Peatlands as Filters for Polluted Mine Water?—A Case Study from an Uranium-Contaminated Karst System in South Africa—Part II: Examples from Literature and a Conceptual Filter Model

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Project Background
1.2. Case Studies of Natural U-Accumulation in Peat
2. Conceptual Model for Peat as a Uranium Filter
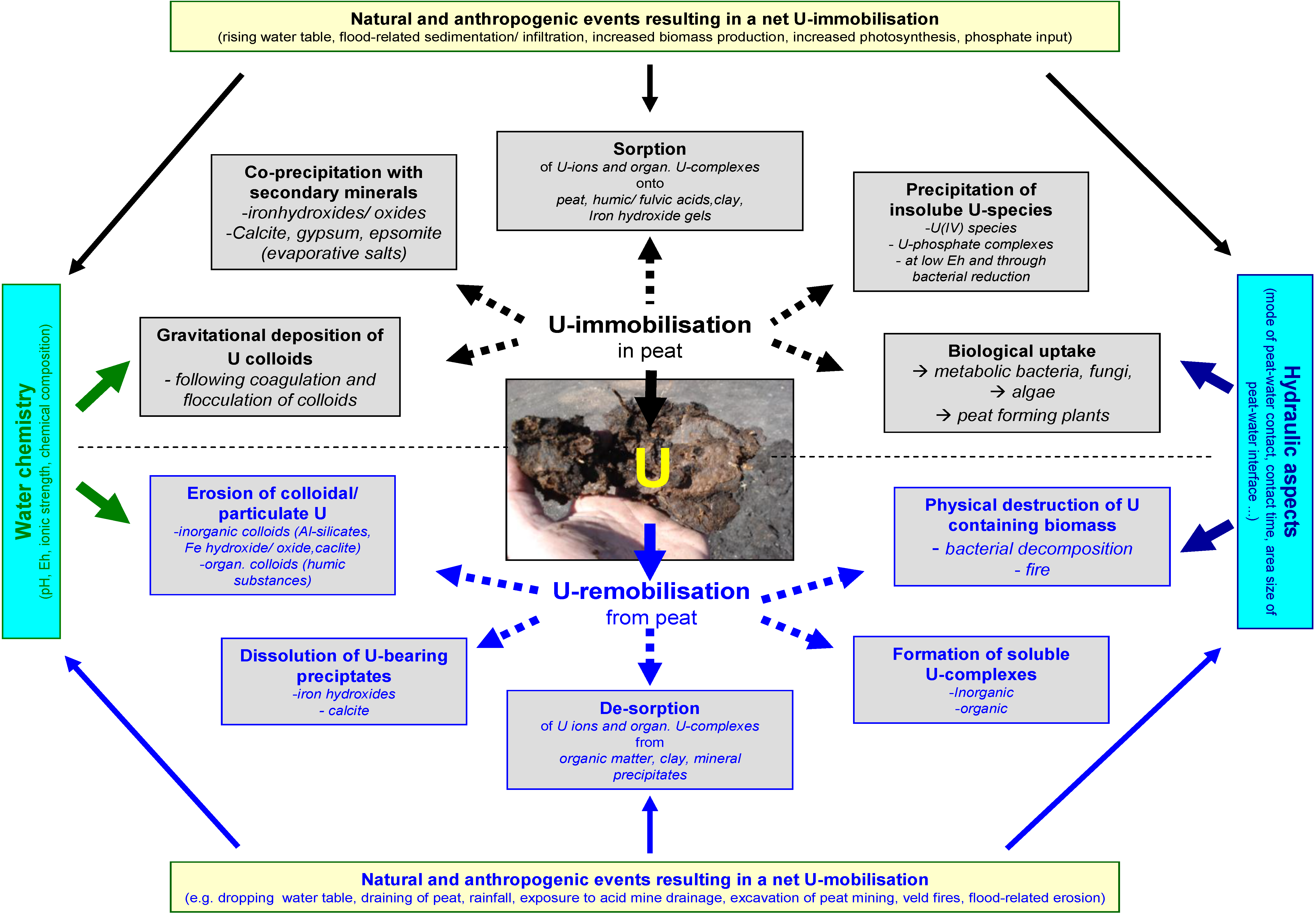
2.1. The Chemical Filter Component
2.1.1. Mechanisms of U-Immobilization
2.1.2. Mechanisms of U-Remobilization
2.1.3. Summary of the Chemical Sub-Model
- (i)
- the identification of physico-chemical processes that lead to the immobilization and remobilization of U in/from peat;
- (ii)
- the identification of factors such as water chemistry (pH, Eh, ionic strength etc.) and hydraulic aspects that govern the occurrence and intensity of the above processes; and
- (iii)
- the determination of consequences natural and anthropogenic events in peatlands may have for the attenuation of U, with special reference to the site specific conditions at the GMB peatland.

