Genome-Wide Comparative Analysis of Aspergillus fumigatus Strains: The Reference Genome as a Matter of Concern

Abstract
:1. Introduction
2. Materials and Methods
2.1. Aspergillus fumigatus Isolates
2.2. DNA Extraction and Aspergillus fumigatus Identification
2.3. DNA Quality and Quantity Assessment
2.4. Screening of cyp51A Changes: PCR Amplification and Sequence Analysis
2.5. DNA Library Preparation and Illumina Whole-Genome Sequencing
2.6. Whole-Genome Sequencing Alignment
2.7. Phylogenetic Analysis and Single Nucleotide Variant Comparison
2.8. Analysis of Genetic Diversity
2.9. Determining Modifications in Genes of Interest
2.10. Visualization of Depth of Coverage
2.11. Mating Type Analysis
3. Results
3.1. Species Identification
3.2. Whole-Genome Sequencing Analysis
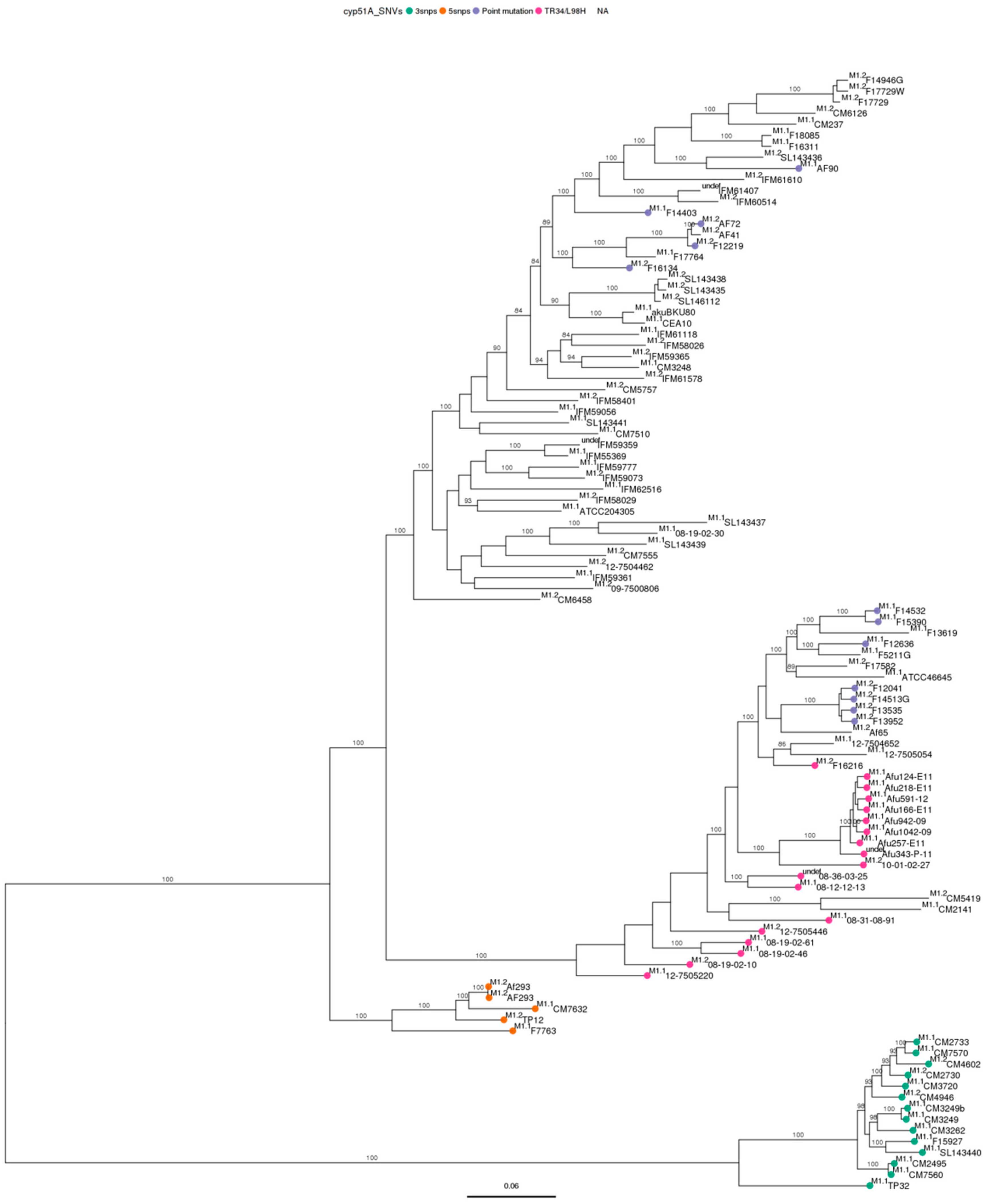
3.3. Phylogenetic Analysis
3.4. Single Nucleotide Variant Analysis against Both References
3.5. Genetic Diversity
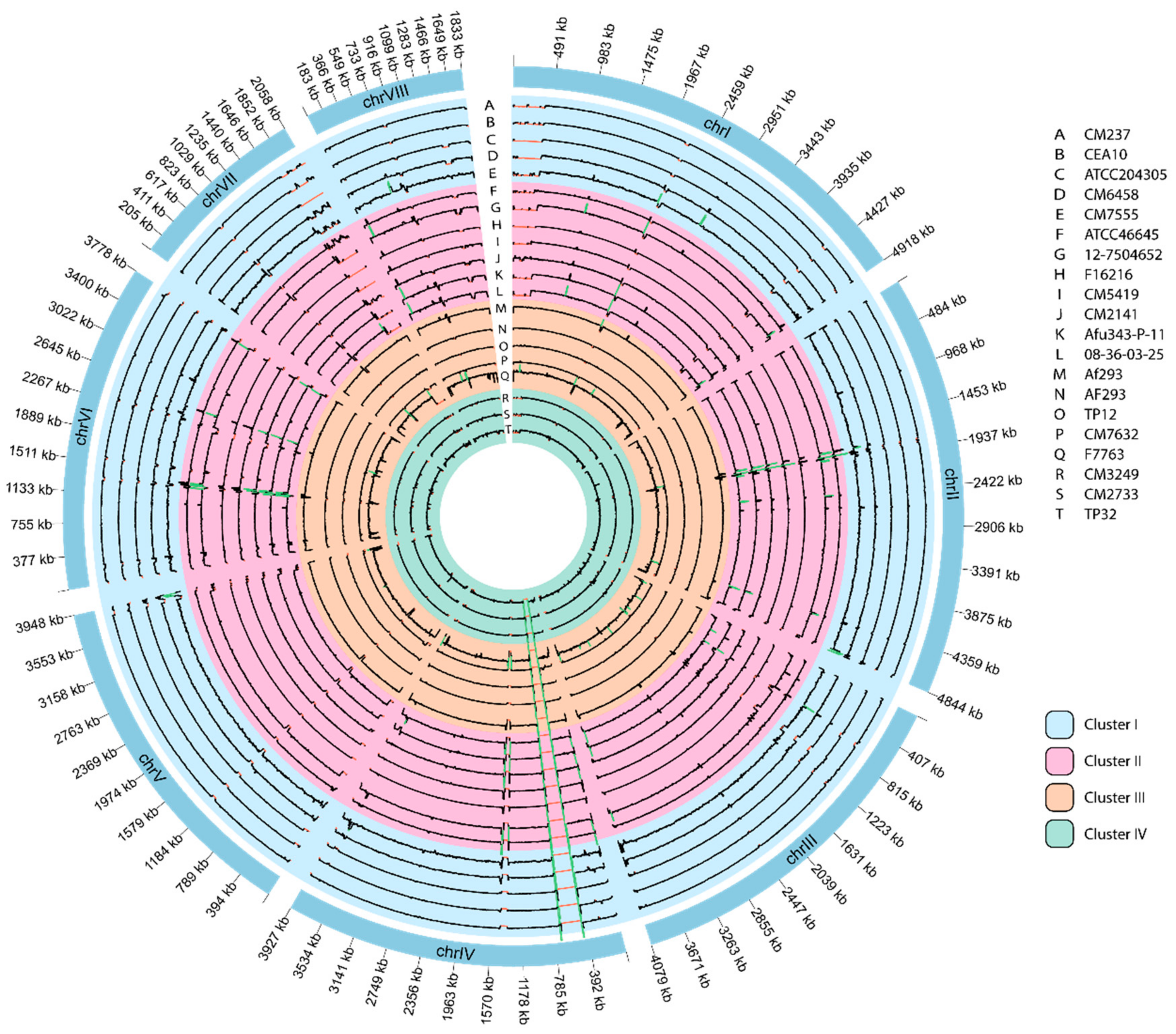
3.6. Genome-Wide Visualization of Depth of Coverage
3.7. Genome Comparisons Based on Their Mating Type
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kontoyiannis, D.P.; Marr, K.A.; Park, B.J.; Alexander, B.D.; Anaissie, E.J.; Walsh, T.J.; Ito, J.; Andes, D.R.; Baddley, J.W.; Brown, J.M.; et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: Overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin. Infect. Dis. 2010, 50, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G.; Alexander, B.D.; Andes, D.R.; Hadley, S.; Kauffman, C.A.; Freifeld, A.; Anaissie, E.J.; Brumble, L.M.; Herwaldt, L.; Ito, J.; et al. Invasive fungal infections among organ transplant recipients: Results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin. Infect. Dis. 2010, 50, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W. Invasive aspergillosis. Clin. Infect. Dis. 1998, 26, 781–805. [Google Scholar] [CrossRef] [PubMed]
- Latgé, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [PubMed]
- Kosmidis, C.; Denning, D.W. The clinical spectrum of pulmonary aspergillosis. Thorax 2015, 70, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Verweij, P.E.; Chowdhary, A.; Melchers, W.J.G.; Meis, J.F. Azole Resistance in Aspergillus fumigatus: Can We Retain the Clinical Use of Mold-Active Antifungal Azoles? Clin. Infect. Dis. 2016, 62, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Köser, C.U.; Ellington, M.J.; Peacock, S.J. Whole-genome sequencing to control antimicrobial resistance. Trends Genet. 2014, 30, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S. Bring microbial sequencing to hospitals. Nature 2014, 509, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; McCallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Snelders, E.; Verweij, P.E.; Melchers, W.J.G. Next-Generation Sequencing in the Mycology Lab. Curr. Fungal Infect. Rep. 2016, 10, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, R. Towards the genotyping of fungi: Methods, benefits and challenges. Curr. Fungal Infect. Rep. 2014, 8, 203–210. [Google Scholar] [CrossRef]
