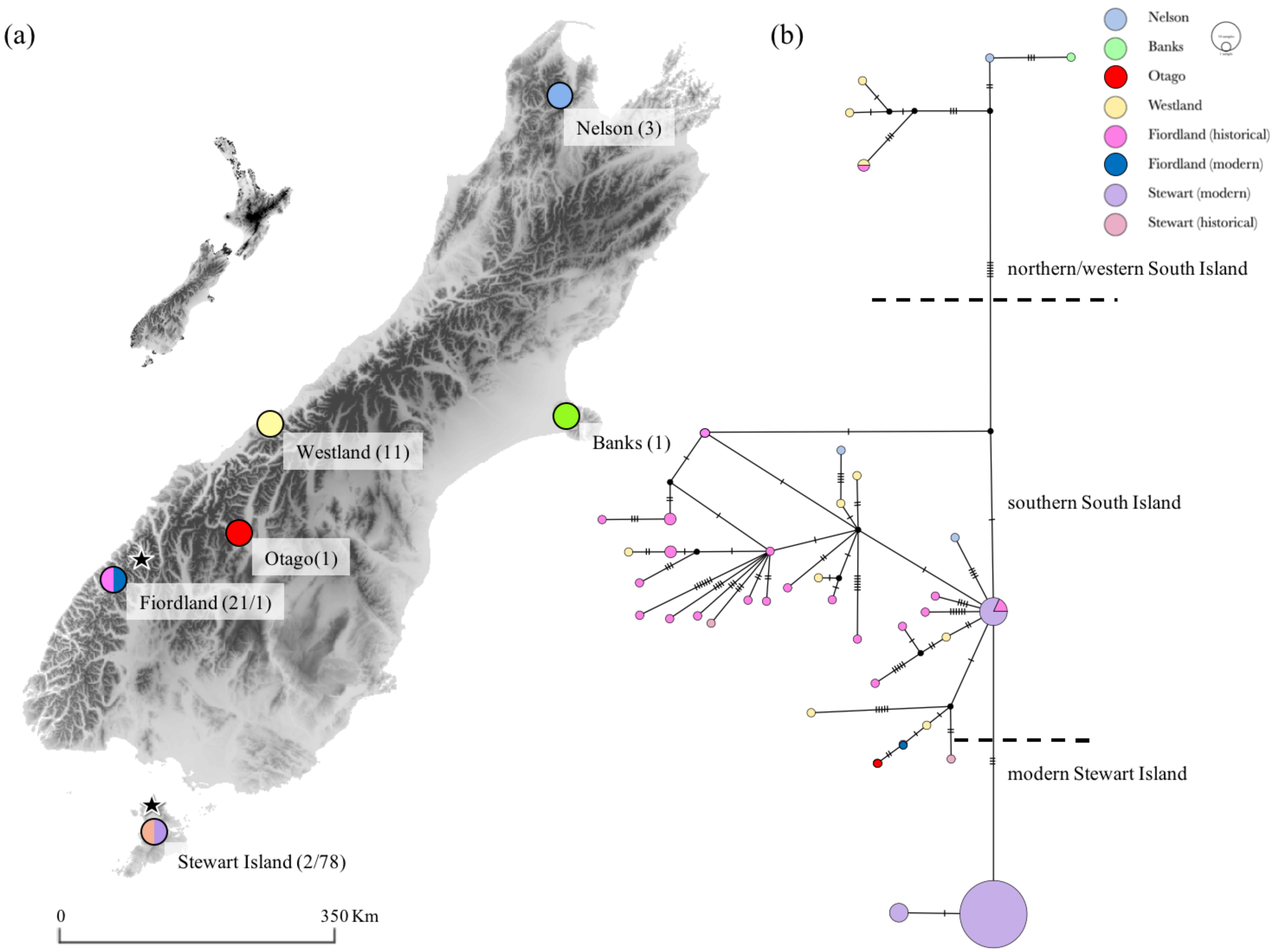
3.1. Genetic Diversity and Phylogeny
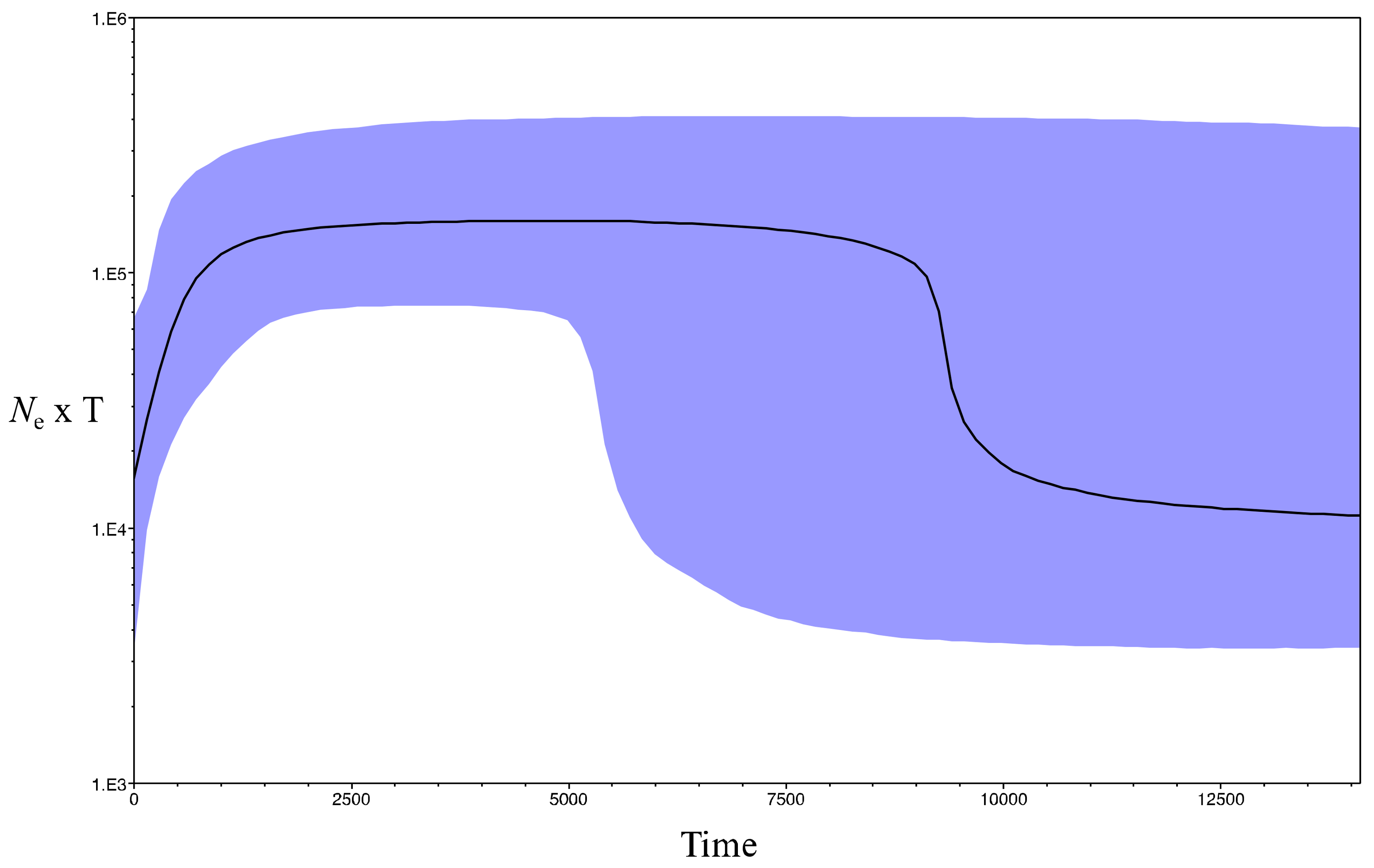
3.2. Recent and Past Demography
4. Discussion
4.1. Genetic Diversity and Phylogeny
4.2. Recent and Past Demography
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jamieson, I.G.; Wallis, G.P.; Briskie, J.V. Inbreeding and endangered species management: Is New Zealand out of step with the rest of the world? Conserv. Biol. 2006, 20, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, I.G. Has the debate over genetics and extinction of island endemics truly been resolved? Anim. Conserv. 2007, 10, 139–144. [Google Scholar] [CrossRef]
- Palkopoulou, E.; Mallick, S.; Skoglund, P.; Enk, J.; Rohland, N.; Li, H.; Omrak, A.; Vartanyan, S.; Poinar, H.; Götherström, A.; et al. Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr. Biol. 2015, 25, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, I.G.; Allendorf, F.W. How does the 50/500 rule apply to MVPs? Trends Ecol. Evol. 2012, 27, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.L.; Slatkin, M. Excess of genomic defects in a woolly mammoth on Wrangel island. PLOS Genet. 2017, 13, e1006601. [Google Scholar] [CrossRef] [PubMed]
- Pečnerová, P.; Palkopoulou, E.; Wheat, C.W.; Skoglund, P.; Vartanyan, S.; Tikhonov, A.; Nikolskiy, P.; van der Plicht, J.; Díez-del-Molino, D.; Dalén, L. Mitogenome evolution in the last surviving woolly mammoth population reveals neutral and functional consequences of small population size. Evol. Lett. 2017, 1, 292–303. [Google Scholar] [CrossRef]
