Characterization and Modeling of Reversible Antibody Self-Association Provide Insights into Behavior, Prediction, and Correction

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Dynamic Light Scattering (DLS)
2.3. Affinity-Capture Self-Interaction Nanoparticle Spectroscopy (AC-SINS)
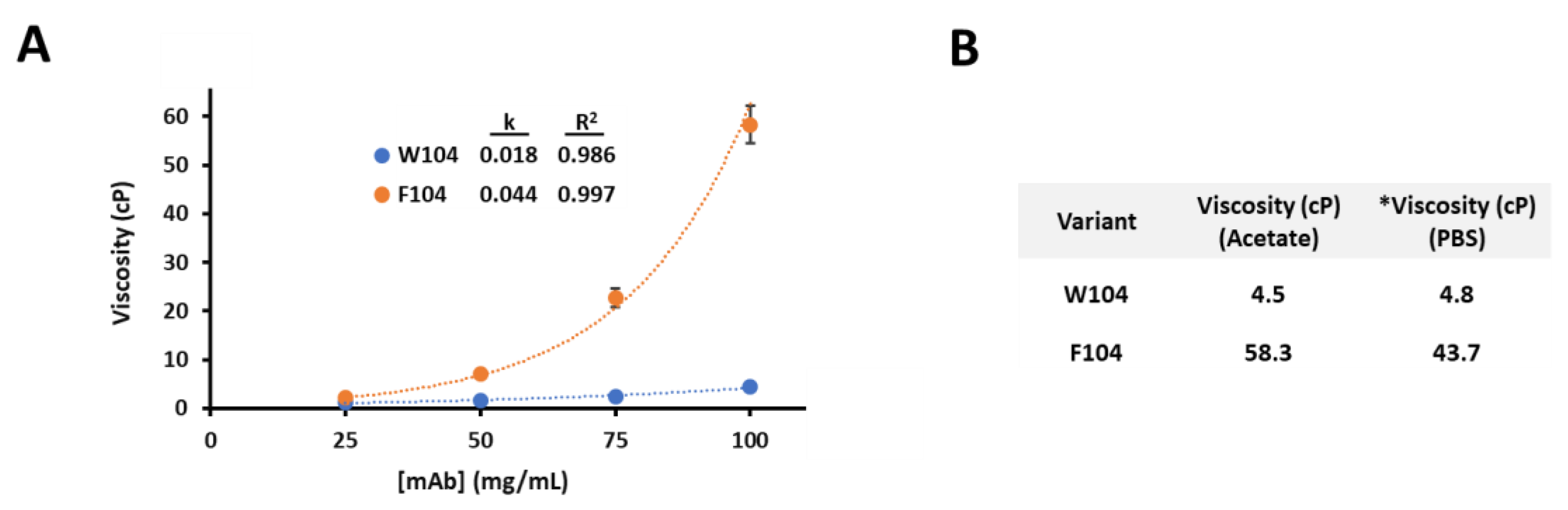
2.4. Viscosity Measurements
2.5. Modeling
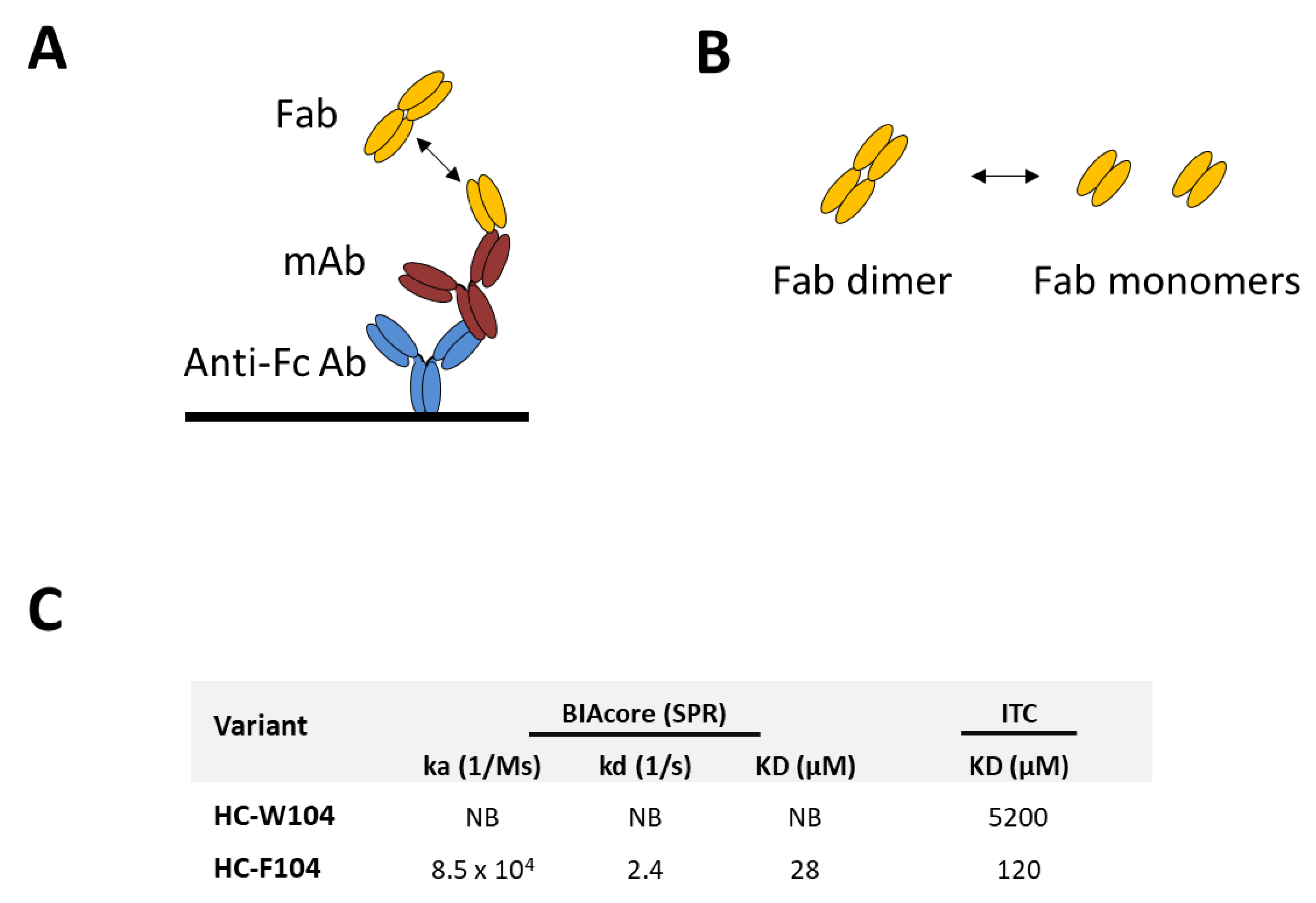
2.6. Isothermal Titration Calorimetry (ITC)