- Hagiwara, D.; Takahashi, H.; Watanabe, A.; Takahashi-Nakaguchi, A.; Kawamoto, S.; Kamei, K.; Gonoi, T. Whole-genome comparison of Aspergillus fumigatus strains serially isolated from patients with aspergillosis. J. Clin. Microbiol. 2014, 52, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Abdolrasouli, A.; Rhodes, J.; Beale, M.A.; Hagen, F.; Rogers, T.R.; Chowdhary, A.; Meis, J.F.; Armstrong-James, D.; Fisher, M.C. Genomic context of azole resistance mutations in Aspergillus fumigatus determined using whole-genome sequencing. MBio 2015, 6, e00536-15. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Chen, P.; Gao, R.; Li, Y.; Zhang, A.; Liu, F.; Lu, L. Screening and characterization of a non-cyp51A mutation in an Aspergillus fumigatus cox10 strain conferring azole resistance. Antimicrob. Agents Chemother. 2017, 61, e02101-16. [Google Scholar] [CrossRef] [PubMed]
- Keller, N. Heterogeneity Confounds Establishment of “a” Model Microbial Strain. mBio 2017, 8, e00135-17. [Google Scholar] [CrossRef] [PubMed]
- Nierman, W.C.; Pain, A.; Anderson, M.J.; Wortman, J.R.; Kim, H.S.; Arroyo, J.; Berriman, M.; Abe, K.; Archer, D.B.; Bermejo, C.; et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 2005, 438, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, N.D.; Khaldi, N.; Joardar, V.S.; Maiti, R.; Amedeo, P.; Anderson, M.J.; Crabtree, J.; Silva, J.C.; Badger, J.H.; Albarraq, A.; et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008, 4, e1000046. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Payne, G.; Fedorova, N.D.; Baker, S.E.; Machida, M.; Yu, J.; Georgianna, D.R.; Dean, R.A.; Bhatnagar, D.; Cleveland, T.E.; et al. What can comparative genomics tell us about species concepts in the genus Aspergillus? Stud. Mycol. 2007, 59, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rubio, R.; Alcazar-Fuoli, L.; Monteiro, M.C.; Monzon, S.; Cuesta, I.; Pelaez, T.; Mellado, E. Insight into the significance of Aspergillus fumigatus cyp51A polymorphisms. Antimicrob. Agents Chemother. 2018, 62, e00241-18. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Ferreira, M.E.; Kress, M.R.V.Z.; Savoldi, M.; Goldman, M.H.S.; Härtl, A.; Heinekamp, T.; Brakhage, A.A.; Goldman, G.H. The akuBKU80 mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 2006, 5, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Cerar, D.; Anderson, M.J.; Albarrag, A.; Fisher, M.C.; Pasqualotto, A.C.; Laverdiere, M.; Arendrup, M.C.; Perlin, D.S.; Denning, D.W. Frequency and evolution of Azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg. Infect. Dis. 2009, 15, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Nakaguchi, A.; Muraosa, Y.; Hagiwara, D.; Sakai, K.; Toyotome, T.; Watanabe, A.; Kawamoto, S.; Kamei, K.; Gonoi, T.; Takahashi, H. Genome sequence comparison of Aspergillus fumigatus strains isolated from patients with pulmonary aspergilloma and chronic necrotizing pulmonary aspergillosis. Med. Mycol. 2015, 53, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.M.; Cohen, J.; Holden, D.W. An Aspergillus fumigatus alkaline protease mutant constructed by gene disruption is deficient in extracellular elastase activity. Mol. Microbiol. 1992, 6, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fuoli, L.; Mellado, E.; Alastruey-Izquierdo, A.