- Dirzo, R.; Young, H.S.; Galetti, M.; Ceballos, G.; Isaac, N.J.B.; Collen, B. Defaunation in the Anthropocene. Science 2014, 345, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Powlesland, R.G.; Merton, D.V.; Cockrem, J.F. A parrot apart: The natural history of the kakapo (Strigops habroptilus), and the context of its conservation management. Notornis 2006, 53, 3–26. [Google Scholar]
- Worthy, T.H.; Holdaway, R.H. The Lost World of the Moa: Prehistoric Life of New Zealand; Indiana University Press: Bloomington, IN, USA, 2002. [Google Scholar]
- Digby, A.; Department of Conservation, Invercargill, New Zealand. Personal communication, 2018.
- Millener, P.R. The Quaternary Avifauna of the North Island, New Zealand. Ph.D. Thesis, University of Auckland, Auckland, New Zealand, 1981. [Google Scholar]
- Worthy, T.H.; Holdaway, R.N. Quaternary fossil faunas from caves in Takaka Valley and on Takaka Hill, northwest Nelson, South Island, New Zealand. J. R. Soc. N. Z. 1994, 24, 297–391. [Google Scholar] [CrossRef]
- Worthy, T.H.; Holdaway, R.N. Quaternary fossil faunas from caves in the Punakaiki area, West Coast, South Island, New Zealand. J. R. Soc. N. Z. 1993, 23, 147–254. [Google Scholar] [CrossRef]
- Wood, J.R. Subfossil kakapo (Strigops habroptilus) remains from near Gibraltar Rock, Cromwell Gorge, Central Otago, New Zealand. Notornis 2006, 53, 191–193. [Google Scholar]
- Worthy, T.H.; Holdaway, R.N. Quaternary fossil faunas, overlapping taphonomies, and palaeofaunal reconstruction in North Canterbury, South Island, New Zealand. J. R. Soc. N. Z. 1996, 26, 275–361. [Google Scholar] [CrossRef]
- Worthy, T.H.; Holdaway, R.N. Quaternary fossil faunas from caves on Mt-Cookson, North-Canterbury, South-Island, New-Zealand. J. R. Soc. N. Z. 1995, 25, 333–370. [Google Scholar] [CrossRef]