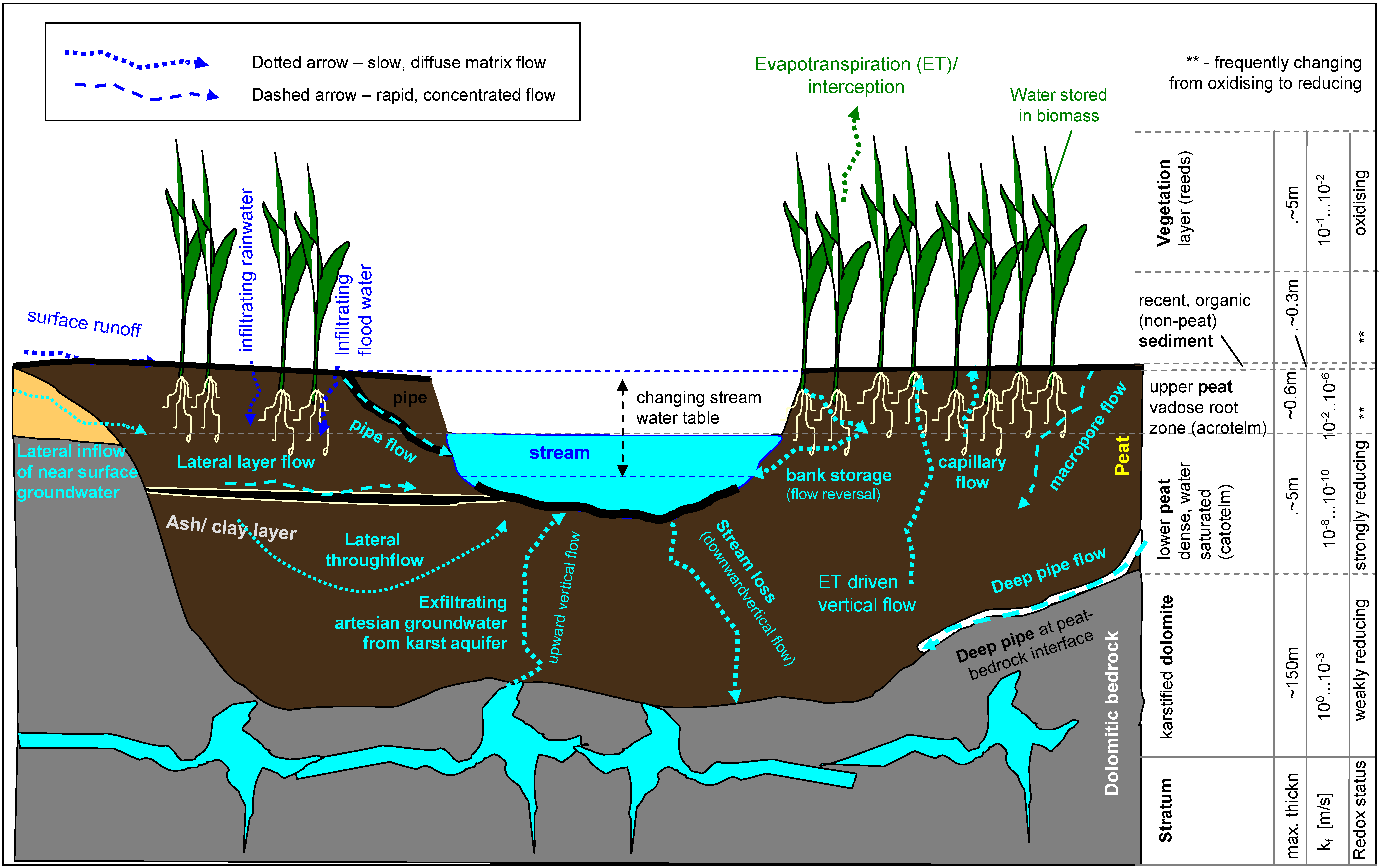
2.2. The Hydraulic Filter Component
2.2.1. Contaminated Surface Water Flows Past the Peat Deposit Without Penetrating to Any Significant Degree (Skimming Over the Surface):
2.2.2. Contaminated Surface Water Infiltrates into the Peat (Downward Matrix Flow)
2.2.3. Rapid, Downward Concentrated Flow through Peat (Non-Matrix Macropore- and Pipe-Flow)
2.2.4. Lateral Water Flow through the Peat (Through-Flow)
2.2.5. Upward (Non Gradient) Water Flow through Overlying Peat
2.2.6. Significance of the Hydraulic Processes for the Chemical Model Component
2.2.7. Summary of the Hydraulic Model Component
- -
- stream water infiltrating from the stream channel into the bank peat;
- -
- exfiltrating groundwater moving laterally from shallow aquifers into peat at the edges of the wetland;
- -
- surface runoff from the surrounding catchment entering the margin of the peatland;
- -
- dolomitic groundwater exfiltrating under artesian pressure from the underlying karst aquifer; and
- -
- rainwater directly infiltrating into the acrotelm.