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Aspergillus section Fumigati: Antifungal susceptibility patterns and sequence-based identification. Antimicrob. Agents Chemother. 2008, 52, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Mellado, E.; Garcia-Effron, G.; Alcazar-Fuoli, L.; Melchers, W.J.; Verweij, P.E.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob. Agents Chemother. 2007, 51, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis toolkit: A MapReduce framework for analyzing nextgeneration DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; ISBN 9780195135855. [Google Scholar]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashu, E.E.; Hagen, F.; Chowdhary, A.; Meis, J.F.; Xu, J. Global population genetic analysis of Aspergillus fumigatus. mSphere 2017, 2, e00019-17. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, C.H.; Beattie, S.R.; Fuller, K.K.; McGurk, E.A.; Tang, Y.-W.; Hohl, T.M.; Obar, J.J.; Cramer, R.A., Jr. Heterogeneity among isolates reveals that fitness in low oxygen correlates with Aspergillus fumigatus virulence. mBio 2016, 7, e01515-16. [Google Scholar] [CrossRef] [PubMed]
- Popovich, K.J.; Snitkin, E.S. Whole Genome Sequencing—Implications for Infection Prevention and Outbreak Investigations. Curr. Infect. Dis. Rep. 2017, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Wortman, J.R.; Fedorova, N.; Crabtree, J.; Joardar, V.; Maiti, R.; Haas, B.J.; Amedeo, P.; Lee, E.; Angiuoli, S.V.; Jiang, B.; et al. Whole genome comparison of the A. fumigatus family. Med. Mycol. 2006, 44 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef]
- Fuller, K.K.; Cramer, R.A.; Zegans, M.E.; Dunlap, J.C.; Loros, J.J. Aspergillus fumigatus photobiology illuminates the marked heterogeneity between isolates. mBio 2016, 7, e01517-16. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, L.; Giovannini, G.; Bromley, M.; Bowyer, P.; Romani, L.; Cavalieri, D. Strain dependent variation of immune responses to A. fumigatus: Definition of pathogenic species. PLoS ONE 2013, 8, e56651. [Google Scholar] [CrossRef] [PubMed]
- Losada, L.; Sugui, J.A.; Eckhaus, M.A.; Chang, Y.C.; Mounaud, S.; Figat, A.; Joardar, V.; Pakala, S.B.; Pakala, S.; Venepally, P. Genetic Analysis Using an Isogenic Mating Pair of Aspergillus fumigatus Identifies Azole Resistance Genes and Lack of MAT Locus’s Role in Virulence. PLoS Pathog. 2015, 11, e1004834. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.S.; O’Gorman, C.M. Sexual development and cryptic sexuality in fungi: Insights from Aspergillus species. FEMS Microbiol. Rev. 2012, 36, 165–192. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.S.; Inderbitzin, P.; Debuchy, R. Mating-type structure, function, regulation and evolution in the Pezizomycotina. In Growth, Differentiation and Sexuality. The Mycota (A Comprehensive Treatise on Fungi as Experimental Systems for Basic and Applied Research); Wendland, J., Ed.; Springer International Publishing: Basel, Switzerland, 2016; Volume I, pp. 351–385. ISBN 978-3-319-25844-7. [Google Scholar]
