
A Theoretical Study of the N2 + H2 Reactive Collisions for High Vibrational and Translational Energies


1
Centro Interdisciplinar de Ciências da Natureza, Universidade Federal da Integração Latino-Americana, Foz do Iguaçu 85866-000, PR, Brazil
2
Departamento de Física, Universidade Federal de Juiz de Fora, Juiz de Fora 36036-330, MG, Brazil
*
Author to whom correspondence should be addressed.
Atmosphere 2021, 12(10), 1349; https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12101349
Submission received: 13 September 2021
/
Revised: 7 October 2021
/
Accepted: 12 October 2021
/
Published: 15 October 2021
(This article belongs to the Special Issue Theoretical Chemistry of Atmospheric Processes)
Abstract
:High translational temperatures appear in the air inside the shock waves layers created by relatively large meteorites, reentry space vehicles, and hypersonic missiles. Under these conditions, reactions between molecular nitrogen and hydrogen are energetically permitted. In the present work, a quasiclassical trajectories study of the reaction for relative translational energies covering the range of translational energy is presented. In the calculations, several values of vibrational quantum numbers and have been considered. To model the interatomic interactions, a six-dimension global potential energy surface for the ground electronic state of was used. The specific initial state reaction cross-sections and rate coefficients are reported. The energy effects produced by the reaction that could influence the shock wave modeling are here considered. An analysis of the possible impact of these processes under the atmospheric composition is also presented.
1. Introduction
Local temperatures in the range of 2000 45,000 can be produced in the Earth’s atmosphere by the movement of relatively large meteorites, reentry space vehicles, and hypersonic missiles and planes [1,2,3,4]. For these temperatures, there is a relatively large number of molecules populating vibrationally excited levels. Then, for such conditions, it is necessary to consider the rate constants of different reactive and nonreactive processes involving vibrationally excited species in works devoted to modeling the movement of the already-mentioned artefacts [2,3,5,6,7]. In particular, the molecular hydrogen, a minor atmospheric constituent (0.000053 mole percent) [8,9,10], shows small reactivity for relative low translational energies. For example, their rate constants, in collisions with important atmospheric components such as and for reactions:
have the values and [11,12] at , respectively. In turn, the atomic hydrogen is involved in the atmospheric reactions:
with rate constant values of , and , respectively, at [11,12], several orders of magnitude higher. It means, that by replacing the molecular hydrogen with atomic hydrogen, it is possible produce a high chemical impact in the atmosphere.
The reaction between molecular nitrogen and hydrogen has not been previously considered in technology [13,14] and shock wave [5] simulations, based upon the small value of the rate constant for temperature below . Nevertheless, preliminary calculations show relatively high values of the rate coefficients for such a reaction at temperatures above [15]. Moreover, the hydrogen atoms produced from title collisions could enter the important reactions (1)–(3) included in the atmospheric ozone cycles [8]. Furthermore, due to the high energy barrier involved, the title reactive collisions could influence the energy balance of the gas. Although an increase in the dissociation with the vibrational energy is expected, it is not clear how the formation of atomic hydrogen could be affected by the competition with other reactive and nonreactive channels, which also may be stimulated by the participation of vibrationally excited molecules.
Thus, the major goal of the present work is to report a detailed theoretical study of the reaction between molecular hydrogen and nitrogen at their corresponding ground electronic states, for several initial vibrationally excited combinations. For such a task, the quasiclassical trajectory (QCT) method and the single-valued double many-body expansion (DMBE) potential energy surface for the electronic ground state of are used. The paper is organized as follows: Section 2 provides a brief survey of the DMBE potential energy surface together with details of the computational method. The results are presented and discussed in Section 3 and Section 4, correspondingly, while the major conclusions are gathered in Section 5.
2. Methodology
To represent the interatomic interaction, a global potential energy surface (PES) for the singlet ground electronic state of is used [16]. This function, depending upon the six interatomic distances, was constructed within the double many-body expansion [17] and calibrated to multireference configuration interaction ab initio energies [16,18]. Such a PES was previously used in dynamic studies of the reaction [19] and the vibrational relaxation in nonreactive collisions of and [15]. For the interest of this work, from the DMBE PES, the reaction enthalpies are:
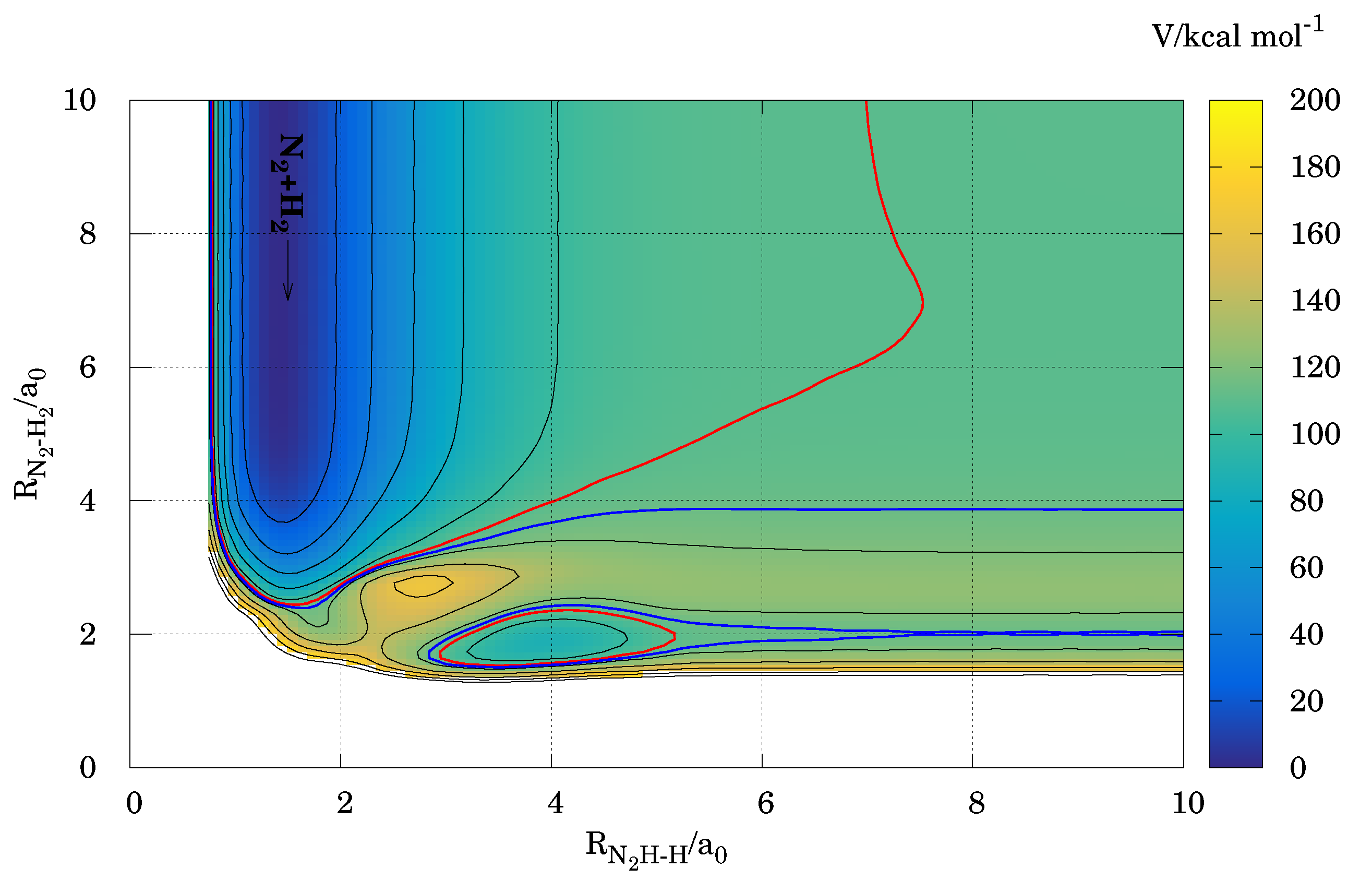
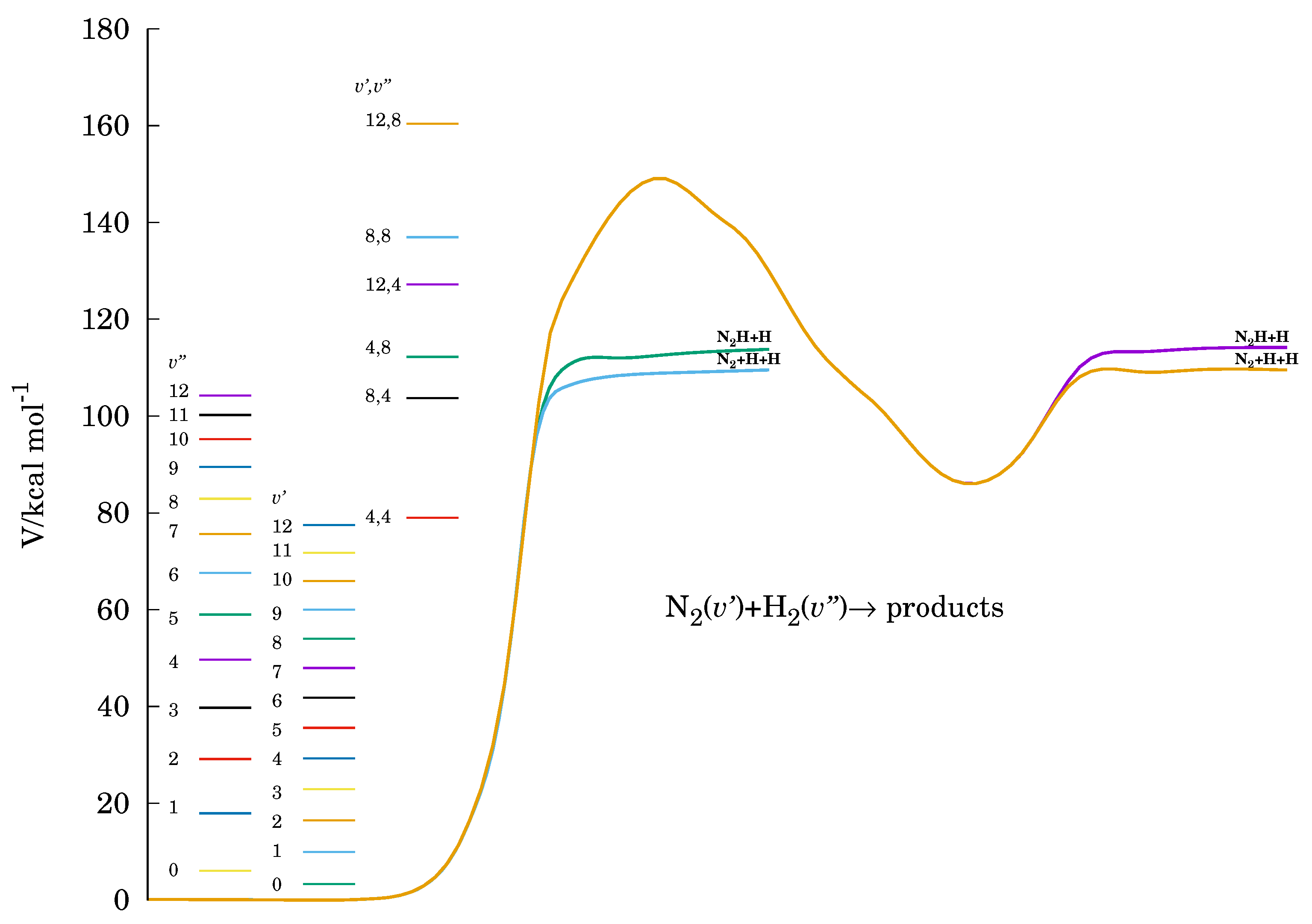
