HDX-MS for Epitope Characterization of a Therapeutic ANTIBODY Candidate on the Calcium-Binding Protein Annexin-A1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
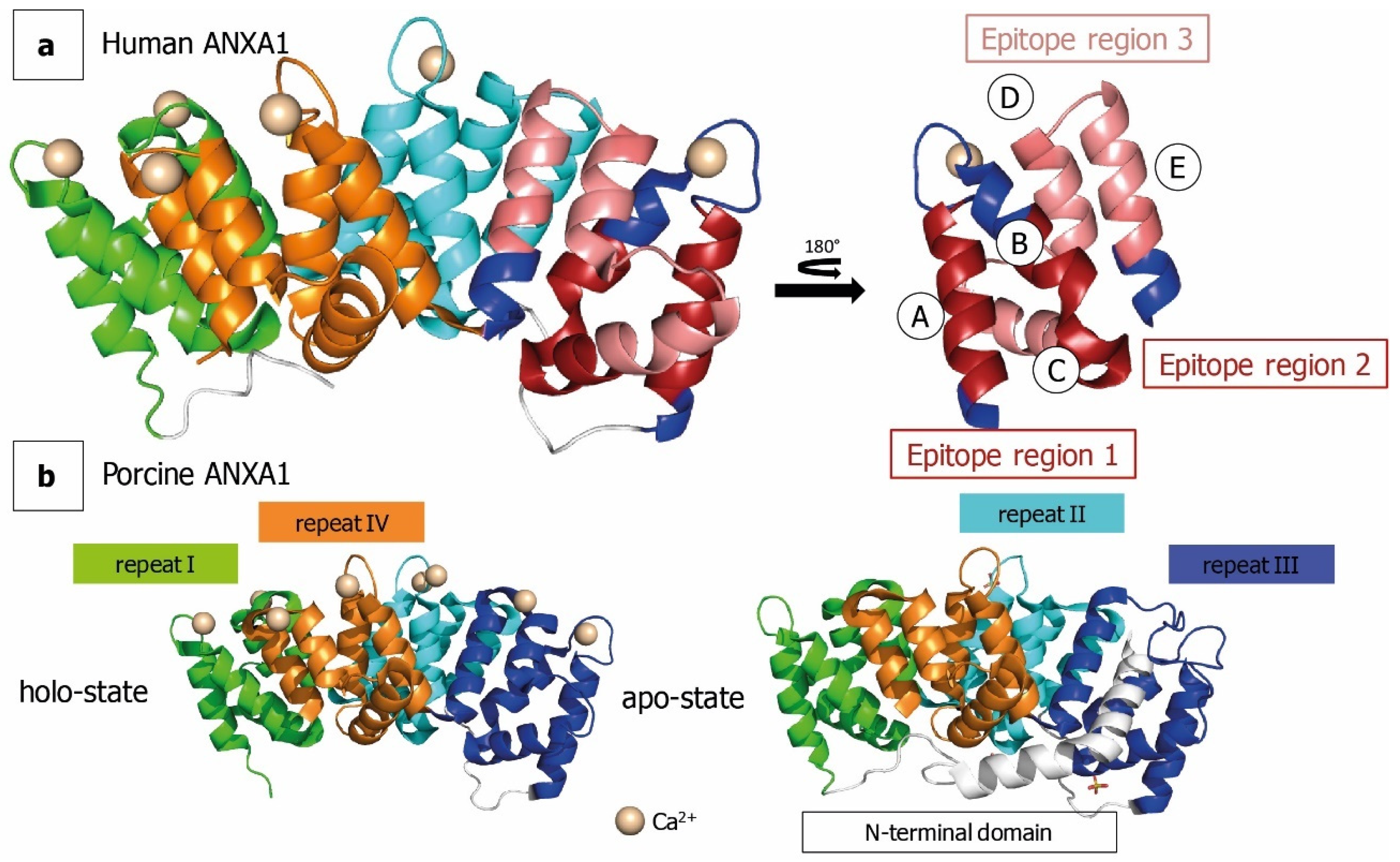
:1. Introduction
2. Results
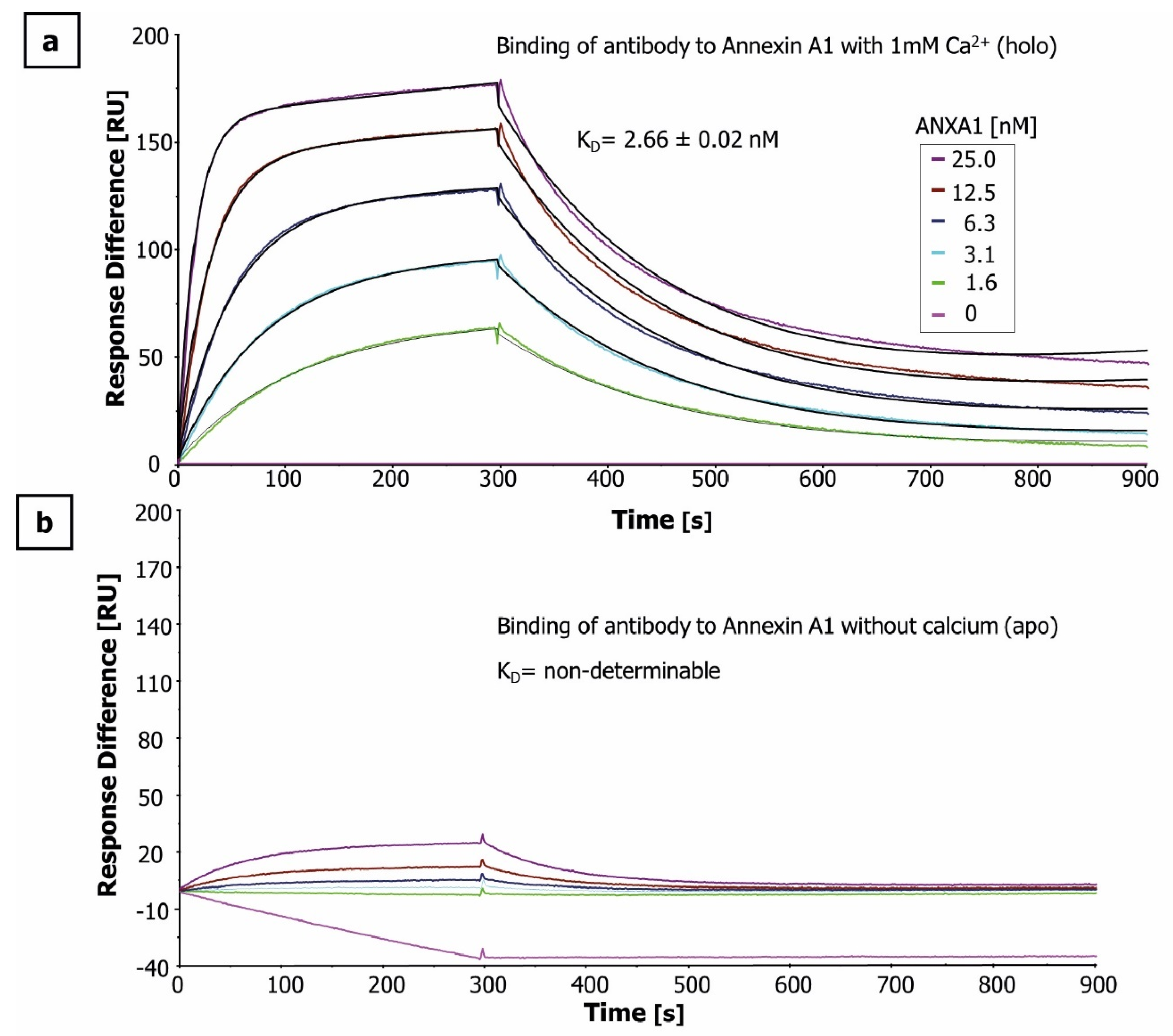
2.1. Antigen and Antibody Characterization
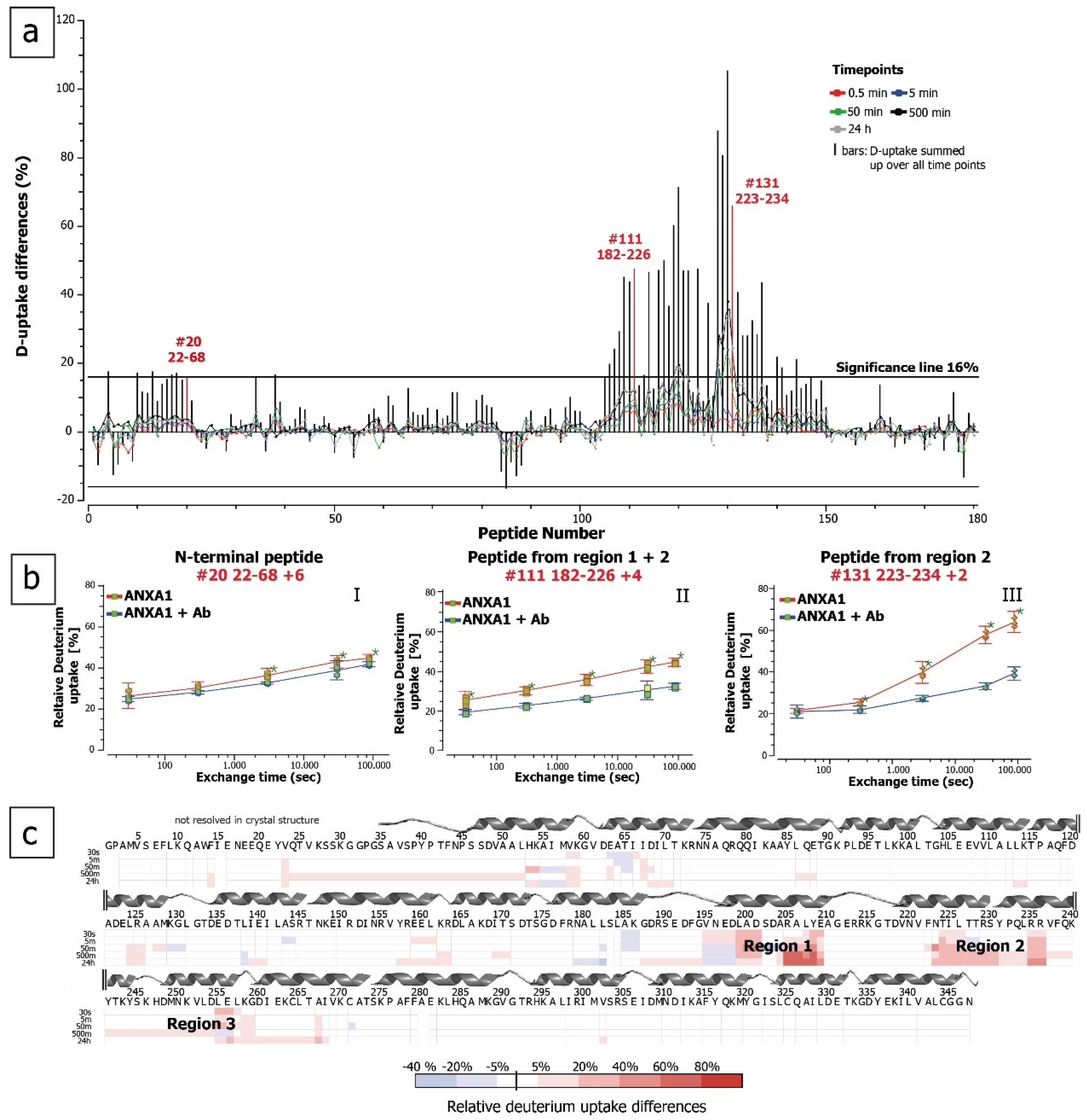
2.2. ANXA1 Deuteration Kinetics
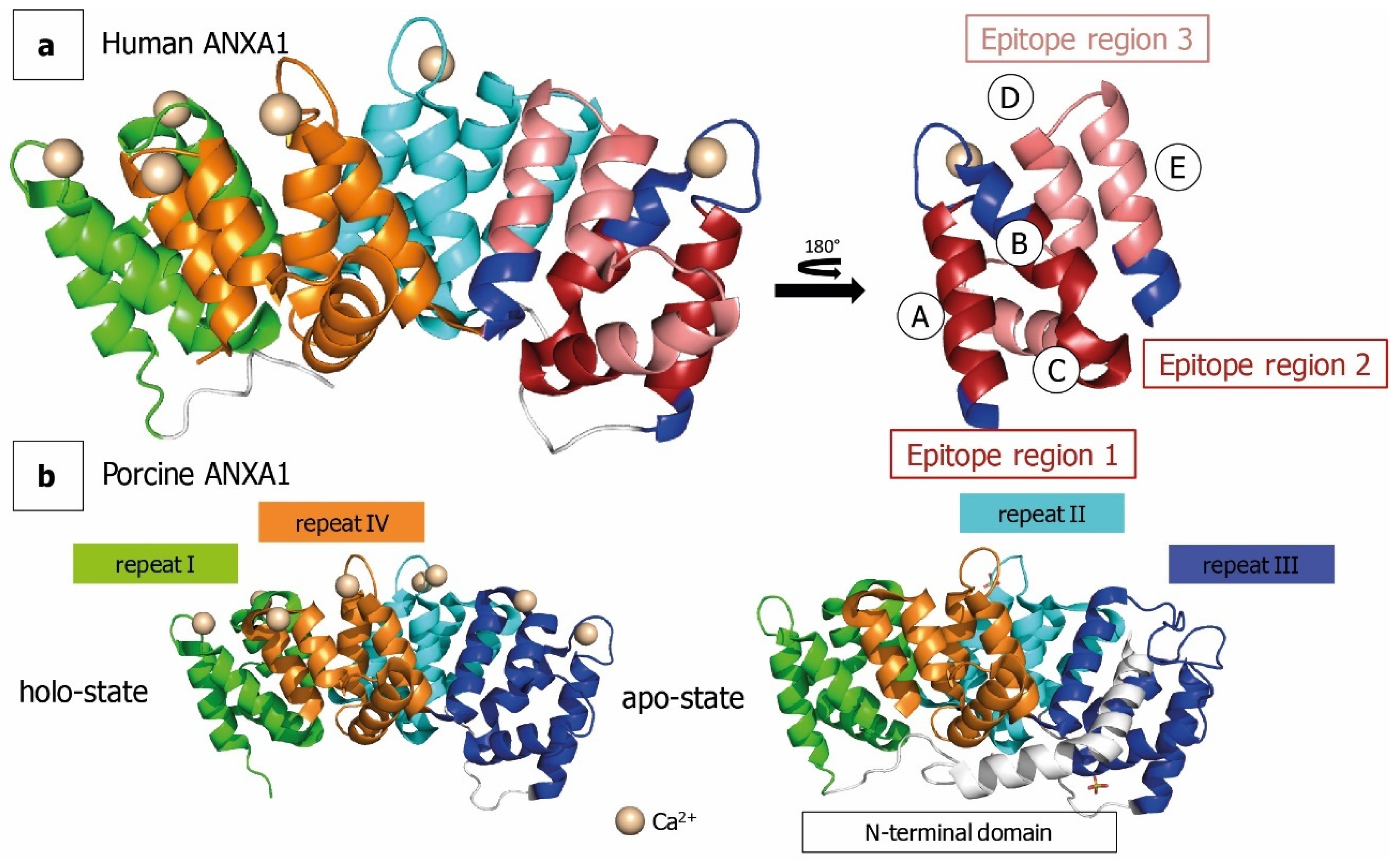
2.3. HDX for Epitope Mapping of Anti-ANXA1 Antibody
3. Discussion
4. Material and Methods