3. Course of Proposed Field and Laboratory Investigations
4. Summary and Conclusions
Acknowledgements
References
- Winde, F.; Erasmus, E. Peatlands as filters for polluted mine water?—A case study from an uranium-contaminated karst system in South Africa. Part I: Hydrogeological setting and U-fluxes. Water 2010, 3, 291–322. [Google Scholar] [CrossRef]
- Owen, D.E.; Otton, J.K. Mountain wetlands: Efficient uranium filters—potential impacts. Ecol. Eng. 1995, 5, 77–93. [Google Scholar] [CrossRef]
- Schöner, A. Hydrogeochemische Prozesse der Uranfixierung in natürlichen Wetlands und deren Anwendbarkeit in der ‘passiven’ Wasserbehandlung (Hydrogeochemical Processes of Uranium Fixation in Natural Wetlands and Their Applicability in Passive’ Water Treatment). Dissertation, Chemisch-Geowissenschaftliche Fakultät, Friedrich-Schiller-Universität Jena, Jena, Germany, 2006; p. 373. [Google Scholar]
- Smuts, W.J. Characteristics of South African Peats and Their Potential Exploitation. PhD Thesis, Faculty of Science, University of Pretoria, Pretoria, South Africa, 1997; p. 222. [Google Scholar]
- Kochenov, A.V.; Zinevyev, V.V.; Lovaleva, S.A. Some features of the accumulation of uranium in peat bogs. Geochem. Int. 1965, 2, 65–70. [Google Scholar]
- Lopatkina, A.P. Conditions of accumulation of uranium in peat. Geochem. Int. 1967, 4, 577–588. [Google Scholar]
- Szalay, A. Accumulation of uranium and other micrometals in coal and organic shales and the role of humic acids in these geochemical enrichments. Ark. Mineral. Geol. 1974, 5, 23–36. [Google Scholar]
- Moore, G.W. Extraction of uranium from aqueous solution by coal and some other materials. Econ. Geol. 1954, 49, 652–658. [Google Scholar] [CrossRef]
- Coggins, A.M.; Jennings, S.G.; Ebinghaus, R. Accumulation rates of the heavy metals lead, mercury and cadmium in ombrotrophic peatlands in the west of Ireland. Atmosph. Env. 2005, 40, 260–278. [Google Scholar] [CrossRef]
- Farmer, J.G.; Anderson, P.; Cloy, J.M.; Graham, M.C.; MacKenzie, A.B.; Cook, G.T. Historical accumulation rates of mercury in four scottish ombrotrophic peat bogs over the past 2,000 years. Sci. Total Environ. 2009, 407, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- Gosset, T.; Trancart, J.L.; Thevenot, D.R. Batch metal removal by peat: Kinetics and thermodynamics. Water Res. 1986, 20, 21–26. [Google Scholar] [CrossRef]
- Denham, M.; Looney, B. Recommended Amendment Mixture for in-Situ Treatment of Waste, Management Unit Groundwater, Ashtabula Closure Project; Report to the US Department of Energy no. 2004, WSRC-TR-2004-00185; Savannah River Site: Aiken, SC, USA, 2004. [Google Scholar]
- Nakashima, S.; Disnar, J.R.; Perruchot, A.; Trichet, J. Experimental study of mechanisms of fixation and reduction of uranium by sedimentary organic matter under diagenetic or hydrothermal conditions. Geochim et Cosmochim Acta 1984, 48, 2321–2329. [Google Scholar] [CrossRef]
- Brown, P.A.; Gill, S.A.; Allen, S.J. Metal removal from wastewater using peat. Water Res. 2000, 34, 3907–3916. [Google Scholar] [CrossRef]
- Winde, F. Urankontamination von Fließgewässern—Prozessdynamik, Mechanismen und Steuerfaktoren. Untersuchungen zum Transport von gelöstem Uran in bergbaulich gestörten Landschaften unterschiedlicher Klimate. (Uranium Contamination of Streams—Process Dynamics, Mechanisms and Governing Factors. Investigations into the Transfer of Dissolved Uranium in Mining-Impacted Landscapes of Different Climatic Conditions); Habilitationsschrift; Chemisch-Geowissenschaftliche Fakultät, Friedrich-Schiller-Universität Jena: Jena, Germany, 2003; p. 375. [Google Scholar]
- Titayeva, N.A. Association of radium and uranium with peat. Geochem. Int. 1967, 4, 1168–1174. [Google Scholar]