- Yu, Y.; Amich, J.; Will, C.; Eagle, C.E.; Dyer, P.S.; Krappmann, S. The novel Aspergillus fumigatus MAT1-2-4 mating-type gene is required for mating and cleistothecia formation. Fungal Genet. Biol. 2017, 108, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guerra, T.M.; Mellado, E.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob. Agents Chemother. 2003, 47, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Mellado, E.; Garcia-Effron, G.; Alcazar-Fuoli, L.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L. Substitutions at methionine 220 in the 14-alpha sterol demethylase (Cyp51A) of Aspergillus fumigatus are responsible for resistance in vitro to azole antifungal drugs. Antimicrob. Agents Chemother. 2004, 48, 2747–2750. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Webster, I.; Moore, C.B.; Gardiner, R.E.; Park, S.; Perlin, D.S.; Denning, D.W. Multi-azole resistance in Aspergillus fumigatus. Int. J. Antimicrob. Agents 2006, 28, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Bellete, B.; Raberin, H.; Morel, J.; Flori, P.; Hafid, J.; Manhsung, R.T. Acquired resistance to voriconazole and itraconazole in a patient with pulmonary aspergilloma. Med. Mycol. 2010, 48, 197–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodiamont, C.J.; Dolman, K.M.; Ten Berge, I.J.; Melchers, W.J.; Verweij, P.E.; Pajkrt, D. Multiple-azole-resistant Aspergillus fumigatus osteomyelitis in a patient with chronic granulomatous disease successfully treated with long-term oral posaconazole and surgery. Med. Mycol. 2009, 47, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, E.; Maertens, J.; Schoemans, H.; Lagrou, K. Azole-resistant Aspergillus fumigatus due to TR46/Y121F/T289A mutation emerging in Belgium, July 2012. Eur. Surveill. 2012, 17, 20326. [Google Scholar]
- Chen, Y.; Li, Z.; Han, X.; Tian, S.; Zhao, J.; Chen, F.; Su, X.; Zhao, J.; Zou, Z.; Gong, Y.; et al. Elevated MIC values to imidazole drugs among Aspergillus fumigatus isolates with TR34/L98H/S297T/F495I mutation. Antimicrob. Agents Chemother. 2018, 62, e01549-17. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Baixench, M.T.; Amsellem, M.; Audureau, E.; Chapron, J.; Kanaan, R.; Honoré, I.; Dupouy-Camet, J.; Dusser, D.; Klaassen, C.H.; et al. High prevalence of azole-resistant Aspergillus fumigatus in adults with cystic fibrosis exposed to itraconazole. Antimicrob. Agents Chemother. 2012, 56, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Kathuria, S.; Xu, J.; Sharma, C.; Sundar, G.; Singh, P.K.; Gaur, S.N.; Hagen, F.; Klaassen, C.H.; Meis, J.F. Clonal expansion and emergence of environmental multiple-triazole-resistant Aspergillus fumigatus strains carrying the TR34/L98H mutations in the cyp51A gene in India. PLoS ONE 2012, 7, e52871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Ashu, E.; Sharma, C.; Kathuria, S.; Chowdhary, A.; Xu, J. Diversity and origins of Indian multi-triazole resistant strains of Aspergillus fumigatus. Mycoses 2016, 59, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, R.; Cuenca-Estrella, M.; Mellado, E. Triazole Resistance in Aspergillus Species: An Emerging Problem. Drugs 2017, 77, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Huang, J.C.; Lin, Y.H.; Chen, Y.H.; Hsieh, M.I.; Choi, P.C.; Lo, H.J.; Liu, W.L.; Hsu, C.S.; Shih, H.I.; et al. Prevalence, mechanisms and genetic relatedness of the human pathogenic fungus Aspergillus fumigatus exhibiting resistance to medical azoles in the environment of Taiwan. Environ. Microbiol. 