2.7. Surface Plasmon Resonance (BIAcore)
3. Results
3.1. Viscosity Characterization for Two Antibody Variants
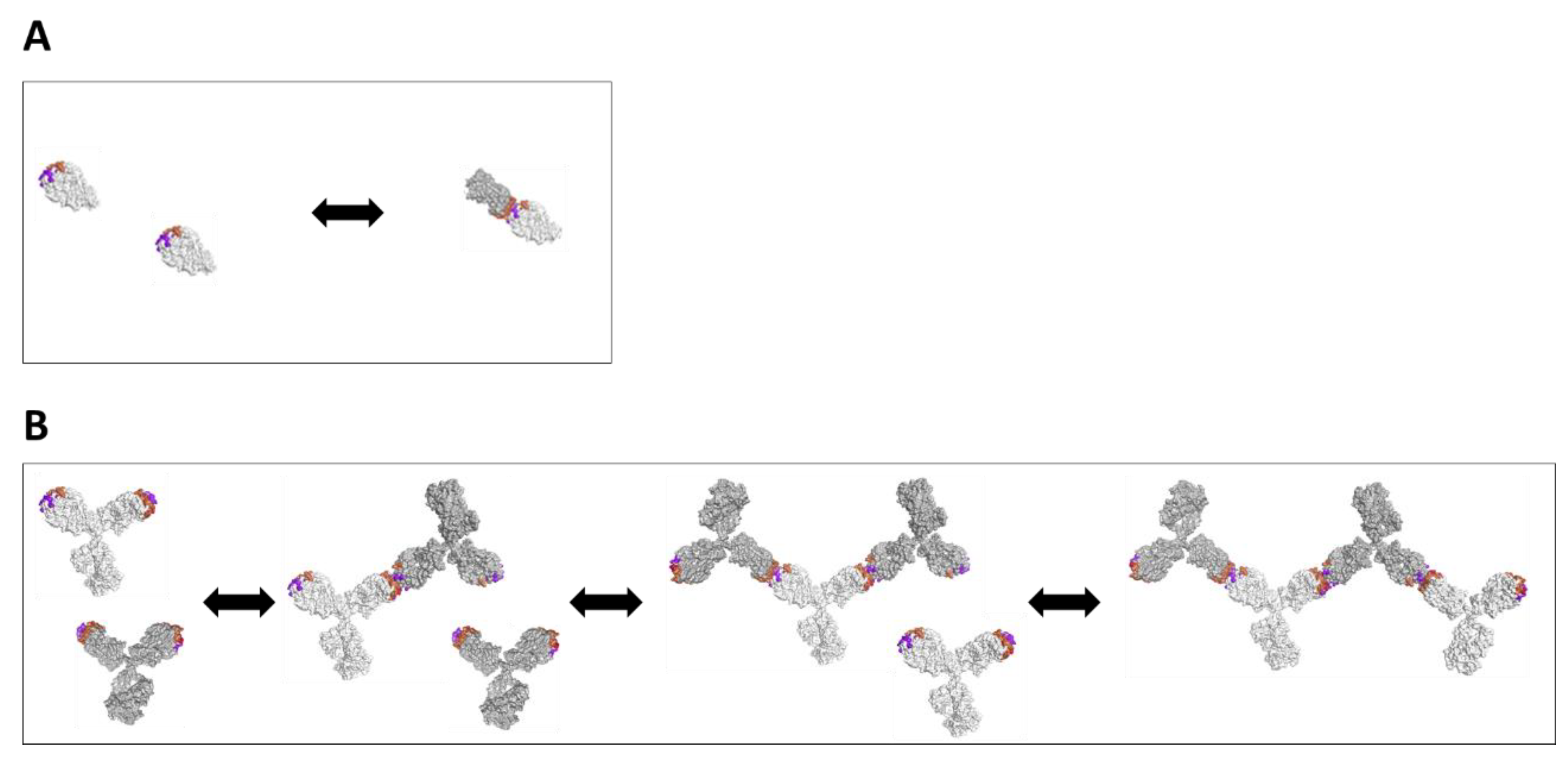
3.2. Biophysical Characterization and Modeling of