Global Long Noncoding RNA and mRNA Expression Changes between Prenatal and Neonatal Lung Tissue in Pigs

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples
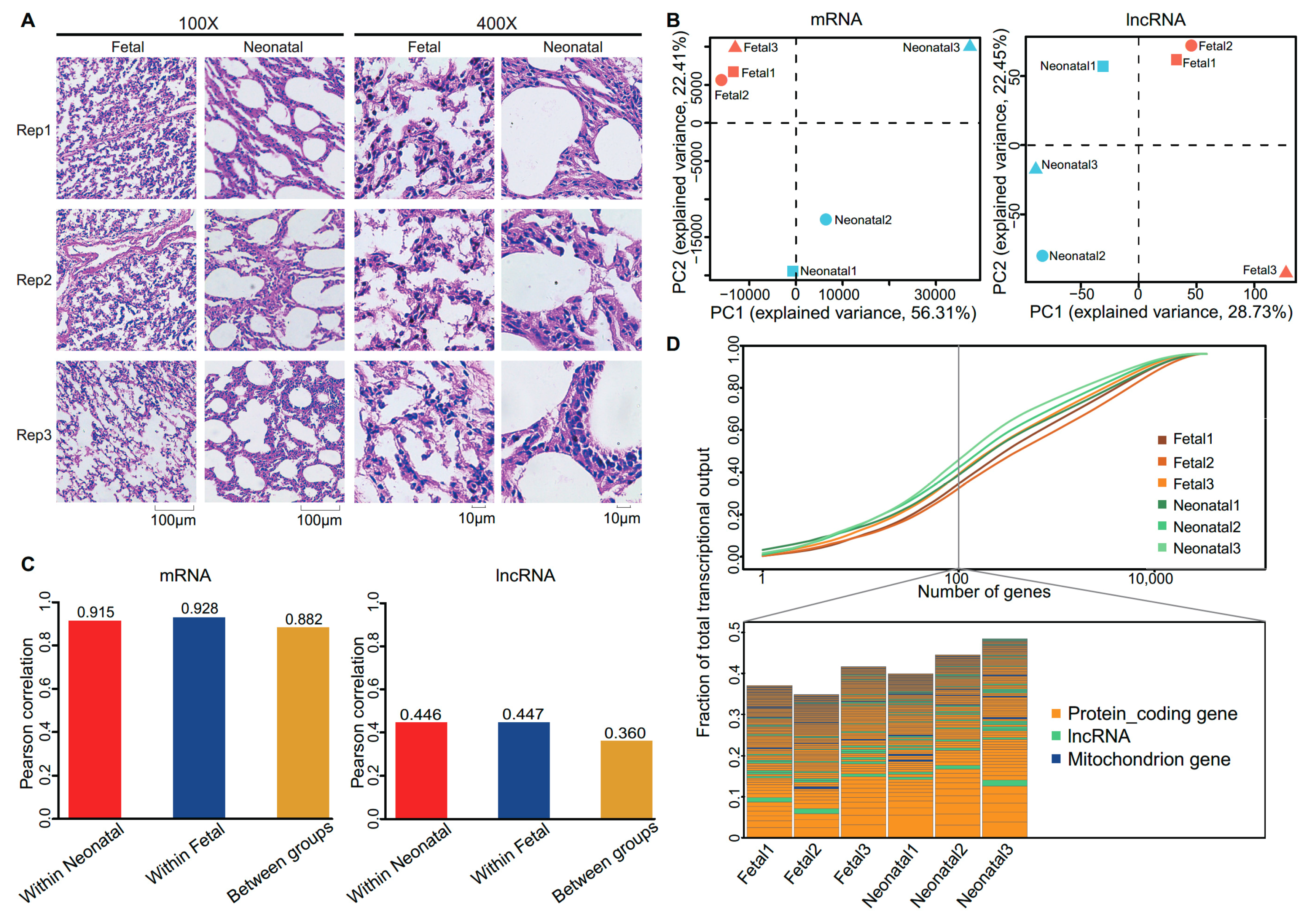
2.2. Morphometry of Lung Tissues
2.3. RNA Isolation, Library Preparation, and Sequencing
2.4. Messenger RNA Expression Analysis
2.5. Long Non-Coding RNA Identification and Expression Analysis
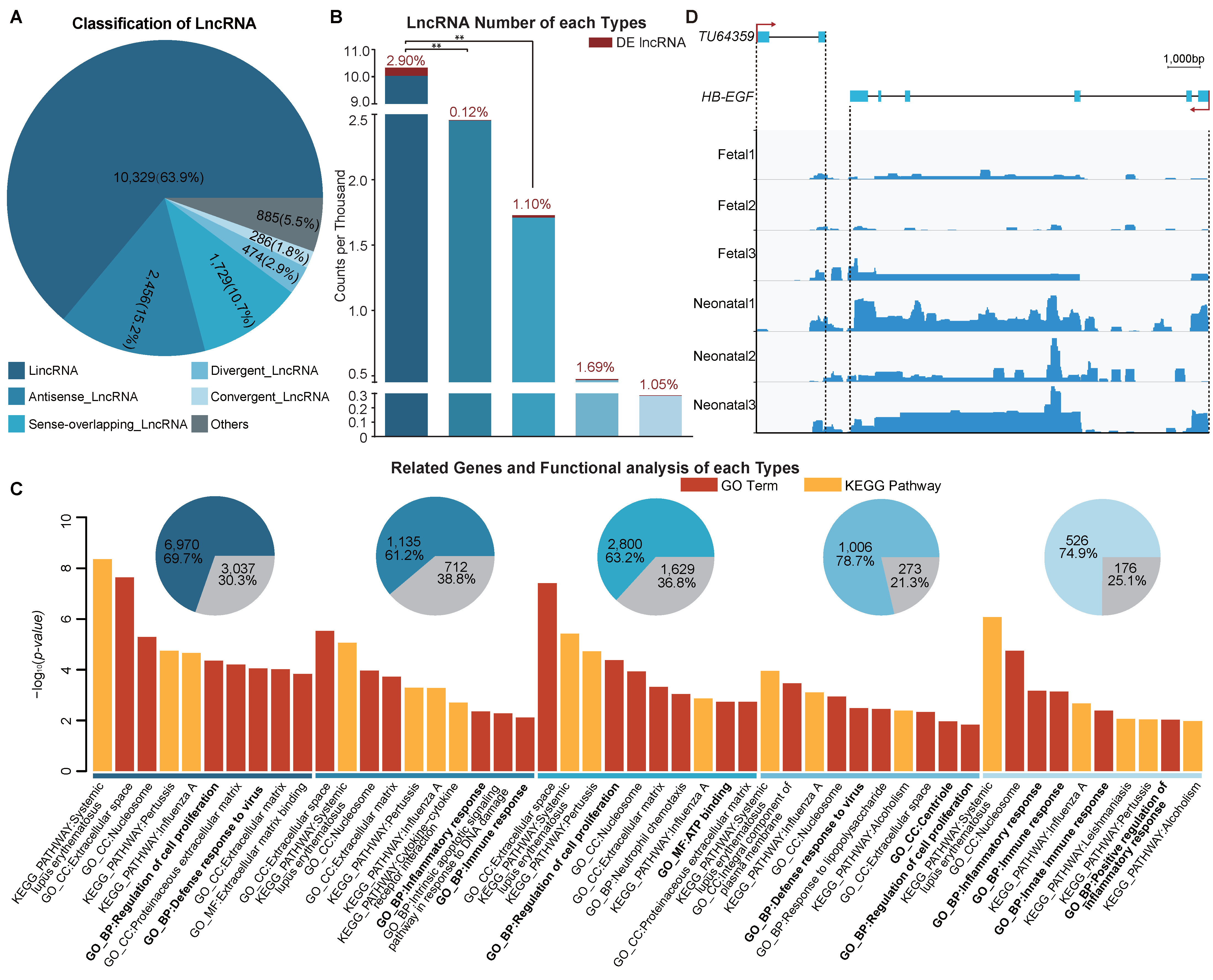
2.6. Long Non-Coding RNA Classification
2.7. Functional Enrichment Analysis
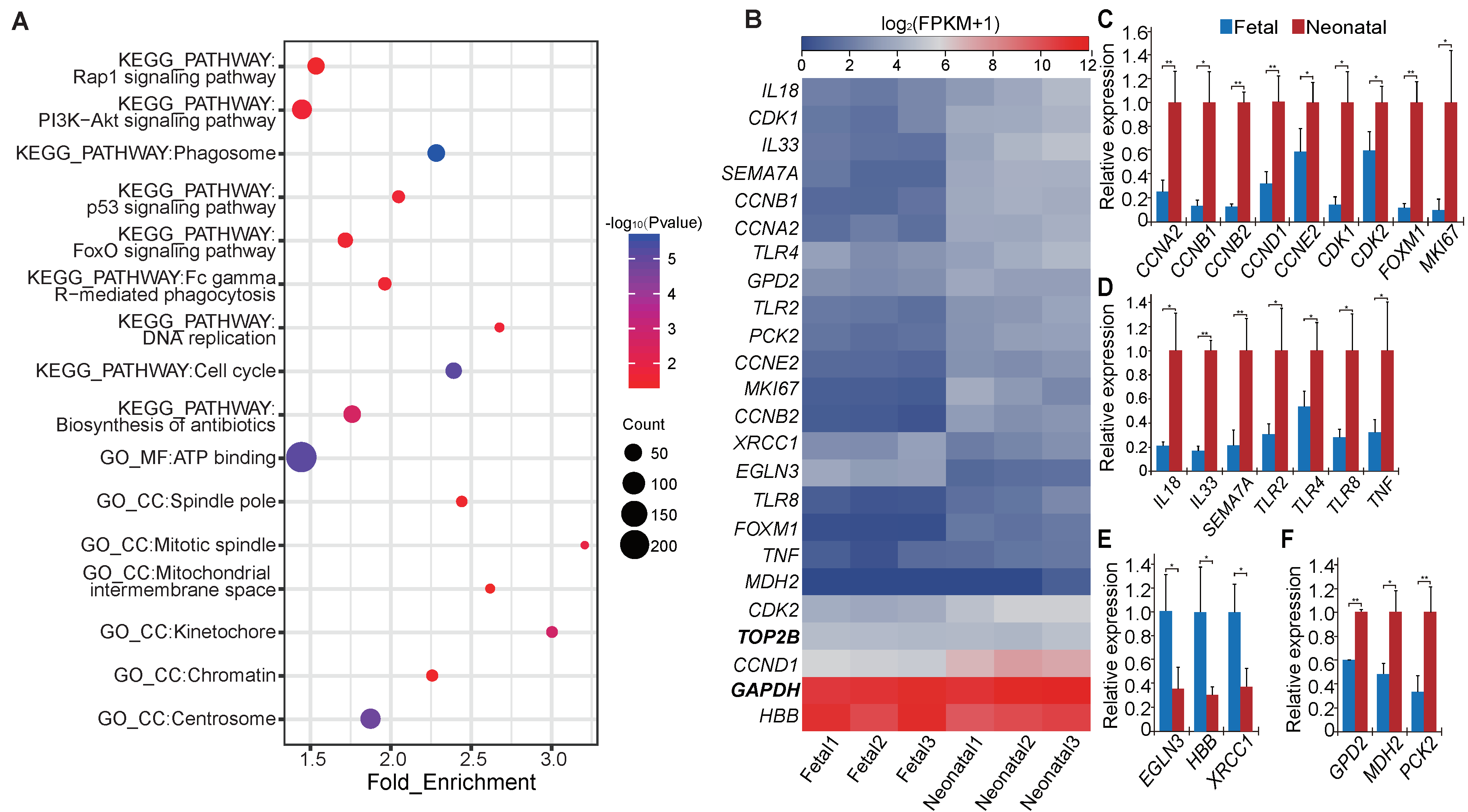
2.8. Quantitative Real-Time PCR Validation
3. Results
3.1. Morphologically Differences between Fetal and Neonatal Pigs
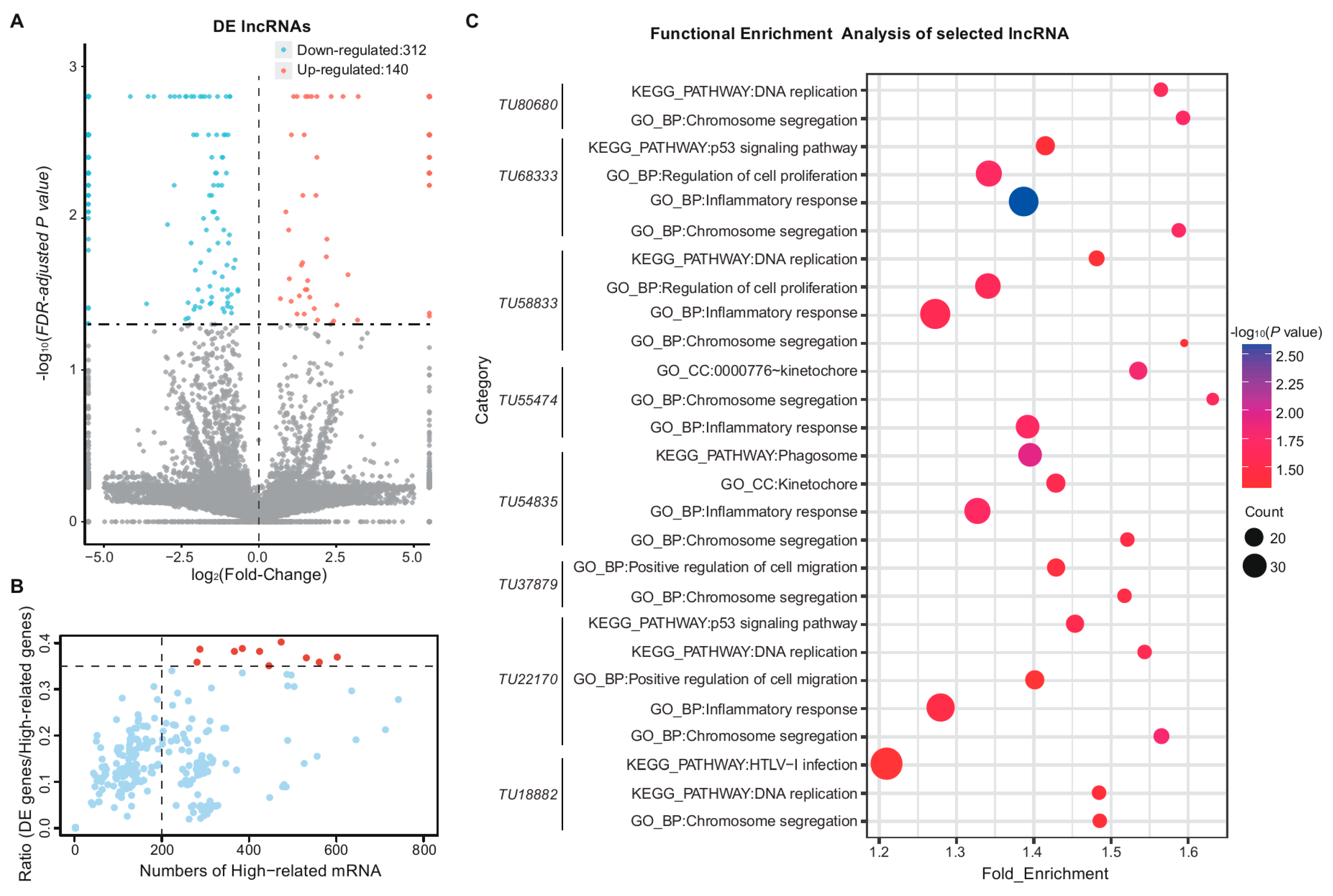
3.2. Expression Profiles of Messenger RNA and Long Non-Coding RNA in Lung
3.3. Characteristics Comparison of Messenger RNAs and Long Non-Coding RNAs
3.4. Functional Enrichment Analysis of Differentially Expressed Messenger RNAs and Long Non-Coding RNAs
3.5. Functional Enrichment Analysis of Different Long Non-Coding RNA Types
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mess, A.M.; Ferner, K.J. Evolution and development of gas exchange structures in Mammalia: The placenta and the lung. Respir. Physiol. Neurobiol. 2010, 173, S74–S82. [Google Scholar] [CrossRef] [PubMed]