Although all the possible reactive channels were considered in the calculations (see later), only the above products were obtained in the energy interval covered in this study. Figure 1 shows a contour plot of the PES for the title reaction. Isoenergy lines identifying the output channels and are also represented. Notice there are direct links between the reactants and the two product valleys here obtained, without passing through the four body moiety configuration region. This is also illustrated in Figure 2.
For molecular dynamic calculations, the quasiclassical trajectories method was used [20,21]. In trajectory calculations, we utilized an adapted version of the VENUS96 [22] code, coupled to the PES and making the appropriate assignment of all possible reactive channels (see Reference [19]). For the problems of interest, mentioned in the Introduction, the chosen relative diatom–diatom translational energy covered the range ; while the selected initial vibrational quantum numbers for and were and , respectively. Figure 2 depicts a relaxed reaction path from the interaction potential together with the vibrational energy levels here considered. The corresponding energies for some combination of both reactants vibrationally excited were also included.
Additional calculations for translational energies in the interval were also carried out for combinations ; and to improve the fitting of the excitation functions. Exploratory calculations were also carried with one or both reactants rotationally excited. Changes produced in the reaction probability due to the rotational effects are similar to the corresponding increase in translational energy. This subject will be addressed in future studies. Here, the initial rotational quantum numbers of the collision partners were fixed at the ground level and are omitted heretofore.
The determination of the step size for numerical integration was performed by trial and error based on accuracy requirements. A value of was found to be sufficient to warrant the conservation of energy to more than 1 part in . In turn, the diatomic–diatomic initial separation was fixed at 15 Å, a value sufficiently large to make the interaction negligible. In turn, the maximum impact parameter , which leads to the r reactive channel [23], was obtained following the usual procedure by computing batches of 800 trajectories for fixed values of b [15,19,24] and decreasing its value until a reactive trajectory was obtained. This procedure should allow for accuracy in of about Å; the calculated values are reported in Table 1. Batches of 10,000 trajectories were then carried out for each translational energy and vibrational combination. Such a number of trajectories were found to be enough to yield reactive cross-sections with an error of typically a few percent.
The specific initial-state reactive cross-section for the channel r is calculated as:
with the corresponding uncertainties:
where is the number of trajectories ending in the corresponding configurations for products r in a total . For a given energetic combination of reactants, the reactive cross-section and the corresponding error for a specific output channel were calculated from relations (6) and (7). In this work, we adopted the following numerical criteria: when the obtained value of is smaller than seven times its corresponding error, the cross-section is negligibly small and the channel is considered as closed.
From the specific initial-state reactive cross-section and assuming a Maxwell–Boltzmann distribution over the translational energy , the specific initial-state reactive thermal rate coefficients are obtained as
where is the Boltzmann constant, is the reduced mass of the colliding molecules, T is the temperature in kelvin, and the electronic degeneracy factor [25,26]. As the electronic states of and the collision partners are singlets, assumes the value of 1.
Classical calculations permit molecular systems in configurations with the vibrational energy below their corresponding quantum minimal value or zero-point energy (ZPE). This ZPE leakage of the classical calculations can be eventually corrected after the trajectories integration. In this work, we selected the intermediate vibrational energy quantum mechanical threshold () [27] to correct the ZPE problem. In the case of the approximation, trajectories ending in products with vibrational energy below a chosen fraction (here, 1/2) of the corresponding ZPE are considered as nonphysical and hence discarded [28]. As the number of rejected trajectories is relatively small, the calculation of new trajectories to replace them can be avoided [28].
3. Results
3.1. Specific Initial-State Reactive Probability and Cross-Section
Table 1 summarized some trajectory results for selected initial states. Within the criteria used to define the closed channels, for the energy range covered in this study, the products formed were only (reaction (4)) and (reaction (5)). However, for the initial vibrational quantum number of less than or equal to 3, both channels remained closed for all studied relative translational energies, independent of the vibrational energies of the , while, in general, the reactive cross-section for formation was negligible. Thus, except when explicitly mentioned, the following discussion refers to reaction (4).
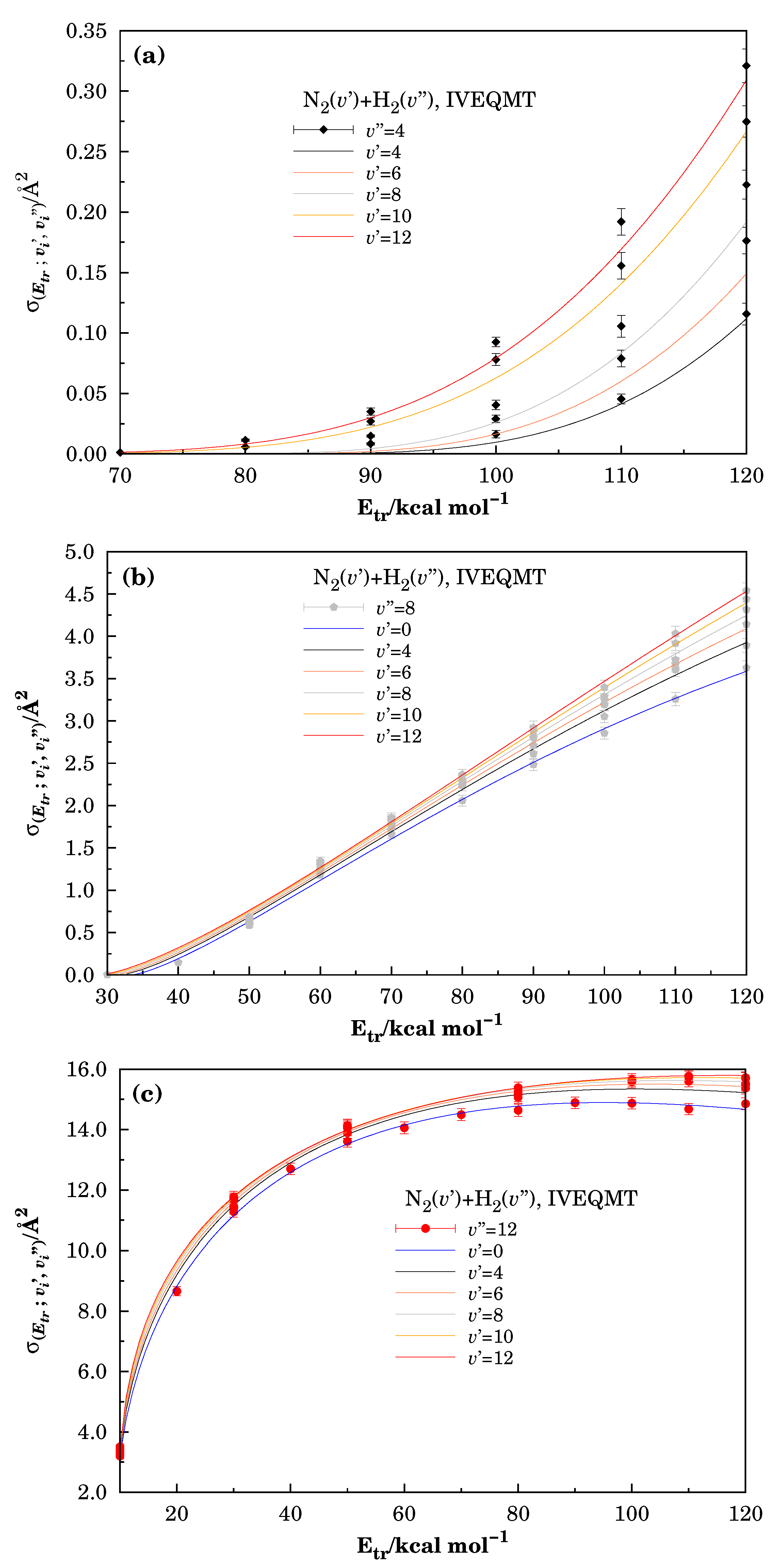
The calculated values of the specific initial-state reactive cross-section for the formation, according to the approach, are collected in Table 2, while Figure 3 shows the calculated points for the specific initial-state reactive cross-section for the formation vs. translational energy together with the associated error bars for the IVEQMT approximation.