4.1. Reagents, Peptides, and Antibody
4.2. ANXA1 Production and Purification
4.3. HPLC-MS Analysis of ANXA1 and Antibody
4.4. Determination of Affinity by Surface Plasmon Resonance (SPR)
4.5. Hydrogen–Deuterium Exchange
ANXA1 Deuteration Kinetics and Epitope Elucidation
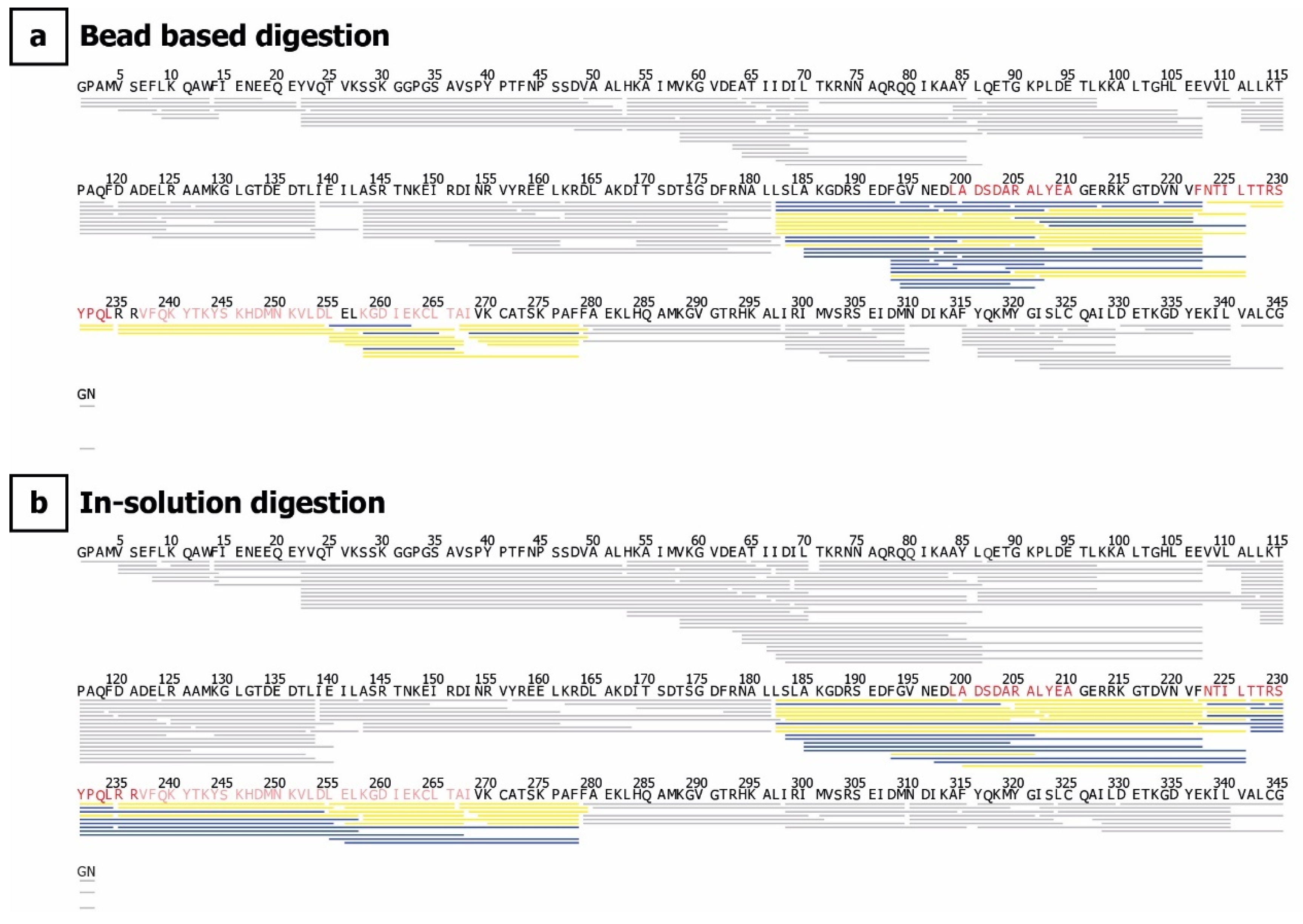
4.6. Chromatography, Mass Spectrometry, and HDX Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosengarth, A.; Luecke, H. Crystallization and preliminary X-ray analysis of full-length annexin i comprising the core and n-terminal domain. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 1459–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosengarth, A.; Gerke, V.; Luecke, H. X-ray structure of full-length annexin 1 and implications for membrane aggregation. J. Mol. Biol. 2001, 306, 489–498. [Google Scholar] [CrossRef]
- Lizarbe, M.A.; Barrasa, J.I.; Olmo, N.; Gavilanes, F.; Turnay, J. Annexin-phospholipid interactions. Functional implications. Int. J. Mol. Sci. 2013, 14, 2652–2683. [Google Scholar] [CrossRef] [Green Version]
- Liemann, S.; Huber, R. Three-dimensional structure of annexins. Cell. Mol. Life Sci. 1997, 53, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, H.M.; Solito, E. Annexin a1: Uncovering the many talents of an old protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef] [Green Version]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-a1: Therapeutic potential in microvascular disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, G.; Zhou, H.; Zhang, Q.; Jin, Y.; Fu, C. Advancements of annexin a1 in inflammation and tumorigenesis. OncoTargets Ther. 2019, 12, 3245–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Acquisto, F.; Perretti, M.; Flower, R.J. Annexin-a1: A pivotal regulator of the innate and adaptive immune systems. Br. J. Pharmacol. 2008, 155, 152–169. [Google Scholar] [CrossRef] [Green Version]
- D’Acquisto, F.; Paschalidis, N.; Sampaio, A.L.; Merghani, A.; Flower, R.J.; Perretti, M. Impaired t cell activation and increased th2 lineage commitment in annexin-1-deficient t cells. Eur. J. Immunol. 2007, 37, 3131–3142. [Google Scholar] [CrossRef]
- D’Acquisto, F.; Merghani, A.; Lecona, E.; Rosignoli, G.; Raza, K.; Buckley, C.D.; Flower, R.J.; Perretti, M. Annexin-1 modulates t-cell activation and differentiation. Blood 2007, 109, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Hays, H.C.W.; Wood, C.B.; Flatau, T.C. Anti Human Annexin a1 Antibody. Patent U.S. 2020/0031911 A1, 2020. Available online: https://www.freepatentsonline.com/y2020/0031911.html (accessed on 20 December 2020).