- Holdaway, R.N. New Zealand’s pre-human avifauna and its vulnerability. N. Z. J. Ecol. 1989, 12, 11–25. [Google Scholar]
- Williams, G.R. The kakapo (Strigops habroptilus, Gray): A review and reappraisal of a near extinct species. Notornis 1956, 7, 29–56. [Google Scholar]
- Wilmshurst, J.M.; Anderson, A.J.; Higham, T.F.G.; Worthy, T.H. Dating the late prehistoric dispersal of Polynesians to New Zealand using the commensal Pacific rat. Proc. Natl. Acad. Sci. USA 2008, 105, 7676–7680. [Google Scholar] [CrossRef] [PubMed]
- Holdaway, R.N.; Worthy, T.H.; Tennyson, A.J.D. A working list of breeding bird species of the New Zealand region at first human contact. N. Z. J. Zool. 2001, 28, 119–187. [Google Scholar] [CrossRef]
- Bergner, L.M.; Dussex, N.; Jamieson, I.G.; Robertson, B.C. European colonization, not polynesian arrival, impacted population size and genetic diversity in the critically endangered New Zealand Kākāpō. J. Hered. 2016, 107, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, B.D.; Powlesland, R.G. The decline of kakapo Strigops habroptilus and attempts at conservation by translocation. Biol. Conserv. 1994, 69, 75–85. [Google Scholar] [CrossRef]
- Powlesland, R.G.; Roberts, A.; Lloyd, B.D.; Merton, D.V. Number, fate, and distribution of kakapo (Strigops habroptilus) found on Stewart Island, New Zealand, 1979–92. N. Z. J. Zool. 1995, 22, 239–248. [Google Scholar] [CrossRef]
- Elliott, G.P.; Eason, D.K.; Jansen, P.W.; Merton, D.V.; Harper, G.A.; Moorhouse, R.J. Productivity of kakapo (Strigops habroptilus) on offshore island refuges. Notornis 2006, 53, 138–142. [Google Scholar]