- Idiz, E.F.; Carlisle, D.; Kaplan, I.R. Interaction between organic matter and trace metals in a uranium rich bog, Kern County, California, USA. Appl. Geochem. 1986, 1, 573–590. [Google Scholar] [CrossRef]
- Ibarra, J.V.; Osacar, J.; Gavilan, J.M. Retention of metallic cations by lignites and humic acids. Fuel 1979, 58, 827–830. [Google Scholar] [CrossRef]
- Crancon, P.; Pili, E.; Charlet, L. Uranium facilitated transport by water-dispersible colloids in field and soil columns. Sci. Total Environ. 2010, 408, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Krepelova, A.; Sachs, S.; Berhard, G. Uranium(VI) sorption onto kaolinite in the presence and absence of humic acid. Radiochim. Acta. 2006, 94, 825–833. [Google Scholar]
- Feng, X.; Simpson, A.J.; Simpson, M.J. Chemical and mineralogical controls on humic acid sorption to clay mineral surfaces. Organ Geochemistr. 2005, 36, 1553–1566. [Google Scholar] [CrossRef]
- Weng, L.; Van Riemsdijk, W.H.; Hiemstra, T. Adsorption of humic acids onto goethite: Effects of molar mass, pH and ionic strength. J. Colloid Interface Sci. 2007, 314, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.J.; Lenhart, J.J.; Honeyman, B.D. The sorption of thorium(IV) and uranium(VI) to hematite in the presence of natural organic matter. Colloid. Surface. Physicochem. Eng. Aspect. 1999, 157, 47–62. [Google Scholar] [CrossRef]
- Ho, C.H.; Miller, N.H. Effect of humic acids on uranium uptake by hematite particles. J. Colloid Interface Sci. 1985, 106, 281–288. [Google Scholar] [CrossRef]
- Lenhart, J.J.; Honeyman, B.D. Uranium(VI) sorption to hematite in the presence of humic acid. Geochim et Cosmochim Acta 1999, 63, 2891–2901. [Google Scholar] [CrossRef]
- Bradley, C. Transient Modelling of Water-Table Variation in a Floodplain Wetland, Narborough Bog, Leicestershire. J. Hydrol. 1996, 185, 87–114. [Google Scholar] [CrossRef]
- Cohen, A.D.; Rollins, M.S.; Zunic, W.M.; Durig, J.R. Effects of chemical and physical differences in peat and their ability to extract hydrocarbons from water. Water Res. 1991, 25, 1047–1060. [Google Scholar] [CrossRef]
- Couillard, D. The use of peat in wastewater treatment. Water Res. 1994, 28, 1261–1274. [Google Scholar] [CrossRef]
- Kao, C.M.; Lei, S.E. Using peat biobarrier to remediate PCE/TCE contaminated aquifers. Water Res. 2000, 34, 835–845. [Google Scholar] [CrossRef]
- Ringquist, L.; Holmgren, A.; Oeborn, I. Poorly humified peat as an adsorbent for metals in wastewater. Water Res. 2001, 36, 2394–2404. [Google Scholar] [CrossRef]
- Ringquist, L.; Oeborn, I. Copper and zinc adsorption onto poorly humified sphagnum and carex peat. Water Res. 2001, 36, 2233–2242. [Google Scholar] [CrossRef]
- Rasmussen, G.; Fremmersvik, G.; Olsen, R.A. Treatment of creosote-contaminated groundwater in a peat-sand permeable barrier—A columns study. J. Hazard. Materials 2002, B93, 285–306. [Google Scholar] [CrossRef]
- Al Faqi, L.; Johnson, P.D.; Allen, S.J. Evaluation of a new peat-based sorbent for metals capture. Bioresour. Technol. 2007, 99, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Boonchayaanant, B.; Nayak, D.; Du, X.; Criddle, C.S. Uranium reduction and resistance to reoxidation under iron-reducing and sulfate-reducing conditions. Water Res. 2009, 43, 4652–4664. [Google Scholar] [CrossRef] [PubMed]
- Nedwell, D.B.; Watson, A. Production, oxidation and emission in a U.K. ombotrophic peat bog: Influence of SO42− from acid rain. Soil Biol. Biochem. 1995, 27, 893–903. [Google Scholar] [CrossRef]
- Jacks, G.; Norrström, A.C. Hydrochemistry and hydrology of forest riparian wetlands. Forest Ecol. Managem. 2004, 196, 187–197. [Google Scholar] [CrossRef]
- Winde, F.; Wade, P.; van der Walt, I.J. Gold tailings as a source of waterborne uranium contamination of streams—The Koekemoerspruit (Klerksdorp goldfield, South Africa) as a case study. Part III: Fluctuations of stream chemistry and their impacts on uranium mobility. Water SA 2004, 30, 233–240. [Google Scholar]