2017, 20, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Snelders, E.; Rijs, A.J.; Kema, G.H.; Melchers, W.J.; Verweij, P.E. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Appl. Environ. Microbiol. 2009, 75, 4053–4057. [Google Scholar] [CrossRef] [PubMed]
- Camps, S.M.T.; Rijs, A.J.M.M.; Klaassen, C.H.W.; Meis, J.F.; O’Gorman, C.M.; Dyer, P.S.; Melchers, W.J.G.; Verweij, P.E. Molecular epidemiology of Aspergillus fumigatus isolates harboring the TR34/L98H azole resistance mechanism. J. Clin. Microbiol. 2012, 50, 2674–2680. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Sharma, C.; Hagen, F.; Meis, J.F. Exploring azole antifungal drug resistance in Aspergillus fumigatus with special reference to resistance mechanisms. Future Microbiol. 2014, 9, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Moye-Rowley, W.S. Multiple mechanisms contribute to the development of clinically significant azole resistance in Aspergillus fumigatus. Front. Microbiol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Fraczek, M.G.; Bromley, M.; Buied, A.; Moore, C.B.; Rajendran, R.; Rautemaa, R.; Ramage, G.; Denning, D.W.; Bowyer, P. The cdr1B efflux transporter is associated with non-cyp51 a mediated itraconazole resistance in Aspergillus fumigatus. J. Antimicrob. Chemother. 2013, 68, 1486–1496. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Samples | Origin | cyp51A Modifications | AZL SC | Mating Type | Source | Lineage |
|---|---|---|---|---|---|---|
| AF293 | SP | 5SNPs | S | M1.2 | CL | 3 |
| akuBKU80 | SP | WT | S | M1.1 | CL | 1.1 |
| ATCC204305 | SP | I242V | S | M1.1 | CL | 1.2 |
| ATCC46645 | SP | WT | S | M1.1 | CL | 2.1 |
| CEA10 | SP | WT | S | M1.1 | CL | 1.1 |
| CM2141 | SP | WT | S | M1.1 | CL | 2.2 |
| CM237 | SP | WT | S | M1.1 | CL | 1.1 |
| CM2495 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM2730 | SP | 3SNPs | S | M1.2 | CL | 4 |
| CM2733 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM3248 | SP | N248K | S | M1.1 | CL | 1.1 |
| CM3249 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM3249b | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM3262 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM3720 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM4602 | SP | 3SNPs | S | M1.2 | CL | 4 |
| CM4946 | SP | 3SNPs | S | M1.2 | CL | 4 |
| CM5419 | SP | WT | S | M1.2 | CL | 2.2 |
| CM5757 | SP | WT | S | M1.2 | CL | 1.1 |
| CM6126 | SP | WT | S | M1.2 | CL | 1.1 |
| CM6458 | SP | WT | S | M1.2 | CL | 1.3 |
| CM7510 | SP | WT | R | M1.1 | CL | 1.1 |
| CM7555 | SP | WT | R | M1.2 | CL | 1.3 |
| CM7560 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM7570 | SP | 3SNPs | S | M1.1 | CL | 4 |
| CM7632 | SP | 5SNPs | S | M1.1 | CL | 3 |
| TP12 | SP | 5SNPs | S | M1.2 | CL | 3 |
| TP32 | SP | 3SNPs | S | M1.1 | CL | 4 |
| Samples | Origin | cyp51A Modifications | AZL SC | Mating Type | Source | Lineage | Ref. |