- White, K.L.; Eason, D.K.; Jamieson, I.G.; Robertson, B.C. Evidence of inbreeding depression in the critically endangered parrot, the kakapo. Anim. Conserv. 2015, 18, 341–347. [Google Scholar] [CrossRef]
- Knapp, M.; Clarke, A.C.; Horsburgh, K.A.; Matisoo-Smith, E.A. Setting the stage—Building and working in an ancient DNA laboratory. Ann. Anat. 2012, 194, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- John, J.S. SeqPrep 1.1. Available online: https://github.com/jstjohn/SeqPrep (accessed on 17 April 2018).
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Prost, S.; Anderson, C.N.K. TempNet: A method to display statistical parsimony networks for heterochronous DNA sequence data. Methods Ecol. Evol. 2011, 2, 663–667. [Google Scholar] [CrossRef]
- Leigh, J.; Bryant, D. PopART: Full-feature software for population genetics. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Nabholz, B.; Lanfear, R.; Fuchs, J. Body mass-corrected molecular rate for bird mitochondrial DNA. Mol. Ecol. 2016, 25, 4438–4449. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Li, W.L.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 2013, 30, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Kass, R.R.E.; Raftery, A.E.A. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Rambaut, A.; Suchard, M.; Xie, D.; Drummond, A. Tracer v1.6. Available online: http://beast.community/tracer (accessed on 17 April 2018).
- Beaumont, M.A.; Zhang, W.Y.; Balding, D.J. Approximate Bayesian computation in population genetics. Genetics 2002, 162, 2025–2035. [Google Scholar] [PubMed]
- Cornuet, J.M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.P.; Department of Conservation, Nelson, New Zealand. Personal communication, 2016.