- Winde, F.; Wade, P.; van der Walt, I.J. Gold tailings as a source of waterborne uranium contamination of streams—The Koekemoerspruit (Klerksdorp goldfield, South Africa) as a case study. Part I: Uranium migration along the aqueous pathway. Water SA 2004, 30, 219–226. [Google Scholar]
- Bhat, S.V.; Melo, J.S.; Chaugule, B.B.; D’Souza, S.F. Biosorption characteristics of uranium(VI) from aqueous medium onto Canella repens, a red alga. J. Hzard. Mat. 2008, 158, 628–635. [Google Scholar]
- Simonsen, J.F.; Harremoes, P. Oxygen and pH fluctuations in rivers. Water Res. 1978, 12, 477–489. [Google Scholar] [CrossRef]
- Jones, C.A.; Nimick, D.A.; McCleskey, R.B. Relative effect of temperature and pH on cycling of dissolved trace elements in prickly pear creek, Montana. Water Air Soil Poll. 2004, 153, 95–113. [Google Scholar] [CrossRef]
- Domínguez-Villar, D.; Arteaga, C.; García-Giménez, R.; Smith, E.A.; Pedraza, J. Diurnal and seasonal water variations of temperature, pH, redox potential and conductivity in gnammas (Weathering Pits): Implications for chemical weathering. Catena 2008, 72, 37–48. [Google Scholar] [CrossRef]
- Mavrocordatos, D.; Mondi-Couture, C.; Atteia, O.; Leppard, G.G.; Perret, D. Formation of a distinct class of Fe-Ca(-Corg)-rich particles in a complex peat-karst system. J. Hydrol. 2000, 237, 234–247. [Google Scholar] [CrossRef]
- Winde, F. Untersuchungen zur Genese, Schwermetallkontamination und hochwassergebundenen Verlagerung rezenter Gerinnebettsedimente in Nebenvorflutern der Halleschen Saaleaue (Investigations into formation, heavy metal contamination and flood-induced re-suspension of recent stream channel sediments in side arms of the River Saale near Halle). In Late Quaternary and Recent Earth Surface Systems in Europe; Aurada, D., Billwitz, K., Lampe, E., Eds.; Martin-Luther-Universität Halle-Wittenberg: Halle, Germany, 1998; pp. 105–122, Supplementband der Zeitschrift für Geomorphologie. [Google Scholar]
- Maggi, F. Biological flocculation of suspended particles in nutrient-rich aqueous ecosystems. J. Hydrol. 2009, 376, 116–125. [Google Scholar] [CrossRef]
- Tsezos, M.; Voleski, B. The mechanism of uranium biosorption by Rhizopus Arrhizus. Biotechnol. Bioengin. 1982, 24, 385–401. [Google Scholar] [CrossRef]
- Tsezos, M.; Voleski, B. Biosorption of uranium and thorium. Biotechnol. Bioengin. 1981, 23, 583–604. [Google Scholar] [CrossRef]
- Sikora, L.J.; Keeney, D.R. Further aspects of soil chemistry under anaerobic conditions. In Ecosystems of the World; Elsevier: New York, NY, USA, 1983. [Google Scholar]
- McNevin, D.; Barford, J.; Hage, J. Adsorption and biological degradation of ammonium and sulfide on peat. Water Res. 1999, 33, 1449–1459. [Google Scholar] [CrossRef]
- Morris, K.; Raiswell, R. Biochemical cycles and remobilisation of actinide elements. In Interactions of Microorganisms with Radionuclids, 1st ed.; Keith-Roach, M.J., Livens, F.R., Eds.; Elsevier Science Ltd.: New York, NY, USA, 2002; Chapter 4. [Google Scholar]
- Van Roy, S.; Vanbroekhoven, K.; Dejonghe, W.; Diels, L. Immobilization of heavy metals in the saturated zone by sorption and in Situ bioprecipitation processes. Hydrometall 2006, 83, 195–203. [Google Scholar]
- Dienemann, C.; Dienemann, H.; Dudel, E.G.; Schurig, C. Uranium fixation by Cladophera spec. In Uranium, Mining and Hydrogeology; Merkel, B.J., Hache-Berger, A., Eds.; Springer Verlag: Berlin and Heidelberg, Germany, 2008; pp. 103–110. [Google Scholar]
- Bhat, S.V.; Melo, J.S.; Chaugule, B.B.; D’Souza, S.F. Biosorption characteristics of uranium(VI) from aqueous medium onto Canella repens, a red alga. J. Hzard. Mat. 2008, 158, 628–635. [Google Scholar] [CrossRef]