- D’Acquisto, F.; Perretti, M. Annexin 1 Antibody. Patent WO 2011/154705 A1, 31 March 2011. [Google Scholar]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen-antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Zoll, S.; Schlag, M.; Shkumatov, A.V.; Rautenberg, M.; Svergun, D.I.; Gotz, F.; Stehle, T. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J. Bacteriol. 2012, 194, 3789–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, D.R.; Cohen, G.H. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. USA 1996, 93, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, S.; Tailford, L.E.; Juge, N.; Angulo, J. Differential epitope mapping by std nmr spectroscopy to reveal the nature of protein-ligand contacts. Angew. Chem. Int. Ed. 2017, 129, 15491–15495. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Bhattiprolu, K.C.; Gubensak, N.; Zangger, K. Investigating protein-ligand interactions by solution nuclear magnetic resonance spectroscopy. Chemphyschem 2018, 19, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.; Turner, H.L.; Nogal, B.; Cottrell, C.A.; Oyen, D.; Pauthner, M.; Bastidas, R.; Nedellec, R.; McCoy, L.E.; Wilson, I.A.; et al. Electron-microscopy-based epitope mapping defines specificities of polyclonal antibodies elicited during hiv-1 bg505 envelope trimer immunization. Immunity 2018, 49, 288–300 e288. [Google Scholar] [CrossRef] [Green Version]
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Remigy, H.W.; Stark, H.; Wiesmann, C. Cryo-em in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a sars-cov-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef]
- Pandit, D.; Tuske, S.J.; Coales, S.J.; Yen E, S.; Liu, A.; Lee, J.E.; Morrow, J.A.; Nemeth, J.F.; Hamuro, Y. Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J. Mol. Recognit. 2012, 25, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (hdx-ms) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Luecke, H.; Song, I.S.; Kang, D.S.; Kim, S.H.; Huber, R. Crystal structure of human annexin i at 2.5 a resolution. Protein Sci. 1993, 2, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Rosengarth, A.; Luecke, H. A calcium-driven conformational switch of the n-terminal and core domains of annexin a1. J. Mol. Biol. 2003, 326, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Hao, G.; Wesolowski, J.S.; Jiang, X.; Lauder, S.; Sood, V.D. Epitope characterization of an anti-pd-l1 antibody using orthogonal approaches. J. Mol. Recognit. 2015, 28, 269–276. [Google Scholar] [CrossRef]
- Puchades, C.; Kukrer, B.; Diefenbach, O.; Sneekes-Vriese, E.; Juraszek, J.; Koudstaal, W.; Apetri, A. Epitope mapping of diverse influenza hemagglutinin drug candidates using hdx-ms. Sci. Rep. 2019, 9, 4735. [Google Scholar] [CrossRef] [Green Version]
- Kielkopf, C.S.; Ghosh, M.; Anand, G.S.; Brown, S.H.J. Hdx-ms reveals orthosteric and allosteric changes in apolipoprotein-d structural dynamics upon binding of progesterone. Protein Sci. 2019, 28, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Y.C.; Krystek, S.R.; Felix, N.; Graziano, R.F.; Srinivasan, M.; Pashine, A.; Chen, G. Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/tl1a interaction. mAbs 2017, 10, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kochert, B.A.; Iacob, R.E.; Wales, T.E.; Makriyannis, A.; Engen, J.R. Hydrogen-deuterium exchange mass spectrometry to study protein complexes. Methods Mol. Biol. 2018, 1764, 153–171. [Google Scholar] [PubMed]
- Wagner, T.R.; Kaiser, P.D.; Gramlich, M.; Ostertag, E.; Ruetalo, N.; Junker, D.; Haering, J.; Traenkle, B.; Becker, M.; Dulovic, A.; et al. Neutrobodyplex—nanobodies to monitor a sars-cov-2 neutralizing immune response. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hamuro, Y.; Coales, S.J. Optimization of feasibility stage for hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 623–629. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gramlich, M.; Hays, H.C.W.; Crichton, S.; Kaiser, P.D.; Heine, A.; Schneiderhan-Marra, N.; Rothbauer, U.; Stoll, D.; Maier, S.; Zeck, A. HDX-MS for Epitope Characterization of a Therapeutic ANTIBODY Candidate on the Calcium-Binding Protein Annexin-A1. Antibodies 2021, 10, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010011
Gramlich M, Hays HCW, Crichton S, Kaiser PD, Heine A, Schneiderhan-Marra N, Rothbauer U, Stoll D, Maier S, Zeck A. HDX-MS for Epitope Characterization of a Therapeutic ANTIBODY Candidate on the Calcium-Binding Protein Annexin-A1. Antibodies. 2021; 10(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010011
Chicago/Turabian StyleGramlich, Marius, Henry C. W. Hays, Scott Crichton, Philipp D. Kaiser, Anne Heine, Nicole Schneiderhan-Marra, Ulrich Rothbauer, Dieter Stoll, Sandra Maier, and Anne Zeck. 2021. "HDX-MS for Epitope Characterization of a Therapeutic ANTIBODY Candidate on the Calcium-Binding Protein Annexin-A1" Antibodies 10, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010011