- Fagundes, N.J.; Ray, N.; Beaumont, M.; Neuenschwander, S.; Salzano, F.M.; Bonatto, S.L.; Excoffier, L. Statistical evaluation of alternative models of human evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 17614–17619. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.-M.; Ravigné, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, G.P.; Trewick, S.A. New Zealand phylogeography: Evolution on a small continent. Mol. Ecol. 2009, 18, 3548–3580. [Google Scholar] [CrossRef] [PubMed]
- Suggate, R.P. Late Pliocene and Quaternary Glaciations of New-Zealand. Quat. Sci. Rev. 1990, 9, 175–197. [Google Scholar] [CrossRef]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.X.; Hostetler, S.W.; McCabe, A.M. The Last Glacial Maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Alloway, B.V.; Almond, P.C.; Augustinus, P.C.; Barrell, D.J.A.; Bertler, N.A.N.; Carter, L.; Litchfield, N.J.; Lowe, D.J.; Mcglone, M.S.; Newnham, R.M.; et al. Towards a Climate Event Stratigraphy for New Zealand over the past 30,000 years. J. Quat. Sci. 2007, 22, 9–35. [Google Scholar] [CrossRef]
- Golledge, N.R.; Mackintosh, A.N.; Anderson, B.M.; Buckley, K.M.; Doughty, A.M.; Barrell, D.J.A.; Denton, G.H.; Vandergoes, M.J.; Andersen, B.G.; Schaefer, J.M. Last Glacial Maximum climate in New Zealand inferred from a modelled Southern Alps icefield. Quat. Sci. Rev. 2012, 46, 30–45. [Google Scholar] [CrossRef]
- Trewick, S.A.; Wallis, G.P. Bridging the “beech-gap”: New Zealand invertebrate phylogeography implicates Pleistocene glaciation and Pliocene isolation. Evolution 2001, 55, 2170–2180. [Google Scholar] [PubMed]
- Boessenkool, S.; Star, B.; Scofield, R.P.; Seddon, P.J.; Waters, J.M. Lost in translation or deliberate falsification? Genetic analyses reveal erroneous museum data for historic penguin specimens. Proc. Biol. Sci. 2010, 277, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Wallis, G.P.; Waters, J.M.; Upton, P.; Craw, D. Transverse alpine speciation driven by glaciation. Trends Ecol. Evol. 2016, 31, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Weston, K.A.; Robertson, B.C. Population structure within an alpine archipelago: Strong signature of past climate change in the New Zealand rock wren (Xenicus gilviventris). Mol. Ecol. 2015, 24, 4778–4794. [Google Scholar] [CrossRef] [PubMed]
- Weir, J.T.; Haddrath, O.; Robertson, H.A.; Colbourne, R.M.; Baker, A.J. Explosive ice age diversification of kiwi. Proc. Natl. Acad. Sci. USA 2016, 113, E5580–E5587. [Google Scholar] [CrossRef] [PubMed]
- McGlone, M.S. Plant biogeography and the late Cenozoic history of New Zealand. N. Z. J. Bot. 1985, 23, 723–749. [Google Scholar] [CrossRef]
- McGlone, M.S.; Newnham, R.M.; Moar, N.T. The vegetation cover of New Zealand during the Last Glacial Maximum: Do pollen records under-represent woody vegetation? Quat. Sci. Rev. 2013, 74, 202–214. [Google Scholar]
- Dussex, N.; Wegmann, D.; Robertson, B.C. Postglacial expansion and not human influence best explains the population structure in the endangered kea (Nestor notabilis). Mol. Ecol. 2014, 23, 2193–2209. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, L.D.; Worthy, T.H.; Tennyson, A.J.D.; Scofield, R.P.; Ramstad, K.M.; Lambert, D.M. Ancient DNA analyses reveal contrasting phylogeographic patterns amongst kiwi (Apteryx spp.) and a recently extinct lineage of spotted kiwi. PLoS ONE 2012, 7, e42384. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A. Pre-European hunting dogs in the South Island, New Zealand. N. Z. J. Archaeol. 1981, 3, 15–20. [Google Scholar]