- Cerne, M.; Smodis, B.; Strok, M. Uptake of radionuclides by a common reed (Phragmites australis Cav. Trin. Ex Steud.) grown in the vicinity of the former uranium mine at Zirovski vrh. Nucl. Eng. Des. 2010. [Google Scholar] [CrossRef]
- Twardowska, I.; Kyziol, J. Soprtion of metals onto natural organic matter as a function of complexation and adsorbent-adsorbate contact mode. Environ. Int. 2003, 28, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tipping, E.; Smith, E.J.; Lawlor, A.J.; Hughes, S.; Stevens, P.A. Predicting the release of metals from ombotrophic peat due to drought-induced acidification. Environ. Poll. 2003, 123, 239–253. [Google Scholar] [CrossRef]
- Dellwig, O.; Böttcher, M.E.; Lipinski, M.; Brumsack, H.J. Trace metals in holocene coastal peats and their relation to pyrite formation (NW Germany). Chem. Geol. 2002, 182, 423–442. [Google Scholar] [CrossRef]
- Mandernack, K.W.; Lynch, L.; Krouse, H.R.; Morgan, M.D. Sulfur cycling in wetland peat of the New Jersey pinelands and its effect on stream water chemistry. Geochim et Cosmochim 2000, 64, 3949–3964. [Google Scholar] [CrossRef]
- Eimers, C.M.; Dillon, P.J.; Schiff, S.L.; Jeffries, D.S. The effects of drying and re-wetting and increased temperature on sulphate release from upland and wetland material. Soil Biol. Biochem. 2003, 35, 1663–1673. [Google Scholar] [CrossRef]
- Wade, P.W.; Woodbourne, S.; Morris, W.M.; Vos, P.; Jarvis, N.V. Tier 1 Risk Assessment of Radionuclids in Selected Sediments of the Mooi River; WRC-Project, K5/1095; Water Research Commission: Pretoria, South Africa, 2002. [Google Scholar]
- Francis, C.W.; Timpson, M.E.; Wilson, J.H. Bench- and pilot-scale studies relating to the removal of uranium from uranium-contaminated soils using carbonate and citrite lixivants. J. Hazard. Mater. 1999, 66, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Schulze, G. Bestandsaufnahme und Charakterisierung der dtofflichen Auswirkungen des Uranerzbergbaus und der Uranaufbereitung (Standort Seelingstädt) am Beispiel des Wasserpfades. (Stock-Taking and Characterisation of Substance-Related Impacts of Uranium Mining and Milling (Site Seelingstädt) using the Aquatic Pathway as an Example.). Veröff. Museum Gera, Naturwiss. Reihe 1993, 20, 40–73. [Google Scholar]
- Baeza, A.; Salas, A.; Legarda, F. Determining factors in the elimination of uranium and radium from groundwaters during standard potabilization process. Sci. Total Environ. 2008, 406, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Quin, F.; Wen, B.; Shan, X.Q.; Xie, Y.N.; Liu, T.; Zhang, S.Z.; Khan, S.U. Mechanisms of competitive adsorption of Pb, Cu, and Cd on peat. Environ. Pollut. 2006, 144, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Boelter, D.H. Hydraulic conductivity of peats. Soil Sci. 1965, 100, 227–231. [Google Scholar] [CrossRef]
- Ingram, H.A.P.; Rycroft, D.W.; Williams, D.J.A. Anomalous transmission of water through certain peats. J. Hydrol. 1974, 22, 213–218. [Google Scholar] [CrossRef]
- Dooge, J. The water balance of bogs and fens. In Hydrology of Marsh-Ridden Areas: Proceedings of the Minsk Symposium; UNESCO: Paris, France, 1975; pp. 233–271. [Google Scholar]
- Dasberg, S.; Neuman, S.P. Peat hydrology in the hula basin, Israel: I. Properties of peat. J. Hydrol. 1977, 32, 219–239. [Google Scholar] [CrossRef]
- Chanson, D.B.; Siegel, D.I. Hydraulic conductivity and related physical properties of peat, lost river peatland, Northern Minnesota. Soil Sci. 1986, 142, 91–99. [Google Scholar] [CrossRef]
- Boeye, D.; Verheyen, R.F. The hydrological balance of a groundwater discharge fen. J. Hydrol. 1992, 137, 149–163. [Google Scholar] [CrossRef]
- Gilman, K. Hydrology and Wetland Conservation; John Wiley & Sons, Ltd.: New York, NY, USA, 1994. [Google Scholar]