|---|---|---|---|---|---|---|---|
| 08-12-12-13 | NT | TR34/L98H, S297T, F495I | R | M1.1 | CL | 2.2 | [13] |
| 08-19-02-10 | NT | TR34/L98H | R | M1.2 | ENV | 2.2 | [13] |
| 08-19-02-30 | NT | WT | S | M1.1 | ENV | 1.3 | [13] |
| 08-19-02-46 | NT | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| 08-19-02-61 | NT | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| 08-31-08-91 | NT | TR34/L98H | R | M1.1 | CL | 2.2 | [13] |
| 08-36-03-25 | NT | TR34/L98H, S297T, F495I | R | Unclear | CL | 2.2 | [13] |
| 09-7500806 | UK | WT | S | M1.2 | CL | 1.3 | [13] |
| 10-01-02-27 | NT | TR34/L98H | R | M1.2 | CL | 2.2 | [13] |
| 12-7504462 | UK | WT | S | M1.2 | CL | 1.3 | [13] |
| 12-7504652 | UK | WT | S | M1.1 | CL | 2.1 | [13] |
| 12-7505054 | UK | WT | S | M1.1 | CL | 2.1 | [13] |
| 12-7505220 | UK | TR34/L98H | R | M1.1 | CL | 2.2 | [13] |
| 12-7505446 | UK | TR34/L98H | R | M1.2 | CL | 2.2 | [13] |
| Af293 | UK | 5SNPs | S | M1.2 | CL | 3 | [13] |
| AF41 | UK | WT | S | M1.2 | CL | 1.1 | [13] |
| Af65 | UK | WT | S | M1.2 | CL | 2.1 | [13] |
| AF72 | UK | G54E | R | M1.2 | CL | 1.1 | [13] |
| AF90 | UK | M220V | R | M1.1 | CL | 1.1 | [13] |
| Afu1042-09 | IN | TR34/L98H | R | M1.1 | CL | 2.2 | [13] |
| Afu124-E11 | IN | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| Afu166-E11 | IN | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| Afu218-E11 | IN | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| Afu257-E11 | IN | TR34/L98H | R | M1.1 | ENV | 2.2 | [13] |
| Afu343-P-11 | IN | TR34/L98H | R | Unclear | CL | 2.2 | [13] |
| Afu591-12 | IN | TR34/L98H | R | M1.1 | CL | 2.2 | [13] |
| Afu942-09 | IN | TR34/L98H | R | M1.1 | CL | 2.2 | [13] |
| F12041 | UK | G138C | R | M1.2 | CL | 2.1 | [21] * |
| F12219 | UK | G54R | R | M1.2 | CL | 1.1 | [21] * |
| F12636 | UK | G54E | R | M1.1 | CL | 2.1 | [21] * |
| F13535 | UK | G138C | R | M1.2 | CL | 2.1 | [21] * |
| F13619 | UK | H147Y, G448S | R | M1.1 | CL | 2.1 | [21] * |
| F13952 | UK | G138C | R | M1.2 | CL | 2.1 | [21] * |
| F14403 | UK | G54R | R | M1.1 | CL | 1.1 | [21] * |
| F14513G | UK | G138C | R | M1.2 | CL | 2.1 | [21] * |
| F14532 | UK | M220T | R | M1.1 | CL | 2.1 | [21] * |
| F14946G | UK | WT | S | M1.2 | CL | 1.1 | [21] * |
| F15390 | UK | M220T | R | M1.1 | CL | 2.1 | [21] * |
| F15927 | CN | 3SNPs | S | M1.1 | CL | 4 | [21] * |
| F16134 | DN | M220K | R | M1.2 | CL | 1.1 | [21] * |
| F16216 | UK | TR34/L98H | R | M1.2 | CL | 2.1 | [21] * |
| F16311 | UK | WT | S | M1.1 | CL | 1.1 | [21] * |
| F17582 | UK | WT | S | M1.2 | CL | 2.1 | [21] * |
| F17729 | UK | WT | S | M1.2 | CL | 1.1 | [21] * |
| F17729W | UK | WT | S | M1.2 | CL | 1.1 | [21] * |
| F17764 | CN | WT | S | M1.1 | CL | 1.1 | [21] * |
| F18085 | UK | WT | S | M1.1 | CL | 1.1 | [21] * |
| F5211G | UK | WT | S | M1.1 | CL | 2.1 | [21] * |
| F7763 | UK | 5SNPs | S | M1.1 | CL | 3 | [21] * |
| IFM55369 | JP | WT | S | M1.1 | CL | 1.2 | [22] |
| IFM58026 | JP | N248K | S | M1.2 | CL | 1.1 | [22] |
| IFM58029 | JP | WT | S | M1.2 | CL | 1.2 | [22] |
| IFM58401 | JP | WT | S | M1.2 | CL | 1.1 | [22] |
| IFM59056 | JP | WT | S | M1.1 | CL | 1.1 | [22] |