- Hillman, S.S.; Hancock, T.V.; Hedrick, M.S. A comparative meta-analysis of maximal aerobic metabolism of vertebrates: Implications for respiratory and cardiovascular limits to gas exchange. J. Comp. Physiol. B 2013, 183, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, Z.; Collins, J.E.; Andrews, R.M.; Stemple, D.; Gong, Z. Comparative transcriptome analyses indicate molecular homology of zebrafish swimbladder and mammalian lung. PLoS ONE 2011, 6, e24019. [Google Scholar] [CrossRef] [PubMed]
- Ten Have-Opbroek, A.A. Lung development in the mouse embryo. Exp. Lung Res. 1991, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Burri, P.H. Lung development and pulmonary angiogenesis. In Lung Development; Gaultier, C., Bourbon, J.R., Post, M., Eds.; Clinical Physiology Series; Springer: New York, NY, USA, 1999; pp. 122–151. [Google Scholar]
- Roth-Kleiner, M.; Hirsch, E.; Schittny, J.C. Fetal lungs of tenascin-C-deficient mice grow well, but branch poorly in organ culture. Am. J. Respir. Cell Mol. Biol. 2004, 30, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Popper, H. Pathology of Lung Disease; Springer: Berlin, Germany, 2017; pp. 1–5. [Google Scholar]