In Figure 3, the vibrational excitation of the molecule increases from Panel (a) to Panel (c) (showing examples of small, middle, and high vibrational excitation for the molecule). Note that the threshold for the reaction (related to the translational energy) decreases and the excitation function increases with the vibrational energy of the molecule (in our calculations, we considered the reactive channel open, when the initial-state reactive cross-section was, at least, 6 times greater than the calculated error). Note also that curves in Panel (a) of Figure 3 have values two orders lower than curves in Panel (c). This means that the level of the reactive cross-section depends on the vibrational excitation.
For , the reactive channel remains closed (the threshold for vibrational energy), inside the considered intervals for the translational energies and vibrational energies of the . In the case of , the reaction channel (4) opens for the vibrational quantum numbers of the that satisfy the conditions (determining the total vibrational energy threshold for these cases). The described situation explains the small values of specific initial-state reactive cross-section reported in Panel (a), where graphics corresponding to vibrational combinations near the mentioned vibrational energy threshold have been shown.
All panels in Figure 3 show that the vibrational excitation of the molecule leads to small increases in the initial-state reactive cross-section as expected from the great difference between the vibrational quanta of both molecules.
The analytical representation of the initial-state reactive excitation functions for vibrational levels included in Figure 3 follows the function [29]:
where the parameters , , n, and were here expressed as the linear functions of the variable
where is the vibrational energy of the used as reference. For Panels (b) and (c), the reference is the energy of the level , while in Panel (a), the fitting for the vibrational quantum numbers takes as reference the energy for level and for the case of vibrational quantum number , the energy reference corresponds to the level .
The coefficients in the linear Equations (10)–(13) are reported in Table S1 of the Supplementary Material (SM). They vary with the vibrational energy of the molecule. We verified that the model works for all intermediate vibrational excitation of both molecules inside the intervals and . Additional figures for the excitation functions of other vibrational combinations were included in the Figure S1 in the SM.
In all cases, the initial-state reactive cross-sections are increasing functions of initial translational and vibrational energies. For higher translational energies, the interactions between molecules during collisions are stronger because they could be closer, while the increase in the initial vibrational energies produces greater deformations of the excited molecule, leading to bigger collision areas and small increases in the quadrupole moment. The competition with other reactive channels produces in all cases the formation of a plateau. The onset of this behavior strongly depends on the vibration energy of the . While in Panel (c), it is observed after a translational energy of 60–70 in Panels (a) and (b), it appears after 140–160 . Despite the large values of the relative translational energies, the initial-state cross-sections for reaction have relatively small values, corresponding to the nonpolar character of both colliding molecules.
As mentioned in the first paragraph of this section, the reactive channel (5) is also opened with very small probabilities. To make a comparison with the reactive channel (4), the additional trajectories were calculated for . The calculated specific initial-state reactive cross-sections are displayed in Figure S4 of the SM. These values are at least 40 times smaller than the corresponding for channel (4) for the same energetic initial conditions. Moreover, as was indicated in [19], the molecule, as a rule, immediately produces ; thus, it is possible to consider the reaction (5) as a slight contribution to the atomic hydrogen formation.
3.2. Energetic Features in the Reactive Molecular Ensemble
The discussion in the previous subsection points out the efficiency of the vibrational excitation of the molecule to produce the increase in reaction (4). In particular, it was demonstrated that for vibrational quantum numbers of the , the reaction (4) allows for translational energies greater than a determined value even when the is in the ground vibrational state. Under the conditions of the present work, as was shown in Figure 1 and Figure 2, reactive trajectories could also avoid the minimum of the PES and go directly to the products. Thus, calculations for these cases show that the barrier for the reaction (4) is determined by the energy difference between products and reactants in their ground electronic states. In all cases, the vibrational energy is essentially used to overpass that barrier and break the chemical bond.
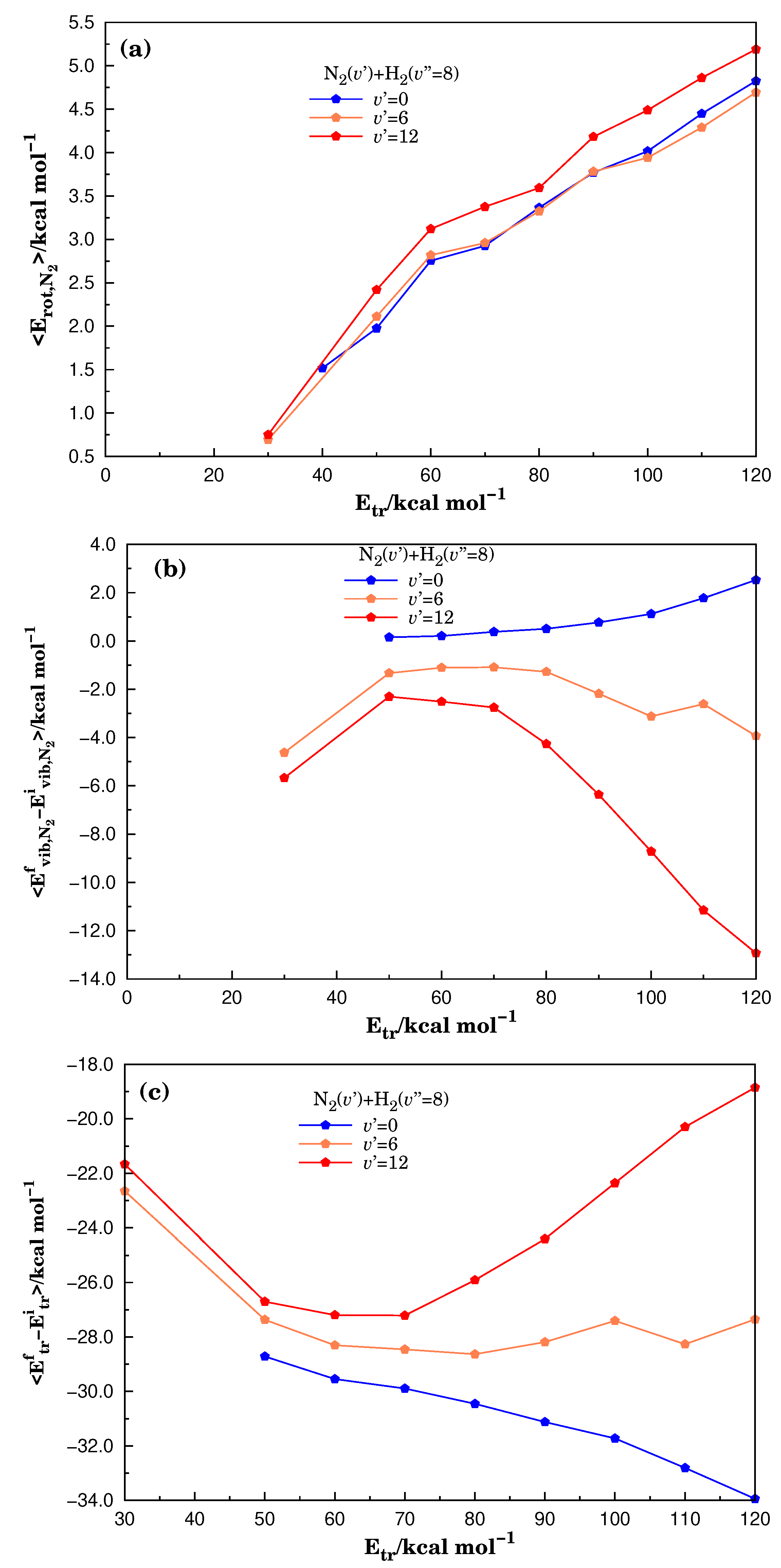
Panel (a) of Figure 4 shows the arithmetic means of the energies corresponding to rotational degrees of freedom of the after reaction (4), while Panel (b) of the same figure reports the difference in the vibrational degrees of freedom of the in the products of the reaction (4) with their initial values. Finally, Panel (c) shows the difference between the arithmetic mean of the relative translational energy of the products and the value of the initial relative translational energy of reactants.