| IFM59073 | JP | WT | S | M1.2 | CL | 1.2 | [22] |
| IFM59359 | JP | WT | S | Unclear | CL | 1.2 | [22] |
| IFM59361 | JP | WT | S | M1.1 | CL | 1.3 | [22] |
| IFM59365 | JP | WT | S | M1.2 | CL | 1.1 | [22] |
| IFM59777 | JP | WT | S | M1.1 | CL | 1.2 | [22] |
| IFM60514 | JP | WT | S | M1.2 | CL | 1.1 | [22] |
| IFM61118 | JP | N248K | S | M1.1 | CL | 1.1 | [22] |
| IFM61407 | JP | WT | S | Unclear | CL | 1.1 | [22] |
| IFM61578 | JP | P216L | S | M1.2 | CL | 1.1 | [22] |
| IFM61610 | JP | WT | S | M1.2 | CL | 1.1 | [22] |
| IFM62516 | JP | P329P | S | M1.1 | CL | 1.2 | [22] |
| SL143435 | PT | N248K | S | M1.2 | UNK | 1.1 | * |
| SL143436 | PT | WT | S | M1.2 | UNK | 1.1 | * |
| SL143437 | PT | WT | S | M1.1 | UNK | 1.3 | * |
| SL143438 | PT | N248K | S | M1.2 | UNK | 1.1 | * |
| SL143439 | PT | WT | S | M1.1 | UNK | 1.3 | * |
| SL143440 | PT | 3SNPs | S | M1.1 | UNK | 4 | * |
| SL143441 | PT | WT | S | M1.1 | UNK | 1.1 | * |
| SL146112 | PT | N248K | S | M1.2 | UNK | 1.1 | * |
| Phylogenetic Groups | SNVs vs. A1163 Reference Genome | SNVs vs. Af293 Reference Genome |
|---|---|---|
| Subcluster I.1 | 34,544 | 75,653 |
| Subcluster I.2 | 40,124 | 65,121 |
| Subcluster I.3 | 46,811 | 79,899 |
| Subcluster II.1 | 81,478 | 98,423 |
| Subcluster II.2 | 80,994 | 97,749 |
| Cluster III | 82,235 | 35,268 |
| Cluster IV | 158,154 | 159,742 |
| Samples | A1163 | Af293 | Samples | A1163 | Af293 | Samples | A1163 | Af293 |
|---|---|---|---|---|---|---|---|---|
| 08-12-12-13 | 80,791 | 91,956 | CM2495 | 151,089 | 148,524 | F17582 | 79,598 | 97,024 |
| 08-19-02-10 | 75,401 | 89,762 | CM2730 | 157,505 | 159,177 | F17729 | 44,054 | 79,106 |
| 08-19-02-30 | 42,487 | 87,831 | CM2733 | 154,361 | 160,494 | F17729W | 43,663 | 78,416 |
| 08-19-02-46 | 78,449 | 96,243 | CM3248 | 22,834 | 74,568 | F17764 | 25,812 | 70,013 |
| 08-19-02-61 | 80,244 | 96,416 | CM3249 | 157,242 | 159,900 | F18085 | 51,241 | 78,553 |
| 08-31-08-91 | 88,972 | 98,911 | CM3249b | 154,510 | 157,780 | F5211G | 82,091 | 99,726 |
| 08-36-03-25 | 83,921 | 102,006 | CM3262 | 161,695 | 160,926 | F7763 | 84,764 | 55,332 |
| 09-7500806 | 45,286 | 74,134 | CM3720 | 156,600 | 158,781 | IFM55369 | 42,849 | 64,003 |
| 10-01-02-27 | 72,487 | 93,299 | CM4602 | 155,381 | 161,767 | IFM58026 | 22,186 | 68,777 |
| 12-7504462 | 48,927 | 75,301 | CM4946 | 158,692 | 161,082 | IFM58029 | 36,521 | 63,106 |
| 12-7504652 | 80,203 | 93,323 | CM5419 | 89,354 | 99,771 | IFM58401 | 26,669 | 63,489 |
| 12-7505054 | 86,952 | 101,471 | CM5757 | 36,027 | 74,420 | IFM59056 | 30,841 | 61,481 |
| 12-7505220 | 75,479 | 100,229 | CM6126 | 55,871 | 86,902 | IFM59073 | 43,230 | 68,775 |
| 12-7505446 | 86,194 | 100,736 | CM6458 | 42,482 | 71,670 | IFM59359 | 39,309 | 67,118 |
| Af293 | 83,486 | 313 | CM7510 | 33,559 | 79,283 | IFM59361 | 42,795 | 73,228 |
| AF293 | 78,045 | 659 | CM7555 | 51,657 | 77,679 | IFM59365 | 21,830 | 74,248 |
| AF41 | 34,879 | 72,914 | CM7560 | 158,657 | 153,769 | IFM59777 | 40,117 | 65,300 |
| Af65 | 90,143 | 103,614 | CM7570 | 160,908 | 162,292 | IFM60514 | 39,629 | 75,765 |
| AF72 | 35,271 | 73,632 | CM7632 | 81,519 | 25,490 | IFM61118 | 21,905 | 70,230 |