- Fraser, C.J.D.; Roulet, N.T.; Lafleur, M. Groundwater flow patterns in a large peatland. J. Hydrol. 2001, 246, 142–154. [Google Scholar] [CrossRef]
- Bragg, O.M. Hydrology of peat-forming wetlands in Scotland. Sci. Total Environ. 2002, 294, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.; Burt, T.P. Piping and pipeflow in a deep peat catchment. Catena 2002, 48, 164–199. [Google Scholar] [CrossRef]
- Holden, J. Going beyond the acrotelm-catotelm model in blanket peat catchments. Geophys. Res. Abstr. 2003, 5, 01233. [Google Scholar]
- Bradley, C.; van Den Berg, J.A. Infiltration mechanisms in a herbaceous peat: Results of a controlled infiltration experiment. Hydrol. Sci. J. 2005, 50, 713–725. [Google Scholar]
- Bradley, C.; Brown, A.G. Shallow groundwater modeling and the over-bank contribution to a small floodplain bog. In Geomorphology and Groundwater, 1st ed.; Brown, A.G., Ed.; Wiley: Chichester, UK, 1995; pp. 37–52. [Google Scholar]
- Daniels, S.M.; Agnew, C.T.; Allot, T.E.H.; Evans, M.G. Water table variability and runoff generation in an eroded peatland, South Pennines, UK. J. Hydrol. 2008, 362, 214–226. [Google Scholar] [CrossRef]
- Holden, J. Flow through macropores of different size classes in blanket peat. J. Hydrol. 2009, 364, 342–348. [Google Scholar] [CrossRef]
- Price, J.S.; Whittington, P.N. Water flow in Sphagnum hummock: Mesocosm measurements and modelling. J. Hydrol. 2010, 381, 333–340. [Google Scholar] [CrossRef]
- Winde, F.; van der Walt, I.J. Gold tailings as a source of waterborne uranium contamination of streams—the Koekemoerspruit (Klerksdorp goldfield, South Africa) as a case study. Part II of III: Dynamics of groundwater-stream interactions. Water SA 2004, 30, 227–232. [Google Scholar]
- Evans, M.G.; Burt, T.P.; Holden, J.; Adamson, J.K. Runoff generation and water table fluctuations in blanket peat: Evidence from UK data spanning the dry summer of 1995. J. Hydrol. 1999, 221, 141–160. [Google Scholar] [CrossRef]
- Wise, W.R.; Annable, M.D.; Walser, J.A.E.; Switt, R.S.; Shaw, D.T. A wetland-aquifer interaction test. J. Hydrol. 2000, 227, 257–272. [Google Scholar] [CrossRef]
- Rycroft, D.W.; Williams, D.J.A.; Ingram, H.A.P. The transmission of water through peat: I. Review. J. Ecol. 1975, 63, 535–556. [Google Scholar] [CrossRef]
- Siegel, D.I. A Review of the Recharge-Discharge Function of Wetlands. In The Ecology and Management of Wetlands: Ecology of Wetlands; Hook, D.D., Ed.; Croom Helm: London, UK, 1988; pp. 59–67. [Google Scholar]
- Price, J.S.; Schlotzhauer, S.M. Importance of shrinkage and compression in determining water storage changes in peat: The case of a mined peatland. Hydrol. Proc. 1999, 13, 2591–2601. [Google Scholar] [CrossRef]
- Reeve, A.S.; Siegel, D.I.; Glaser, P.H. simulating vertical flow in large peatlands. J. Hydrol. 2000, 227, 201–217. [Google Scholar] [CrossRef]
- Van Loon, A.H.; Schot, P.P.; Griffioen, J.; Bierkens, M.F.P.; Wassen, M.J. Throughflow as a determining factor for habitat contiguity in a near-natural fen. J. Hydrol. 2009, 379, 30–40. [Google Scholar] [CrossRef]
- Siegel, D.I.; Reeve, A.S.; Glaser, P.H.; Romanowicz, E.A. Climate-driven flushing of pore water in peatlands. Nature 1995, 374, 531–533. [Google Scholar] [CrossRef]
- Hemond, H.F.; Goldman, J.C. On non-darcian water flow in peat. J. Ecol. 1985, 73, 579–584. [Google Scholar] [CrossRef]
- Blodau, C.; Moore, T.R. Macro porosity affects water movement and pore water sampling in peat soils. Soil Sci. 2002, 167, 98–109. [Google Scholar] [CrossRef]
- Jones, J.A.A. Extending the Hewlett model of stream runoff generation. Area 1979, 11, 110–114. [Google Scholar]
- Jones, J.A.A.; International Geographical Union, Chair Commission on Water Sustainability, retired professor, University of Wales, Aberystwyth. personal communication, July 2009.