Panel (a) of Figure 4 shows an increase in the rotational energy mean value in the interval with the increment of the relative translational energy. Such incremental results are practically independent of the vibrational energy of the molecule; thus, it is determined by the T-R processes. The observed slight increment in the rotational energy of the with the quantum number in the panel is probably due to the variation of inertial molecular properties when the molecular oscillations grow.
From the analysis of Panel (b) of Figure 4, with the exception of the case (for which it is evident that it only has possibility to increase the vibrational energy), the figure shows a loss in the vibrational energy in the interval . This energy loss is conditioned, principally, by the V-T processes but also includes a small transfer of energy used to break the chemical bond. This transferred energy warrants the small increase in the specific initial-state reactive cross-section with vibrational energy reported in Figure 3.
Following the previous analysis, one may rationalize that the energy associated with the relative translational degrees of freedom and the vibrational energy of the are the principal energy sources for the chemical reaction. To carry out an approximated quantitative energy balance, we introduced the following expression obtained from the energy conservation for the different processes discussed above:
where is the initial relative translational energy; is the arithmetic means of translational energies corresponding to the reactive molecular ensemble after the chemical reaction (4); and are the initial vibrational energies of the and molecules before reaction, respectively; is the energy barrier for reaction (4) discussed above; and is the arithmetic mean of the internal energy (the sum of vibrational and rotational energies) after reaction (of course, the arithmetic means were obtained from the trajectory calculations reported in the present work).
From Panel (c) of Figure 4, for the considered initial energy conditions, the molecular reactive ensemble experiences a significant cooling. To compare the results in Figure 4 with others obtained for different initial conditions, Figures S2 and S3 were included in the SM. In these figures, similar curves were reported for the initial conditions corresponding to collisions of (Figures S2) and (Figure S3). The curves in Figure S2 show that the experiences a small excitation when it is initially in its ground vibrational state, while in other situations it shows a loss of vibrational energy. In turn, the curves for the rotational degrees of freedom show a higher excitation than those observed in Figure 4 for . On the other hand, the cooling process reported in the Panels (c) of both figures are less intensive in the case of Figure S2. Moreover, for the collisions , the molecular reactive ensemble is slightly heated.
The comparison of Figure S3 with the other reported cases in Figure 4 and Figure S2 leads to the conclusion that when the reactive processes diminish, the relaxation processes are more relevant, leading to the highest rotational excitation, a stronger loss of vibrational energy, and, in general, the heating of the molecular reactive ensemble.
3.3. Specific Initial-State Reactive Thermal Rate Coefficients
From the specific initial-state reactive cross-sections Equation (9) and using the integral in Equation (8), one then obtains:
for the specific initial-state reactive thermal rate coefficients as a function of the temperature. Here, is the gamma function.
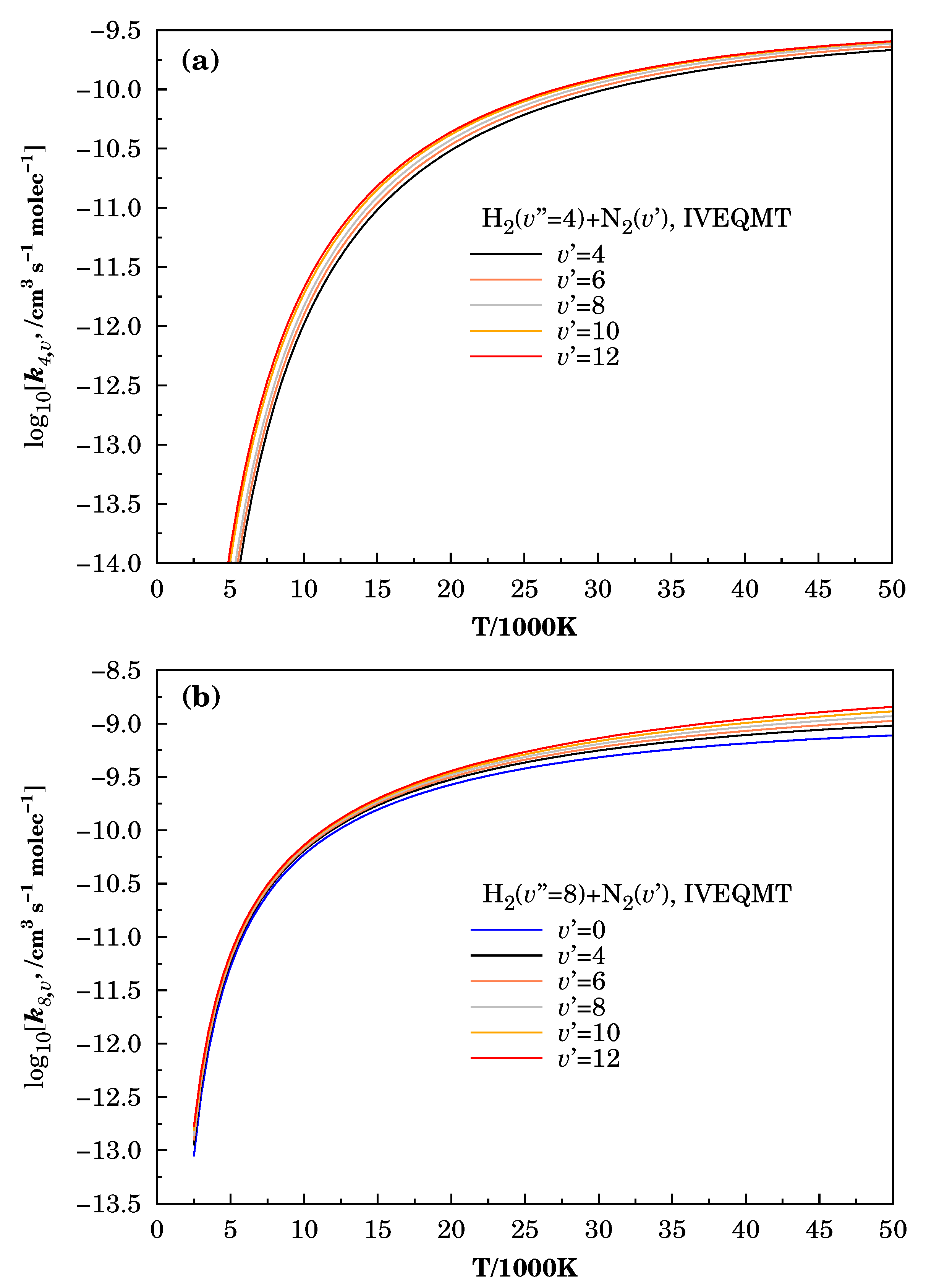
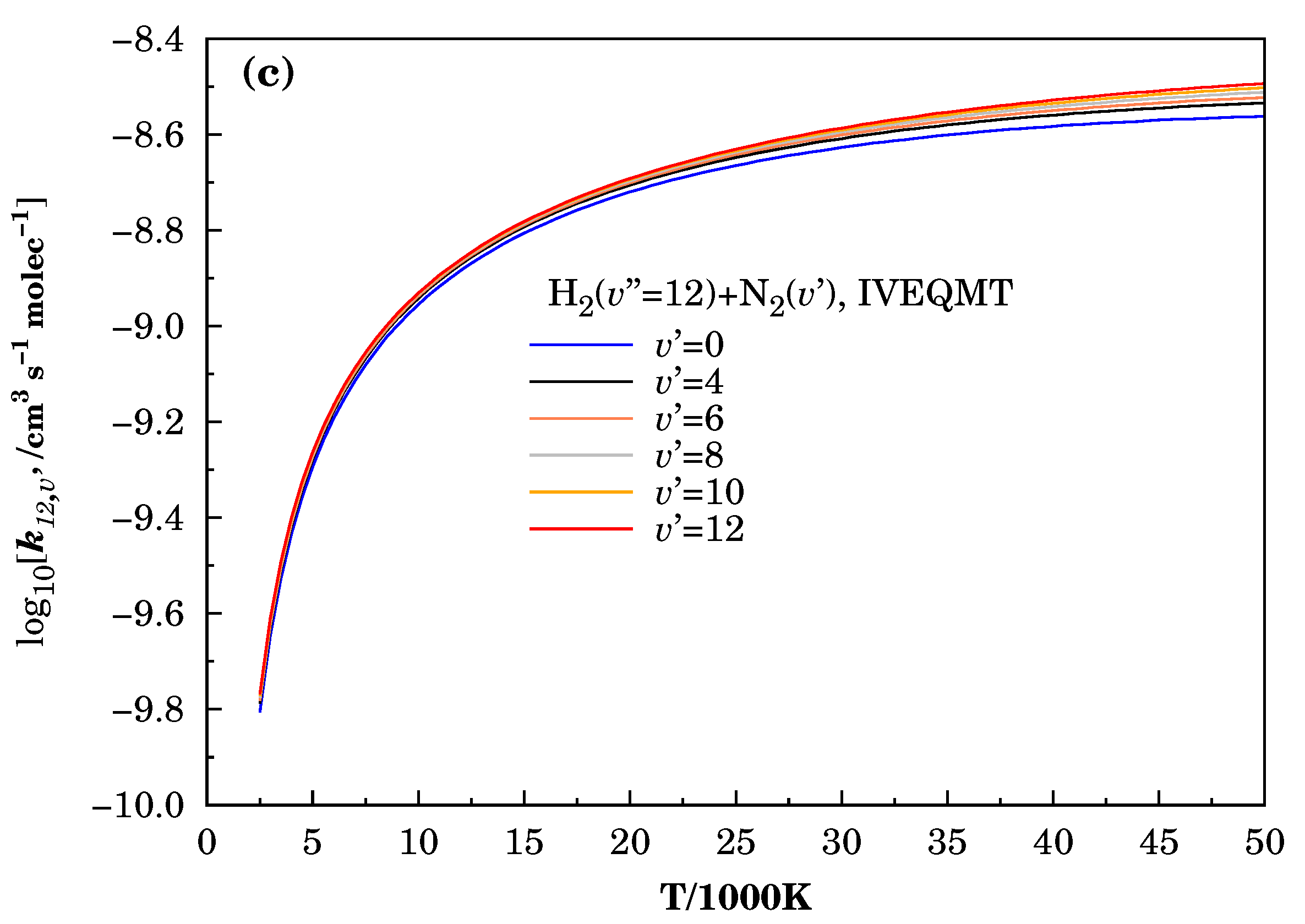
Figure 5 shows the curves of the specific initial-state reactive thermal rate coefficients for the same combinations of vibrational energies used before for the calculations of the corresponding cross-sections within the approaches IVEQMT. As expected from the specific initial-state cross-sections, they are increasing the functions of both vibrational and relative translational energies, and for a fixed vibrational excitation of the , the increase in the vibrational energy produces only slight growths in the specific initial-state thermal rate coefficients.