| AF90 | 40,262 | 73,395 | F12041 | 78,812 | 93,712 | IFM61407 | 32,025 | 76,034 |
| Afu1042-09 | 80,346 | 96,348 | F12219 | 35,440 | 73,872 | IFM61578 | 26,201 | 67,946 |
| Afu124-E11 | 81,115 | 97,509 | F12636 | 80,359 | 98,501 | IFM61610 | 41,690 | 74,102 |
| Afu166-E11 | 80,948 | 97,350 | F13535 | 78,741 | 93,777 | IFM62516 | 41,229 | 67,364 |
| Afu218-E11 | 80,571 | 96,955 | F13619 | 85,412 | 107,985 | SL143435 | 22,037 | 81,031 |
| Afu257-E11 | 80,499 | 97,008 | F13952 | 78,466 | 93,926 | SL143436 | 52,604 | 84,188 |
| Afu343-P11 | 73,797 | 101,305 | F14403 | 27,476 | 74,454 | SL143437 | 61,289 | 91,394 |
| Afu591-12 | 80,781 | 97,039 | F14513G | 78,647 | 94,213 | SL143438 | 22,676 | 81,649 |
| Afu942-09 | 80,827 | 97,149 | F14532 | 79,931 | 102,436 | SL143439 | 39,562 | 87,955 |
| akuBKU80 | 1043 | 78,152 | F14946G | 44,442 | 79,719 | SL143440 | 162,993 | 165,788 |
| ATCC 204305 | 37,614 | 60,180 | F15390 | 79,743 | 101,967 | SL143441 | 38,498 | 72,994 |
| ATCC 46645 | 84,994 | 103,941 | F15927 | 161,157 | 164,076 | SL146112 | 21,782 | 81,137 |
| CEA10 | 1009 | 75,854 | F16134 | 19,660 | 77,718 | TP12 | 83,363 | 24,982 |
| CM2141 | 88,704 | 107,238 | F16216 | 78,073 | 90,736 | TP32 | 163,367 | 162,032 |
| CM237 | 48,402 | 83,874 | F16311 | 51,387 | 78,630 | F17582 | 79,598 | 97,024 |
| Variants | A1163 Reference Genome | Af293 Reference Genome | ||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | I | II | III | IV | |
| Frameshift variants | 256 | 512 | 477 | 828 | 446 | 587 | 133 | 819 |
| Inframe deletions | 149 | 278 | 264 | 511 | 246 | 330 | 94 | 527 |
| Inframe insertions | 191 | 369 | 291 | 561 | 325 | 447 | 91 | 556 |
| Intergenic variants | 6405 | 11,107 | 12,819 | 6730 | 13,428 | 16,394 | 3,785 | 8792 |
| Intron variants | 1568 | 3668 | 3418 | 9066 | 3120 | 4322 | 960 | 8670 |
| Missense variants | 4342 | 10,251 | 10,008 | 23,107 | 8766 | 11,973 | 2562 | 22,746 |
| Protein altering | 8 | 19 | 12 | 18 | 16 | 22 | 4 | 21 |
| Start lost | 9 | 16 | 23 | 57 | 21 | 24 | 6 | 57 |
| Stop gained | 95 | 230 | 264 | 298 | 141 | 218 | 43 | 288 |
| Stop lost | 13 | 27 | 32 | 61 | 25 | 33 | 9 | 61 |
| Stop retained | 10 | 22 | 21 | 52 | 17 | 24 | 5 | 52 |
| Synonymous variants | 3675 | 9026 | 9005 | 26,080 | 8189 | 11,058 | 2365 | 25,418 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Rubio, R.; Monzon, S.; Alcazar-Fuoli, L.; Cuesta, I.; Mellado, E. Genome-Wide Comparative Analysis of Aspergillus fumigatus Strains: The Reference Genome as a Matter of Concern. Genes 2018, 9, 363. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9070363
Garcia-Rubio R, Monzon S, Alcazar-Fuoli L, Cuesta I, Mellado E. Genome-Wide Comparative Analysis of Aspergillus fumigatus Strains: The Reference Genome as a Matter of Concern. Genes. 2018; 9(7):363. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9070363
Chicago/Turabian StyleGarcia-Rubio, Rocio, Sara Monzon, Laura Alcazar-Fuoli, Isabel Cuesta, and Emilia Mellado. 2018. "Genome-Wide Comparative Analysis of Aspergillus fumigatus Strains: The Reference Genome as a Matter of Concern" Genes 9, no. 7: 363. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9070363