- Tipa, R. Kakapo in Maori lore. Notornis 2006, 53, 191–193. [Google Scholar]
- Holdaway, R.N.; Jacomb, C. Rapid extinction of the moas (Aves: Dinornithiformes): Model, test, and implications. Science 2000, 287, 2250–2254. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.P.; Blackburn, T.M. Extinction and endemism in the New Zealand avifauna. Glob. Ecol. Biogeogr. 2004, 13, 509–517. [Google Scholar] [CrossRef]
- Duncan, R.P.; Blackburn, T.M.; Worthy, T.H. Prehistoric bird extinctions and human hunting. Proc. R. Soc. B Biol. Sci. 2002, 269, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Merton, D.V.; Morris, R.B.; Atkinson, I.A.E. Lek behaviour in a parrot: The kakapo Strigops habroptilus of New Zealand. IBIS 1984, 126, 277–283. [Google Scholar] [CrossRef]
- Dussex, N.; Robertson, B.C. Contemporary effective population size and predicted maintenance of genetic diversity in the endangered kea (Nestor notabilis). N. Z. J. Zool. 2017, 45, 13–28. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Period | n | h | S | Hd | π | D | FS |
|---|---|---|---|---|---|---|---|
| Modern | 79 | 4 | 7 | 0.0331 (0.024) | 0.00005 (0.00002) | −0.944 | 0.859 |
| Historical | 39 | 36 | 100 | 0.996 (0.0064) | 0.00059 (0.00006) | −2.16 ** | −24.48 ** |
| (a) | Parameters | Prior | Posterior Mode | 5% HPD | 95% HPD |
| Ne-modern | Uniform (1–200) | 17.6 | 7.65 | 96.5 | |
| Ne-pre-European | Uniform (5 × 103–6 × 105) | 5.67 × 105 | 1.80 × 105 | 5.86 × 105 | |
| Ne-pre-glaciation | Uniform (103–3 × 105) | 3.90 × 103 | 2.61 × 103 | 5.75 × 104 | |
| t-bottleneck-Eu | Uniform (1–10) | 4.47 | 2.44 | 5.56 | |
| t-post-glaciation | Uniform (300–600) | 600 | 354 | 600 | |
| μ rate | Uniform (10−8–10−7) | 1 × 10−7 | 6.45 × 10−8 | 1 × 10−7 | |
| (b) | |||||
| Ne-modern | Uniform (1–200) | 16.8 | 6.38 | 88.6 | |
| Ne-pre-human | Uniform (5 × 103–6 × 105) | 5.76 × 105 | 2.28 × 105 | 5.90 × 105 | |
| Ne-pre-European | Uniform (5 × 103–6 × 105) | 2.66 × 105 | 4.62 × 104 | 5.17 × 105 | |
| Ne-pre-glaciation | Uniform (103–3 × 105) | 5.91 × 103 | 2.87 × 103 | 6.22 × 104 | |
| t-bottleneck-Pol | Uniform (20–30) | 20.1 | 20 | 30 | |
| t-bottleneck-Eu | Uniform (1–10) | 4.24 | 2.32 | 5.42 | |
| t-post-glaciation | Uniform (300–600) | 600 | 345 | 594 | |
| μ rate | Uniform (10−8–10−7) | 1 × 10−7 | 6.25 × 10−8 | 1 × 10−7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dussex, N.; Von Seth, J.; Robertson, B.C.; Dalén, L. Full Mitogenomes in the Critically Endangered Kākāpō Reveal Major Post-Glacial and Anthropogenic Effects on Neutral Genetic Diversity. Genes 2018, 9, 220. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040220
Dussex N, Von Seth J, Robertson BC, Dalén L. Full Mitogenomes in the Critically Endangered Kākāpō Reveal Major Post-Glacial and Anthropogenic Effects on Neutral Genetic Diversity. Genes. 2018; 9(4):220. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040220
Chicago/Turabian StyleDussex, Nicolas, Johanna Von Seth, Bruce C. Robertson, and Love Dalén. 2018. "Full Mitogenomes in the Critically Endangered Kākāpō Reveal Major Post-Glacial and Anthropogenic Effects on Neutral Genetic Diversity" Genes 9, no. 4: 220. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9040220