4. Discussion
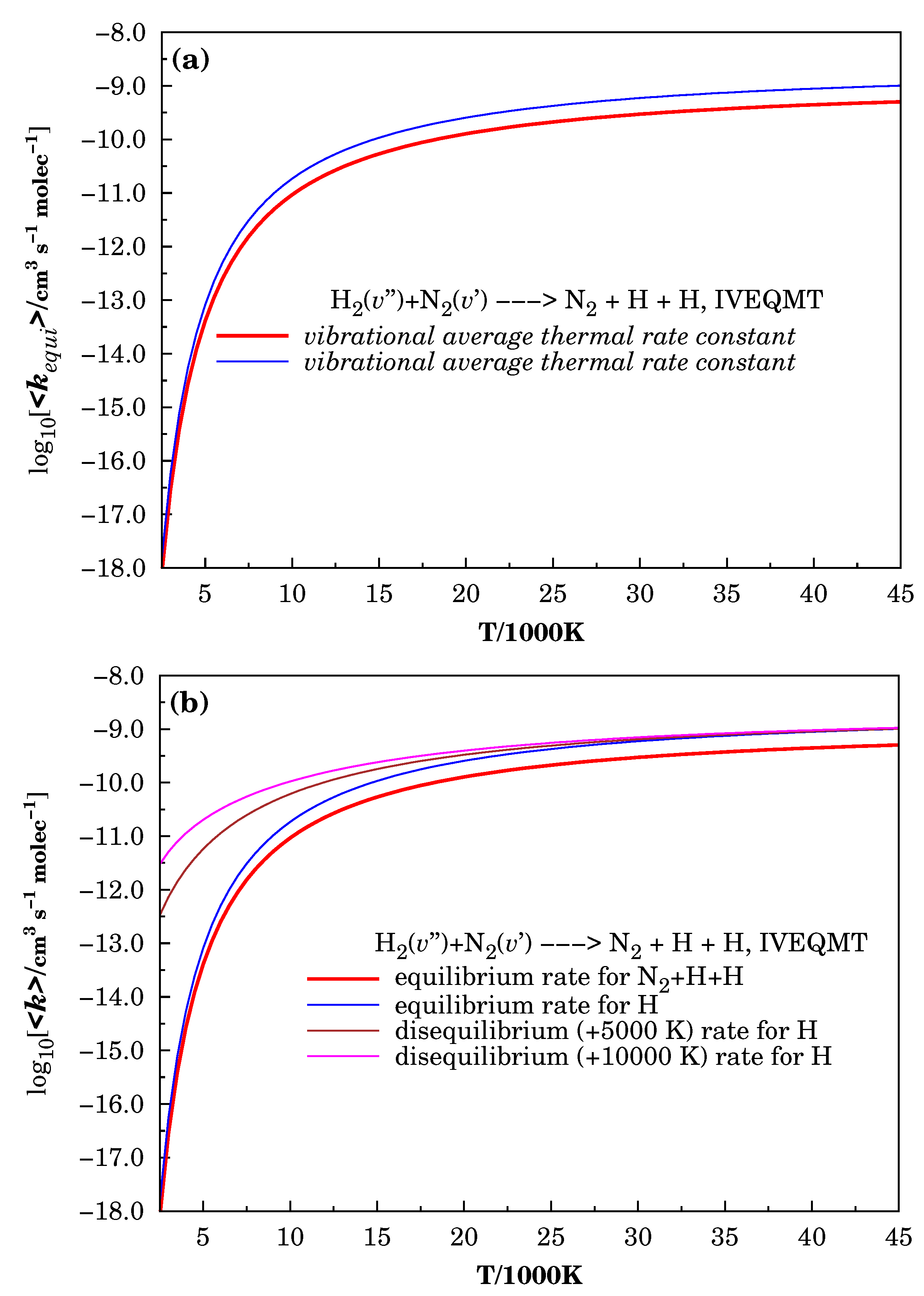
The analysis conducted in the subsection “Energetic Features in the Reactive Molecular Ensemble” points out that the majority of the initial energy combinations leading to reaction produce a decrease in the translational energy; thus, considering the values of the vibrational average thermal rate coefficients for the reaction (4) here reported, it could be necessary to account for this decrease in the translational energy to calculate the temperature of the gas behind the shock wave in the modeling works devoted to the hypersonic flights.
Figure 6 shows the vibrational average thermal rate coefficients for different situations. The red curves in this figure corresponds to the vibrational average thermal rate coefficients for reaction (4) calculated using the equation
where and are the vibrational populations of and , respectively, while the blue curves represent the vibrational average thermal rate coefficients that determine the temporal variation of the H concentration following the equation:
Both curves experience a very rapid increase until the formation of a high-value plateau after the translational temperature of 15,000 K. Such temperatures, and higher, are obtained in the modeling studies reported in [1,2,3] for the shock wave produced by hypersonic objects in the atmosphere; thus, the formation of abundant atomic hydrogen considering the high chemical rate predicted by Equation (17) is a real possibility in the air around hypersonic objects.
In a recent paper [30], a QCT study of the reaction (18) for a wide range of temperatures ( 20,000) was presented. The total rate for the reaction (18) was reported, including also the weighted contribution of the
and surfaces. Independently of the quantitative differences which are expected for different systems, the qualitative similarities between the rate constants are remarkable.
As expected, from the rapid expansion of the gas behind the shock wave, high thermal nonequilibrium conditions in the gas layer near the moving objects are reported in the mentioned papers [1,2,3]. To make a qualitative evaluation of the effect of the disequilibrium conditions on the atomic hydrogen formation, the curves calculated considering the hypothetical hot Boltzmann distribution shifted in 5000 (brown curve) and 10,000 K (magenta curve) are represented in Panel (b) of the Figure 6. These curves show that, under nonequilibrium conditions, their plateau behavior is retained until the translational temperature around 10,000 , and the values of the average thermal rate coefficients for temperatures below 7500 are, at least, one order of magnitude higher than those obtained under thermal equilibrium conditions. Considering the high values of the average thermal rate coefficients in Equation (16), it is possible to expect the conversion of a considerable quantity of molecular hydrogen in atomic hydrogen that could enter important atmospheric reactions as (1)–(3), leading to a relatively significant atmospheric impact. Finally, we should mention that no comparison with experimental data was possible since, to our knowledge, no such data exist in the literature for the interval of temperature considered in the present work.