- Reeve, A.S.; Siegel, D.I.; Glaser, P.H. Simulating dispersive mixing in large peatlands. J. Hydrol. 2001, 242, 103–114. [Google Scholar] [CrossRef]
- Reeve, A.S.; Siegel, D.I.; Glaser, P.H. Simulating vertical flow in large peatlands. J. Hydrol. 2000, 227, 201–217. [Google Scholar] [CrossRef]
- Fraser, C.J.D.; Roulet, N.T.; Lafleur, M. Groundwater flow patterns in a large peatland. J. Hydrol. 2001, 246, 142–154. [Google Scholar] [CrossRef]
- Sade, R.; Litaor, M.I.; Shenker, M. Evaluation of groundwater and phosphorous transport in fractured altered wetland soils. J. Hydrol. 2010, 393, 133–142. [Google Scholar] [CrossRef]
- Lowry, C.S.; Fratta, D.; Anderson, M.P. Ground penetrating radar and spring formation in a groundwater dominated peat wetland. J. Hydrol. 2009, 373, 68–79. [Google Scholar] [CrossRef]
- Bauer, P.; Thabeng, G.; Stauffer, F.; Kinzelbach, W. Estimation of the evapotranspiration rate from diurnal groundwater fluctuations in the Okavango Delta, Botswana. J. Hydrol. 2004, 288, 344–355. [Google Scholar] [CrossRef]
- Bradley, C.; Brown, A.G. Shallow Groundwater Modeling and the Overbank Contribution to a Small Floodplain Bog. In Geomorphology and Groundwater; Brown, A.G., Ed.; Wiley: Wiltshire, UK, 1995; pp. 37–52. [Google Scholar]
- Cloke, H.L.; Anderson, M.G.; McDonnell, J.J.; Renaud, J.-P. Using numerical modelling to evaluate the capillary fringe groundwater ridging hypothesis of streamflow generation. J. Hydrol. 2006, 316, 141–162. [Google Scholar] [CrossRef]
- Winde, F. Peatlands as filters for polluted mine water?—A case study from an uranium-contaminated karst system in South Africa. Part III: Quantifying the hydraulic filter component. Water 2010, 3, 356–390. [Google Scholar]
- Winde, F. Peatlands as filters for polluted mine water?—A case study from an uranium-contaminated karst system in South Africa. Part IV: Quantifying the chemical filter component. Water 2010, 3, 391–423. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Winde, F. Peatlands as Filters for Polluted Mine Water?—A Case Study from an Uranium-Contaminated Karst System in South Africa—Part II: Examples from Literature and a Conceptual Filter Model. Water 2011, 3, 323-355. https://0-doi-org.brum.beds.ac.uk/10.3390/w3010323
Winde F. Peatlands as Filters for Polluted Mine Water?—A Case Study from an Uranium-Contaminated Karst System in South Africa—Part II: Examples from Literature and a Conceptual Filter Model. Water. 2011; 3(1):323-355. https://0-doi-org.brum.beds.ac.uk/10.3390/w3010323
Chicago/Turabian StyleWinde, Frank. 2011. "Peatlands as Filters for Polluted Mine Water?—A Case Study from an Uranium-Contaminated Karst System in South Africa—Part II: Examples from Literature and a Conceptual Filter Model" Water 3, no. 1: 323-355. https://0-doi-org.brum.beds.ac.uk/10.3390/w3010323