- Hooper, S.B.; Polglase, G.R.; Roehr, C.C. Cardiopulmonary changes with aeration of the newborn lung. Paediatr. Respir. Rev. 2015, 16, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, A.M. Fetal and neonatal pulmonary circulation. Annu. Rev. Physiol. 1979, 41, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.B.; Te Pas, A.B.; Lang, J.; van Vonderen, J.J.; Roehr, C.C.; Kluckow, M.; Gill, A.W.; Wallace, E.M.; Polglase, G.R. Cardiovascular transition at birth: A physiological sequence. Pediatr. Res. 2015, 77, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Enders, A. Reasons for diversity of placental structure. Placenta 2009, 30, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Olver, R.E.; Walters, D.V.; Wilson, S.M. Developmental regulation of lung liquid transport. Annu. Rev. Physiol. 2004, 66, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.B.; Harding, R. Role of aeration in the physiological adaptation of the lung to air-breathing at birth. Curr. Respir. Med. Rev. 2005, 1, 185–195. [Google Scholar] [CrossRef]
- Mortola, J.P. Respiratory Physiology of Newborn Mammals: A Comparative Perspective; John Hopkins University Press: Baltimore, MD, USA, 2001. [Google Scholar]
- Hooper, S.B.; Kitchen, M.J.; Wallace, M.J.; Yagi, N.; Uesugi, K.; Morgan, M.J.; Hall, C.; Siu, K.K.; Williams, I.M.; Siew, M.; et al. Imaging lung aeration and lung liquid clearance at birth. FASEB J. 2007, 21, 3329–3337. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.B.; Kitchen, M.J.; Siew, M.L.; Lewis, R.A.; Fouras, A.; te Pas, A.B.; Siu, K.K.; Yagi, N.; Uesugi, K.; Wallace, M.J. Imaging lung aeration and lung liquid clearance at birth using phase contrast X-ray imaging. Clin. Exp. Pharmacol. Physiol. 2009, 36, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Bassols, A.; Costa, C.; Eckersall, P.D.; Osada, J.; Sabrià, J.; Tibau, J. The pig as an animal model for human pathologies: A proteomics perspective. Proteomics Clin. Appl. 2014, 8, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Meurens, F.; Summerfield, A.; Nauwynck, H.; Saif, L.; Gerdts, V. The pig: A model for human infectious diseases. Trends Microbiol. 2012, 20, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.S.; Abraham, W.M.; Brogden, K.A.; Engelhardt, J.F.; Fisher, J.T.; McCray, P.B., Jr.; McLennan, G.; Meyerholz, D.K.; Namati, E.; Ostedgaard, L.S.; et al. The porcine lung as a potential model for cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L240–L263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennai, S.; Souilamas, R.; Maignan, M.; Brouta, A.; Pison, C.; Fontaine, E.; Briot, R. Effects of cyclosporine a in ex vivo reperfused pig lungs. Microcirculation 2014, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Kelvin, D.J.; Li, L.; Zheng, Z.; Yoon, S.W.; Wong, S.S.; Farooqui, A.; Wang, J.; Banner, D.; et al. Infectivity, transmission, and pathology of human-isolated H7N9 influenza virus in ferrets and pigs. Science 2013, 341, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, S.; Iwamoto, R.; Mekada, E. HB-EGF decelerates cell proliferation synergistically with TGFα in perinatal distal lung development. Dev. Dyn. 2008, 237, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Chan, C.K. Analysis of RNA-seq data using TopHat and Cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assemblyline. Available online: https://code.google.com/archive/p/assemblyline/ (accessed on 4 September 2018).