5. Conclusions
In this work, we have reported a quasiclassical dynamic study of the reaction for several vibrational states of the reactants () and a wide range of relative translational energy (). According to our calculation, the most probable product was . The formation of was also observed, with a negligible contribution to total reaction. As expected, the vibrational energy in the molecule has a determinant role in the dissociation process. The initial vibrational energy content in is partially used in dissociating the molecular hydrogen. The reaction excitation functions exhibit typical shapes for barrier-like processes, and the models were correspondingly presented. Specific initial-state thermal rate coefficients reaction and vibrationally averaged rate constants were reported. The relatively high values of these rates for the considered conditions indicate the role of the title reaction in atmospheric issues involving high temperatures might be important, particularly as a source of atomic hydrogen. Of course, such a verification requires considering the reaction in the kinetic models. The energetic analysis of the calculated trajectories points toward a cooling effect when transforming the molecular hydrogen in the dissociated form by collisions with vibrationally excited molecular nitrogen.
Supplementary Materials
The following are available at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/atmos12101349/s1, Table S1, Figures S1–S4.
Author Contributions
Conceptualization, J.d.D.G. and M.Y.B.; data curation, J.d.D.G.; writing—review and editing, J.d.D.G. and M.Y.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Data Availability Statement
The data presented in this study are available alongside the article and the Supplementary Material.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PES | Potential Energy Surface |
| QCT | Quasiclassical Trajectories |
| IVEQMT | Intermediate Vibrational Energy Quantum Mechanical Threshold |
| ZPE | Zero-Point Energy |
| V-T processes | Energy transfer from vibrational to translational degrees of freedom |
| T-R processes | nergy transfer from translational to rotational degrees of freedom |
References
- Keidar, M.; Kim, M.; Boyd, I.D. Electromagnetic Reduction of Plasma Density During Atmospheric Reentry and Hypersonic Flights. J. Spacecr. Rocket. 2008, 45, 445. [Google Scholar] [CrossRef] [Green Version]
- Josyula, E.; Suchyta, C.J.; Vedula, P.; Burt, J.M. Multiquantum Transitions in Oxygen and Nitrogen Molecules in Hypersonic Non-equilibrium Flows. J. Thermophys. Heat Transf. 2019, 33, 378–391. [Google Scholar] [CrossRef]
- Shoev, G.; Oblapenko, G.; Kunova, O.; Mekhonoshina, M.; Kustova, E. Validation of vibration-dissociation coupling models in hypersonic non-equilibrium separated flows. Acta Astronaut. 2018, 144, 147–159. [Google Scholar] [CrossRef]
- Koner, D.; Bemish, R.J.; Meuwly, M. Dynamics on Multiple Potential Energy Surfaces: Quantitative Studies of Elementary Processes Relevant to Hypersonics. J. Phys. Chem. A 2020, 124, 6255–6269. [Google Scholar] [CrossRef] [PubMed]
- Josyula, E. Oxygen Atoms’ Effect on Vibrational Relaxation of Nitrogen in Blunt-Body Flows. J. Thermophys. Heat Trans. 2001, 15, 106–115. [Google Scholar] [CrossRef]
- Kustova, E.V.; Oblapenko, G.P. Reaction and internal energy relaxation rates in viscous thermochemically non-equilibrium gas flows. Phys. Fluids 2015, 27, 016102. [Google Scholar] [CrossRef]
- Kustova, E.V.; Oblapenko, G.P. Mutual effect of vibrational relaxation and chemical reactions in viscous multitemperature flows. Phys. Rev. E 2016, 93, 033127. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.P. Chemistry of Atmospheres; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Jacob, D.J. Introduction to Atmospheric Chemistry; Princeton University Press: Princeton, NJ, USA, 1999. [Google Scholar]
- Mackenzie, F.; Mackenzie, J.A. Our Changing Planet; Prentice-Hall: Upper Saddle River, NJ, USA, 1995. [Google Scholar]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, J.R.F.; Troe, J.A. Evaluated kinetic and photochemical data for atmospheric chemistry. Supplement III. J. Phys. Chem. Ref. Data 1989, 18, 881. [Google Scholar] [CrossRef]
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Hampson, R.F., Jr.; Kerr, J.A.; Rossi, M.J.; Troe, J. Evaluated kinetic, photochemical and heterogeneous data for atmospheric chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 1997, 26, 521–1011. [Google Scholar] [CrossRef]
- Gordiets, B.; Ferreira, C.; Pinheiro, M.; Ricard, A. Self-consistent kinetic model of low-pressure N-2-H-2 flowing discharges: I. Volume processes. Plasma Sources Sci. Technol. 1998, 7, 363–378. [Google Scholar] [CrossRef]
- Gordiets, B.; Ferreira, C.M.; Pinheiro, M.J.; Ricard, A. Self-consistent kinetic model of low-pressure N2-H2 flowing discharges: II Surface processes and densities of N, H, NH3 species. Plasma Sources Sci. Technol. 1998, 7, 379–388. [Google Scholar] [CrossRef]
- de Dios Garrido, J.; Ballester, M.Y. Relaxation processes in non-reactive collisions of H2 and N2 at high translational energies. Mol. Phys. 2021, 119, e1831635. [Google Scholar] [CrossRef]
- Poveda, L.A.; Biczysko, M.; Varandas, A.J.C. Accurate ab initio based DMBE potential energy surface for the ground electronic state of N2H2. J. Chem. Phys. 2009, 131, 044309. [Google Scholar] [CrossRef]
- Varandas, A.J.C. Intermolecular and Intramolecular Potentials: Topographical Aspects, Calculation, and Functional Representation via A Double Many-Body Expansion Method. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; Chapter 2; pp. 255–338. [Google Scholar] [CrossRef]
- Biczysko, M.; Poveda, L.; Varandas, A. Accurate MRCI Study of Ground-State N2H2 Potential Energy Surface. Chem. Phys. Lett. 2006, 424, 46–53. [Google Scholar] [CrossRef] [Green Version]
- de Castro, D.G.; Poveda, L.A.; Crispim, L.W.S.; Ballester, M.Y. Quasi-Classical Trajectory Study of NH(3Σ−) + NH(3Σ−) Reactive Collisions. J. Phys. Chem. A 2019, 123, 9113–9122. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Porter, R.N.; Sharma, R.D. Exchange Reactions with Activation Energy. I. Simple Barrier Potential for (H, H2). J. Chem. Phys. 1965, 43, 3259–3287. [Google Scholar] [CrossRef]
- Karplus, M.; Porter, R.N.; Sharma, R.D. Dynamics of reactive collisions: The H + H2 exchange reaction. J. Chem. Phys. 1964, 40, 2033–2034. [Google Scholar] [CrossRef]
- Hase, W.L.; Duchovic, R.J.; Hu, X.; Komornik, A.; Lim, K.F.; Lu, D.H.; Peslherbe, G.H.; Swamy, K.N.; van de Linde, S.R.; Varandas, A.J.C.; et al. VENUS 96. Quantum Chem. Program Exch. Bull. 1996, 16, 43. [Google Scholar]
- Levine, R.D. Molecular Reaction Dynamics; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Garrido, J.d.D.; Ellakkis, S.; Ballester, M.Y. Relaxation of Vibrationally Excited OH Radical by SO. J. Phys. Chem A. 2019, 123, 8994–9007. [Google Scholar] [CrossRef]
- Truhlar, D.G. Multiple Potential Energy Surfaces for Reactions of Species in Degenerate Electronic States. J. Chem. Phys. 1972, 56, 3189–3190. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Newton, M.D. Comment on “Multiple Potential Energy Surfaces for Reactions of Species in Degenerate Electronic States” by D. G. Truhlar. J. Chem. Phys. 1972, 56, 3191–3192. [Google Scholar] [CrossRef]
- Varandas, A.J.C. Excitation function for H+O2: A study of zero-point energy effects and rotational distributions in trajectory calculations. J. Chem. Phys. 1993, 99, 1076. [Google Scholar] [CrossRef]
- Varandas, A.J.C. A novel nonactive model to account for the leak of zero-point energy in trajectory calculations. Application to H+O2 reaction near threshold. Chem. Phys. Lett. 1994, 225, 18. [Google Scholar] [CrossRef]
- LeRoy, R.L. Relationships between Arrhenius activation energies and excitation functions. J. Phys. Chem. 1969, 73, 4338. [Google Scholar] [CrossRef]
- Veliz, J.C.S.V.; Koner, D.; Schwilk, M.; Bemish, R.J.; Meuwly, M. The N(4S)+O2(X3)→O(3P)+NO(X2Π) reaction: Thermal and vibrational relaxation rates for the 2A′, 4A′ and 2A″ states. Phys. Chem. Chem. Phys. 2020, 22, 3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Contour plot of the DMBE PES for the reaction (black lines). The N–N bond distance, the , and the angles are partially relaxed. Red line contours stand for the dissociation limit, while blue line contours correspond to the channel.
Figure 1.
Contour plot of the DMBE PES for the reaction (black lines). The N–N bond distance, the , and the angles are partially relaxed. Red line contours stand for the dissociation limit, while blue line contours correspond to the channel.

Figure 2.
Energetics of a partially relaxed reaction path. Vibrational levels of the reactants and some combinations of both reactants vibrationally excited are also represented.
Figure 2.
Energetics of a partially relaxed reaction path. Vibrational levels of the reactants and some combinations of both reactants vibrationally excited are also represented.

Figure 3.
Specific initial-state reactive cross-section for the formation considering the reactive collisions inside the IVEQMT approach. In Panel (a) , in Panel (b) , and in Panel (c) .
Figure 3.
Specific initial-state reactive cross-section for the formation considering the reactive collisions inside the IVEQMT approach. In Panel (a) , in Panel (b) , and in Panel (c) .

Figure 4.
Comparison of the arithmetic mean of energies for different degrees of freedom in the reaction (4) within the IVEQMT approach versus translational energies when the reactants are the and the in the vibrational excited states . In Panel (a), the mean values of the rotational energy for the product are displayed. In Panel (b), the differences between the mean values of the vibrational energy for the after reaction and the corresponding initial value are shown. In Panel (c), the differences between mean values of the relative translational energy of the molecular system after reaction and the corresponding initial values are presented.
Figure 4.
Comparison of the arithmetic mean of energies for different degrees of freedom in the reaction (4) within the IVEQMT approach versus translational energies when the reactants are the and the in the vibrational excited states . In Panel (a), the mean values of the rotational energy for the product are displayed. In Panel (b), the differences between the mean values of the vibrational energy for the after reaction and the corresponding initial value are shown. In Panel (c), the differences between mean values of the relative translational energy of the molecular system after reaction and the corresponding initial values are presented.

Figure 5.
Specific initial-state reactive thermal rate coefficients for the formation considering the reactive collisions inside the IVEQMT approach. In Panel (a) , in Panel (b) , and in Panel (c) .
Figure 5.
Specific initial-state reactive thermal rate coefficients for the formation considering the reactive collisions inside the IVEQMT approach. In Panel (a) , in Panel (b) , and in Panel (c) .


Figure 6.
Calculated vibrational average thermal rate coefficients for studied processes. In Panel (a) for the formation (red curve) and atomic hydrogen formation (blue curve) considering a Boltzmann distribution for the population of vibrationally excited reactants at the calculated temperature. In Panel (b), together with the curves previously described in Panel (a), the vibrational average thermal rate coefficients for atomic hydrogen formation considering thermal disequilibrium conditions for the vibrational populations of reactants are shown (see text for description).
Figure 6.
Calculated vibrational average thermal rate coefficients for studied processes. In Panel (a) for the formation (red curve) and atomic hydrogen formation (blue curve) considering a Boltzmann distribution for the population of vibrationally excited reactants at the calculated temperature. In Panel (b), together with the curves previously described in Panel (a), the vibrational average thermal rate coefficients for atomic hydrogen formation considering thermal disequilibrium conditions for the vibrational populations of reactants are shown (see text for description).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the trajectory calculations for the reactive collisions + within the IVEQMT approach. in Å. Translational energies () are given in . The symbol “-” indicates reactive channels remain closed.
Table 1.
Summary of the trajectory calculations for the reactive collisions + within the IVEQMT approach. in Å. Translational energies () are given in . The symbol “-” indicates reactive channels remain closed.
| 70.0 | 4 | 4 | - | - | - | 4 | 6 | - | - | - |
| 80.0 | - | - | - | - | - | - | ||||
| 90.0 | - | - | - | 1.0 | 9996 | 27 | ||||
| 100.0 | 1.1 | 9986 | 43 | 1.1 | 9984 | 76 | ||||
| 110.0 | 1.1 | 9980 | 119 | 1.4 | 9982 | 128 | ||||
| 120.0 | 1.4 | 9970 | 182 | 1.5 | 9980 | 249 | ||||
| 70.0 | 4 | 8 | - | - | - | 4 | 10 | - | - | - |
| 80.0 | - | - | - | 0.5 | 9993 | 73 | ||||
| 90.0 | 0.8 | 9986 | 74 | 1.1 | 9990 | 71 | ||||
| 100.0 | 1.1 | 9985 | 110 | 1.1 | 9982 | 205 | ||||
| 110.0 | 1.6 | 9977 | 131 | 1.6 | 9971 | 193 | ||||
| 120.0 | 1.5 | 9971 | 314 | 1.5 | 9974 | 388 | ||||
| 40.0 | 4 | 12 | - | - | - | 8 | 0 | 1.6 | 9846 | 182 |
| 50.0 | - | - | - | 2.0 | 9786 | 456 | ||||
| 60.0 | - | - | - | 2.3 | 9944 | 710 | ||||
| 70.0 | 0.4 | 9999 | 25 | 2.4 | 9674 | 890 | ||||
| 80.0 | 0.7 | 9997 | 74 | 2.6 | 9664 | 937 | ||||
| 90.0 | 1.1 | 9991 | 92 | 2.7 | 9660 | 1048 | ||||
| 100.0 | 0.9 | 9976 | 363 | 2.5 | 9537 | 1387 | ||||
| 110.0 | 1.4 | 9977 | 311 | 2.6 | 9509 | 1459 | ||||
| 120.0 | 1.5 | 9973 | 453 | 2.6 | 9460 | 1616 | ||||
| 30.0 | 8 | 4 | - | - | - | 8 | 6 | 0.6 | 10,000 | 7 |
| 50.0 | 2.2 | 10,000 | 412 | 2.2 | 10,000 | 405 | ||||
| 60.0 | 2.3 | 9999 | 752 | 2.4 | 10,000 | 723 | ||||
| 70.0 | 2.4 | 9999 | 952 | 2.5 | 9999 | 902 | ||||
| 80.0 | 2.5 | 9998 | 1139 | 2.5 | 10,000 | 1163 | ||||
| 90.0 | 2.5 | 9997 | 1330 | 2.6 | 9995 | 1258 | ||||
| 100.0 | 2.6 | 9989 | 1436 | 2.6 | 9988 | 1469 | ||||
| 110.0 | 2.7 | 9982 | 1572 | 2.6 | 9987 | 1726 | ||||
| 120.0 | 2.6 | 9980 | 1830 | 2.6 | 9977 | 1946 | ||||
| 30.0 | 8 | 8 | 0.6 | 10,000 | 13 | 8 | 10 | 1.1 | 10,000 | 12 |
| 50.0 | 2.2 | 10,000 | 429 | 2.1 | 10,000 | 488 | ||||
| 60.0 | 2.4 | 10,000 | 711 | 2.4 | 10,000 | 716 | ||||
| 70.0 | 2.5 | 10,000 | 929 | 2.5 | 10,000 | 936 | ||||
| 80.0 | 2.5 | 9999 | 1167 | 2.5 | 10,000 | 1173 | ||||
| 90.0 | 2.7 | 9998 | 1224 | 2.7 | 9997 | 1256 | ||||
| 100.0 | 2.6 | 9996 | 1530 | 2.8 | 9998 | 1335 | ||||
| 110.0 | 2.7 | 9988 | 1623 | 2.7 | 9991 | 1709 | ||||
| 120.0 | 2.7 | 9975 | 1880 | 2.7 | 9989 | 1937 | ||||
| 10.0 | 8 | 12 | - | - | - | 12 | 0 | 2.8 | 9996 | 1297 |
| 20.0 | - | - | - | 3.4 | 9988 | 2381 | ||||
| 30.0 | 0.5 | 10,000 | 26 | 3.5 | 9980 | 2925 | ||||
| 40.0 | - | - | - | 3.6 | 9917 | 3093 | ||||
| 50.0 | 2.0 | 10,000 | 540 | 3.5 | 9862 | 3487 | ||||
| 60.0 | 2.5 | 10,000 | 684 | 3.6 | 9812 | 3388 | ||||
| 70.0 | 2.5 | 10,000 | 945 | 3.6 | 9767 | 3477 | ||||
| 80.0 | 2.5 | 10,000 | 1205 | 3.6 | 9712 | 3492 | ||||
| 90.0 | 2.8 | 9998 | 1185 | 3.5 | 9675 | 3743 | ||||
| 100.0 | 2.9 | 9995 | 1285 | 3.5 | 9684 | 3743 | ||||
| 110.0 | 2.7 | 9992 | 1759 | 3.5 | 9640 | 3677 | ||||
| 120.0 | 2.7 | 9997 | 1982 | 3.6 | 9696 | 3538 | ||||
| 10.0 | 12 | 4 | 3.0 | 10,000 | 1164 | 12 | 6 | 3.0 | 9999 | 1169 |
| 30.0 | 3.6 | 9995 | 2798 | 3.6 | 9995 | 2813 | ||||
| 50.0 | 3.6 | 9976 | 3405 | 3.6 | 9986 | 3411 | ||||
| 80.0 | 3.6 | 9943 | 3680 | 3.6 | 9976 | 3706 | ||||
| 100.0 | 3.7 | 9951 | 3607 | 3.7 | 9980 | 3632 | ||||
| 120.0 | 3.6 | 9924 | 3747 | 3.6 | 9955 | 3785 | ||||
| 10.0 | 12 | 8 | 2.9 | 9999 | 1255 | 12 | 10 | 3.0 | 9998 | 1213 |
| 30.0 | 3.6 | 9999 | 2867 | 3.7 | 9999 | 2722 | ||||
| 50.0 | 3.7 | 9995 | 3286 | 3.6 | 9995 | 3467 | ||||
| 80.0 | 3.4 | 9993 | 4184 | 3.5 | 9989 | 3958 | ||||
| 110.0 | 3.5 | 9992 | 4051 | 3.5 | 9989 | 4086 | ||||
| 120.0 | 3.6 | 9968 | 3800 | 3.6 | 9983 | 3854 | ||||
| 10.0 | 12 | 12 | 3.1 | 10,000 | 1161 | |||||
| 30.0 | 3.6 | 10,000 | 2890 | |||||||
| 50.0 | 3.6 | 9996 | 3452 | |||||||
| 80.0 | 3.5 | 9994 | 3996 | |||||||
| 110.0 | 3.6 | 9988 | 3869 | |||||||
| 120.0 | 3.6 | 9993 | 3852 |
Table 2.