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Primer-BLAST. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/tools/primer-blast/ (accessed on 4 September 2018).
- Loosli, C.G.; Potter, E.L. Pre- and postnatal development of the respiratory portion of the human lung with special reference to the elastic fibers. Am. Rev. Respir. Dis. 1959, 80, 5–23. [Google Scholar] [PubMed]
- Breeze, R.G.; Wheeldon, E.B. The cells of the pulmonary airways. Am. Rev. Respir. Dis. 1977, 116, 705–777. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Morphological basis of alveolar-capillary gas exchange. Physiol. Rev. 1973, 53, 419–495. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.B.; Te Pas, A.B.; Kitchen, M.J. Respiratory transition in the newborn: A three-phase process. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101, F266–F271. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.E.; Dotter, C.P.; Guenzl, P.M.; Gisslinger, H.; Gisslinger, B.; Cleary, C.; Kralovics, R.; Pauler, F.M.; Barlow, D.P. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Zhu, W.; Liu, T.; Wang, W.; Huang, G.; Huang, J.; Zhao, P.; Zhao, Y.; Yao, S. Characterization of long non-coding RNA expression profiles in lymph node metastasis of early-stage cervical cancer. Oncol. Rep. 2016, 35, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisúa Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391–5404. [Google Scholar] [CrossRef] [PubMed]
- Quelle, D.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993, 7, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Bewick, M.A.; Conlon, M.S.; Lafrenie, R.M. Polymorphisms in XRCC1, XRCC3, and CCND1 and survival after treatment for metastatic breast cancer. J. Clin. Oncol. 2006, 24, 5645–5651. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Fung, T.; Poon, R. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Pupo, G.M.; Scolyer, R.A.; Kefford, R.F.; Rizos, H. p16INK4a deficiency promotes DNA hyper-replication and genetic instability in melanocytes. Pigment Cell Melanoma Res. 2013, 26, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Langella, C.; D’Angelo, D.; Mussnich, P.; Wierinckx, A.; Terracciano, L.; Raverot, G.; Lachuer, J.; Rotondi, S.; Jaffrain-Rea, M.L.; et al. Mir-23b and miR-130b expression is downregulated in pituitary adenomas. Mol. Cell. Endocrinol. 2014, 390, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, X.; He, Y.; Yang, Z.; Sun, T.; Wang, J.; Zhu, G.; Xing, W.; You, C. CCNB2 overexpression is a poor prognostic biomarker in Chinese NSCLC patients. Biomed. Pharmacother. 2015, 74, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006, 5, 2555–2556. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.B.; Lavine, J.A.; Suhonen, J.I.; Krautkramer, K.A.; Rabaglia, M.E.; Sperger, J.M.; Fernandez, L.A.; Yandell, B.S.; Keller, M.P.; Wang, I.M.; et al. FoxM1 is up-regulated by obesity and stimulates β-cell proliferation. Mol. Endocrinol. 2010, 24, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Murakami, T.; Kawasaki, M.; Takahashi, M. The cell cycle associated change of the Ki-67 reactive nuclear antigen expression. J. Cell Physiol. 1987, 133, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Hiemstra, P. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med. 2006, 84, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tong, M.; Yi, L.; Cheng, Y.; Zhang, M.; Cao, Z.; Wang, J.; Lin, P.; Cheng, S. Identification and characterization of the toll-like receptor 8 gene in the Chinese raccoon dog (Nyctereutes procyonoides). Immunol. Lett. 2016, 178, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.A.; Guo, R.F.; Yun, E.C.; Sarma, V.; Warner, R.L.; Crouch, L.D.; Senaldi, G.; Ulich, T.R.; Ward, P.A. Role of IL-18 in acute lung inflammation. J. Immunol. 2001, 167, 7060–7068. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef] [PubMed]
- Renz, H.; Brandtzaeg, P.; Hornef, M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 2011, 12, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Schultz, M.J.; van der Poll, T. Pneumonia models and innate immunity to respiratory bacterial pathogens. Shock 2005, 24, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Okuno, T.; Yamamoto, M.; Pasterkamp, R.J.; Takegahara, N.; Takamatsu, H.; Kitao, T.; Takagi, J.; Rennert, P.D.; Kolodkin, A.L.; et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through α1β1 integrin. Nature 2007, 446, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.O.; Del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Chiappe-Gutierrez, M.; Kitzmueller, E.; Labudova, O.; Fuerst, G.; Hoeger, H.; Hardmeier, R.; Nohl, H.; Gille, L.; Lubec, B. mRNA levels of the hypoxia inducible factor (HIF-1) and DNA repair genes in perinatal asphyxia of the rat. Life Sci. 1998, 63, 1157–1167. [Google Scholar] [CrossRef]
- Grek, C.L.; Newton, D.A.; Spyropoulos, D.D.; Baatz, J.E. Hypoxia up-regulates expression of hemoglobin in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Li, M.Z.; Li, Q.H.; Guan, J.Q.; Li, X.W. Differential expression analysis of porcine MDH1, MDH2 and ME1 genes in adipose tissues. Genet. Mol. Res. 2012, 11, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Szaszák, M.; Shima, K.; Käding, N.; Hannus, M.; Solbach, W.; Rupp, J. Host metabolism promotes growth of Chlamydia pneumoniae in a low oxygen environment. Int. J. Med. Microbiol. 2013, 303, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Lucas, A.; Hyroššová, P.; Novellasdemunt, L.; Viñals, F.; Perales, J.C. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J. Biol. Chem. 2014, 289, 22090–22102. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinf. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Burri, P.H. Morphology and respiratory function of the alveolar unit. Int. Arch. Allergy Appl. Immunol. 1985, 76, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Levy, O. Innate immunity of the newborn: Basic mechanisms and clinical correlates. Nat. Rev. Immunol. 2007, 7, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, S.L. Cell proliferation in the mammalian lung. Int. Rev. Exp. Pathol. 1980, 22, 131–191. [Google Scholar] [PubMed]
- Bourbon, J.R.; Boucherat, O.; Boczkowski, J.; Crestani, B.; Delacourt, C. Bronchopulmonary dysplasia and emphysema: In search of common therapeutic targets. Trends Mol. Med. 2009, 15, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Firth, M.A.; Shewen, P.E.; Hodgins, D.C. Passive and active components of neonatal innate immune defenses. Anim. Health Res. Rev. 2005, 6, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, X.; Chen, P.; Yu, S.; Nie, F.; Lu, B.; Zhang, T.; Zhou, Y.; Chen, Q.; Wei, C.; et al. Long non-coding RNA SNHG20 promotes non-small cell lung cancer cell proliferation and migration by epigenetically silencing of P21 expression. Cell Death Dis. 2017, 8, e3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, P.H. Structural aspects of postnatal lung Development—Alveolar formation and growth. Biol. Neonate 2006, 89, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.C.; Cheville, N.F. The