Specific initial-state cross-sections for reaction + within the IVEQMT approach and the corresponding error, both in Å. Translational energies () are given in .
Table 2.
Specific initial-state cross-sections for reaction + within the IVEQMT approach and the corresponding error, both in Å. Translational energies () are given in .
| 4 | 4 | 90.0 | - | - | 4 | 6 | 90.0 | 0.0084 | 0.001 |
| 100.0 | 0.0163 | 0.003 | 100.0 | 0.0289 | 0.233 | ||||
| 110.0 | 0.0452 | 0.004 | 110.0 | 0.0789 | 0.233 | ||||
| 120.0 | 0.1120 | 0.009 | 120.0 | 0.1763 | 0.011 | ||||
| 4 | 8 | 80.0 | - | - | 4 | 10 | 80.0 | 0.0057 | 0.0006 |
| 90.0 | 0.0148 | 0.001 | 90.0 | 0.0270 | 0.003 | ||||
| 100.0 | 0.0418 | 0.004 | 100.0 | 0.0780 | 0.005 | ||||
| 110.0 | 0.1056 | 0.009 | 110.0 | 0.1556 | 0.011 | ||||
| 120.0 | 0.2226 | 0.012 | 120.0 | 0.2749 | 0.013 | ||||
| 4 | 12 | 40.0 | - | - | 8 | 0 | 40.0 | 0.1486 | 0.010 |
| 50.0 | - | - | 50.0 | 0.5855 | 0.026 | ||||
| 60.0 | - | - | 60.0 | 1.1866 | 0.043 | ||||
| 70.0 | 0.0012 | 0.0002 | 70.0 | 1.6647 | 0.053 | ||||
| 80.0 | 0.0114 | 0.001 | 80.0 | 2.0591 | 0.063 | ||||
| 90.0 | 0.0350 | 0.003 | 90.0 | 2.4846 | 0.072 | ||||
| 100.0 | 0.0925 | 0.004 | 100.0 | 2.8555 | 0.070 | ||||
| 110.0 | 0.1920 | 0.011 | 110.0 | 3.2584 | 0.078 | ||||
| 120.0 | 0.3210 | 0.014 | 120.0 | 3.6278 | 0.082 | ||||
| 8 | 4 | 50.0 | 0.6264 | 0.030 | 8 | 6 | 50.0 | 0.6158 | 0.029 |
| 60.0 | 1.2500 | 0.044 | 60.0 | 1.3083 | 0.046 | ||||
| 70.0 | 1.7228 | 0.053 | 70.0 | 1.7712 | 0.056 | ||||
| 80.0 | 2.2368 | 0.062 | 80.0 | 2.2835 | 0.062 | ||||
| 90.0 | 2.6123 | 0.067 | 90.0 | 2.6729 | 0.070 | ||||
| 100.0 | 3.0530 | 0.074 | 100.0 | 3.1234 | 0.075 | ||||
| 110.0 | 3.6067 | 0.083 | 110.0 | 3.6703 | 0.080 | ||||
| 120.0 | 3.8935 | 0.083 | 120.0 | 4.1422 | 0.084 | ||||
| 8 | 8 | 30.0 | 0.0014 | 0.0004 | 8 | 10 | 30.0 | 0.0045 | 0.001 |
| 50.0 | 0.6523 | 0.030 | 50.0 | 0.6761 | 0.029 | ||||
| 60.0 | 1.2866 | 0.046 | 60.0 | 1.2956 | 0.046 | ||||
| 70.0 | 1.8240 | 0.057 | 70.0 | 1.8378 | 0.057 | ||||
| 80.0 | 2.2914 | 0.063 | 80.0 | 2.3031 | 0.063 | ||||
| 90.0 | 2.8037 | 0.075 | 90.0 | 2.8773 | 0.075 | ||||
| 100.0 | 3.2505 | 0.076 | 100.0 | 3.2881 | 0.082 | ||||
| 110.0 | 3.7214 | 0.084 | 110.0 | 3.9175 | 0.086 | ||||
| 120.0 | 4.3164 | 0.089 | 120.0 | 4.4410 | 0.090 | ||||
| 8 | 12 | 10.0 | - | - | 12 | 0 | 10.0 | 3.1958 | 0.082 |
| 20.0 | - | - | 20.0 | 8.6574 | 0.154 | ||||
| 30.0 | 0.0020 | 0.0004 | 30.0 | 11.2792 | 0.175 | ||||
| 40.0 | - | - | 40.0 | 12.6985 | 0.189 | ||||
| 50.0 | 0.6785 | 0.028 | 50.0 | 13.6073 | 0.185 | ||||
| 60.0 | 1.3430 | 0.049 | 60.0 | 14.0585 | 0.195 | ||||
| 70.0 | 1.8555 | 0.057 | 70.0 | 14.4943 | 0.197 | ||||
| 80.0 | 2.3660 | 0.064 | 80.0 | 14.6393 | 0.198 | ||||
| 90.0 | 2.9192 | 0.079 | 90.0 | 14.8886 | 0.190 | ||||
| 100.0 | 3.3957 | 0.087 | 100.0 | 14.8748 | 0.190 | ||||
| 110.0 | 4.0317 | 0.087 | 110.0 | 14.6792 | 0.190 | ||||
| 120.0 | 4.5405 | 0.091 | 120.0 | 14.8566 | 0.199 | ||||
| 12 | 4 | 10.0 | 3.2911 | 0.090 | 12 | 6 | 10.0 | 3.3056 | 0.090 |
| 30.0 | 11.3978 | 0.183 | 30.0 | 11.4588 | 0.183 | ||||
| 50.0 | 13.8968 | 0.193 | 50.0 | 13.9073 | 0.193 | ||||
| 80.0 | 15.0690 | 0.197 | 80.0 | 15.1252 | 0.196 | ||||
| 100.0 | 15.5895 | 0.207 | 100.0 | 15.6519 | 0.207 | ||||
| 120.0 | 15.3727 | 0.198 | 120.0 | 15.4803 | 0.198 | ||||
| 12 | 8 | 10.0 | 3.31614 | 0.087 | 12 | 10 | 10.0 | 3.43036 | 0.092 |
| 30.0 | 11.6741 | 0.184 | 30.0 | 11.7080 | 0.191 | ||||
| 50.0 | 14.1396 | 0.202 | 50.0 | 14.1229 | 0.193 | ||||
| 80.0 | 15.2055 | 0.179 | 80.0 | 15.2489 | 0.188 | ||||
| 110.0 | 15.6025 | 0.190 | 110.0 | 15.7420 | 0.189 | ||||
| 120.0 | 15.5214 | 0.198 | 120.0 | 15.7183 | 0.198 | ||||
| 12 | 12 | 10.0 | 3.50514 | 0.096 | |||||
| 30.0 | 11.7666 | 0.184 | |||||||
| 50.0 | 14.0604 | 0.193 | |||||||
| 80.0 | 15.3876 | 0.188 | |||||||
| 110.0 | 15.7715 | 0.198 | |||||||
| 120.0 | 15.6944 | 0.198 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Garrido, J.d.D.; Ballester, M.Y. A Theoretical Study of the N2 + H2 Reactive Collisions for High Vibrational and Translational Energies. Atmosphere 2021, 12, 1349. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12101349
AMA Style
Garrido JdD, Ballester MY. A Theoretical Study of the N2 + H2 Reactive Collisions for High Vibrational and Translational Energies. Atmosphere. 2021; 12(10):1349. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12101349
Chicago/Turabian StyleGarrido, Juan de Dios, and Maikel Yusat Ballester. 2021. "A Theoretical Study of the N2 + H2 Reactive Collisions for High Vibrational and Translational Energies" Atmosphere 12, no. 10: 1349. https://0-doi-org.brum.beds.ac.uk/10.3390/atmos12101349
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.