neonatal porcine lung: Ultrastructural morphology and postnatal development of the terminal airways and alveolar region. Anat. Rec. 1984, 210, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, J.L.; Gard, A.L.; Gerhold, K.A.; Wilcox, E.C.; Liu, A.; Schwan, J.; Le, A.V.; Baevova, P.; Dimitrievska, S.; Zhao, L.; et al. Comparative biology of decellularized lung matrix: Implications of species mismatch in regenerative medicine. Biomaterials 2016, 102, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, G.C.; Cheville, N.F. Morphometry of postnatal development in the porcine lung. Anat. Rec. 1985, 211, 427–433. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Data (Gb) | Clean Data (Gb) | Proportion of Q30 (%) | Raw Reads | Clean Reads | Mapped Reads | Map Ratio (%) |
|---|---|---|---|---|---|---|---|
| Fetal1 | 15.48 | 14.60 | 91.99 | 103,218,160 | 97,324,642 | 74,846,405 | 76.90 |
| Fetal2 | 15.07 | 13.60 | 91.93 | 100,467,280 | 90,697,458 | 69,307,578 | 76.42 |
| Fetal3 | 14.13 | 13.52 | 92.14 | 94,178,310 | 90,125,098 | 68,548,684 | 76.06 |
| Neonatal1 | 15.53 | 14.44 | 91.56 | 103,521,604 | 96,233,814 | 72,838,975 | 75.69 |
| Neonatal2 | 16.23 | 15.41 | 91.75 | 108,169,398 | 102,757,612 | 78,091,900 | 76.00 |
| Neonatal3 | 16.14 | 14.89 | 91.84 | 107,604,186 | 99,294,804 | 75,662,550 | 76.20 |
| Gene Name | Fetus 1 | Fetus 2 | Fetus 3 | Neonatal1 | Neonatal2 | Neonatal3 | log2(FC) 2 | q Value 3 |
|---|---|---|---|---|---|---|---|---|
| ATP6 | 279.86 | 325.45 | 361.37 | 813.54 | 312.92 | 436.18 | 0.49 | 5.00 × 10−1 |
| ATP8 | 1828.44 | 1283.11 | 1824.56 | 16,615.00 | 6478.03 | 4800.99 | 2.42 | 2.97 × 10−1 |
| COX1 | 1894.03 | 2303.75 | 2511.00 | 2865.40 | 2338.47 | 4216.83 | 0.16 | 7.36 × 10−1 |
| COX2 | 1266.21 | 1274.47 | 1569.65 | 2131.98 | 1818.49 | 2814.47 | 0.36 | 2.30 × 10−1 |
| COX31 | 495.71 | 517.16 | 667.49 | 1252.63 | 778.14 | 1285.44 | 0.79 | 1.85 × 10−2 |
| CYTB1 | 24.26 | 34.00 | 41.33 | 102.45 | 36.26 | 41.20 | 0.82 | 2.90 × 10−3 |
| ND11 | 18.68 | 28.52 | 36.24 | 92.43 | 38.03 | 60.25 | 0.97 | 6.53 × 10−4 |
| ND21 | 10.90 | 19.53 | 22.55 | 70.53 | 18.87 | 35.77 | 1.21 | 6.53 × 10−4 |
| ND31 | 1118.87 | 923.81 | 1311.11 | 2951.35 | 2004.06 | 3626.46 | 0.98 | 6.53 × 10−4 |
| ND41 | 15.15 | 25.23 | 32.48 | 89.11 | 31.83 | 25.04 | 0.99 | 6.53 × 10−4 |
| ND4 | 91.18 | 120.58 | 141.40 | 297.58 | 106.88 | 40.46 | 0.29 | 9.30 × 10−1 |
| ND51 | 17.82 | 34.67 | 35.99 | 125.60 | 34.05 | 30.01 | 1.24 | 6.53 × 10−4 |
| ND6 | 8.56 | 3.81 | 28.28 | 11.18 | 4.98 | 14.80 | −0.78 | 8.50 × 10−1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, L.; Hu, S.; Tu, T.; Huang, Z.; Tang, Q.; Ma, J.; Wang, X.; Li, X.; Zhou, X.; Shuai, S.; et al. Global Long Noncoding RNA and mRNA Expression Changes between Prenatal and Neonatal Lung Tissue in Pigs. Genes 2018, 9, 443. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090443
Jin L, Hu S, Tu T, Huang Z, Tang Q, Ma J, Wang X, Li X, Zhou X, Shuai S, et al. Global Long Noncoding RNA and mRNA Expression Changes between Prenatal and Neonatal Lung Tissue in Pigs. Genes. 2018; 9(9):443. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090443
Chicago/Turabian StyleJin, Long, Silu Hu, Teng Tu, Zhiqing Huang, Qianzi Tang, Jideng Ma, Xun Wang, Xuewei Li, Xuan Zhou, Surong Shuai, and et al. 2018. "Global Long Noncoding RNA and mRNA Expression Changes between Prenatal and Neonatal Lung Tissue in Pigs" Genes 9, no. 9: 443. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090443