Antibody Self-Association Using Individual Antibody Domains for Two Antibody Variants
3.3. Evaluation of Self-Binding by BIAcore and ITC
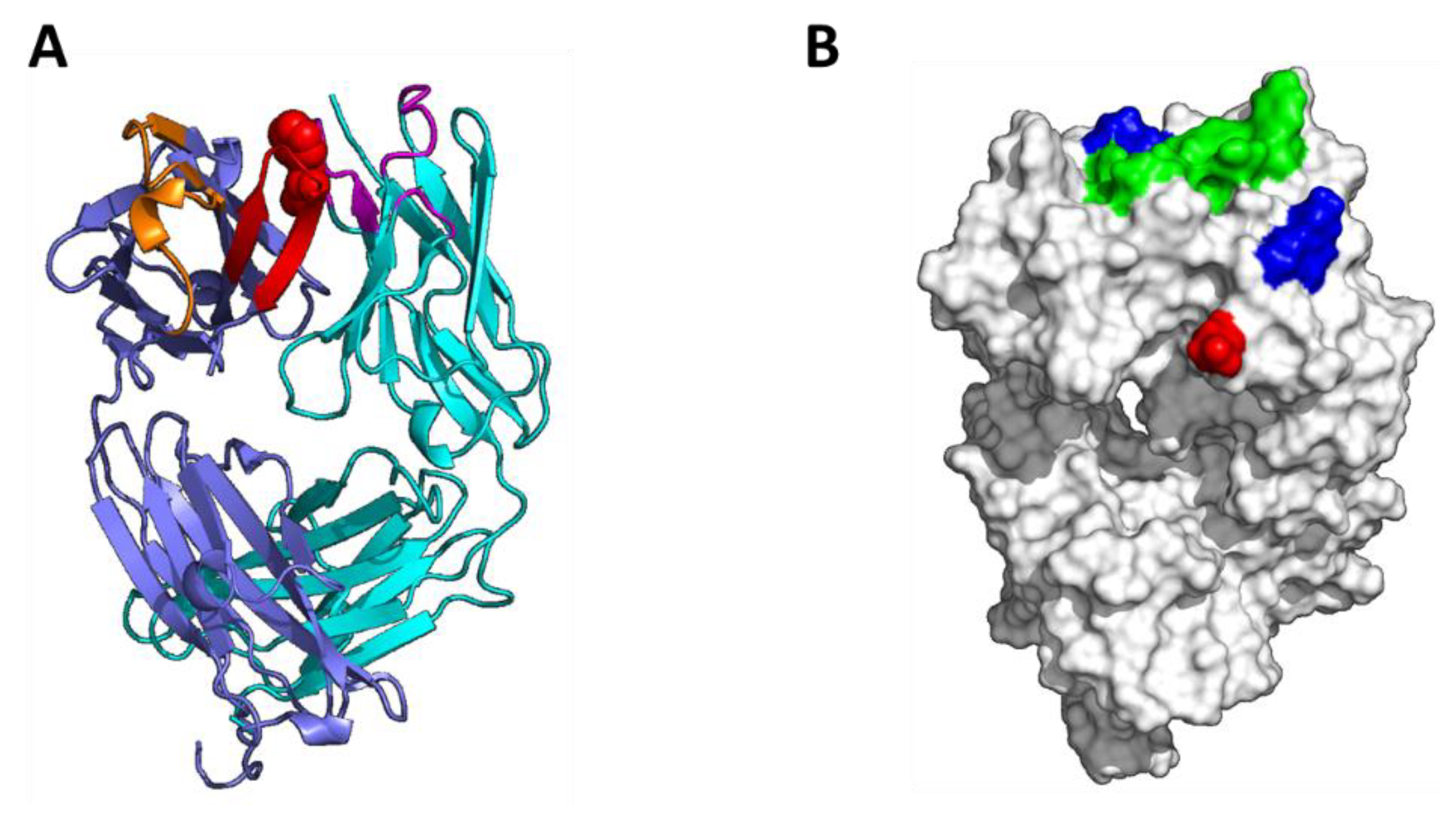
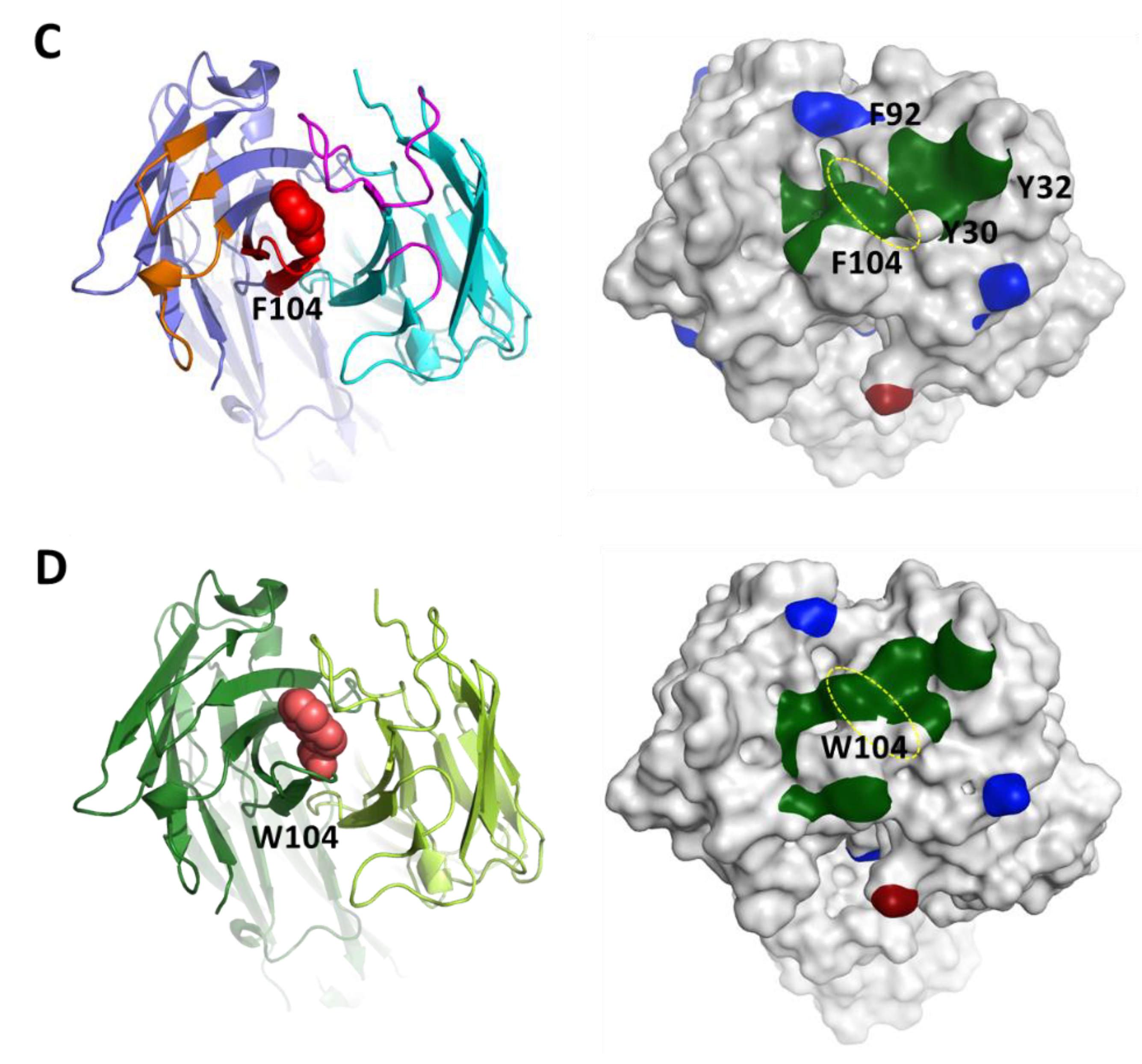
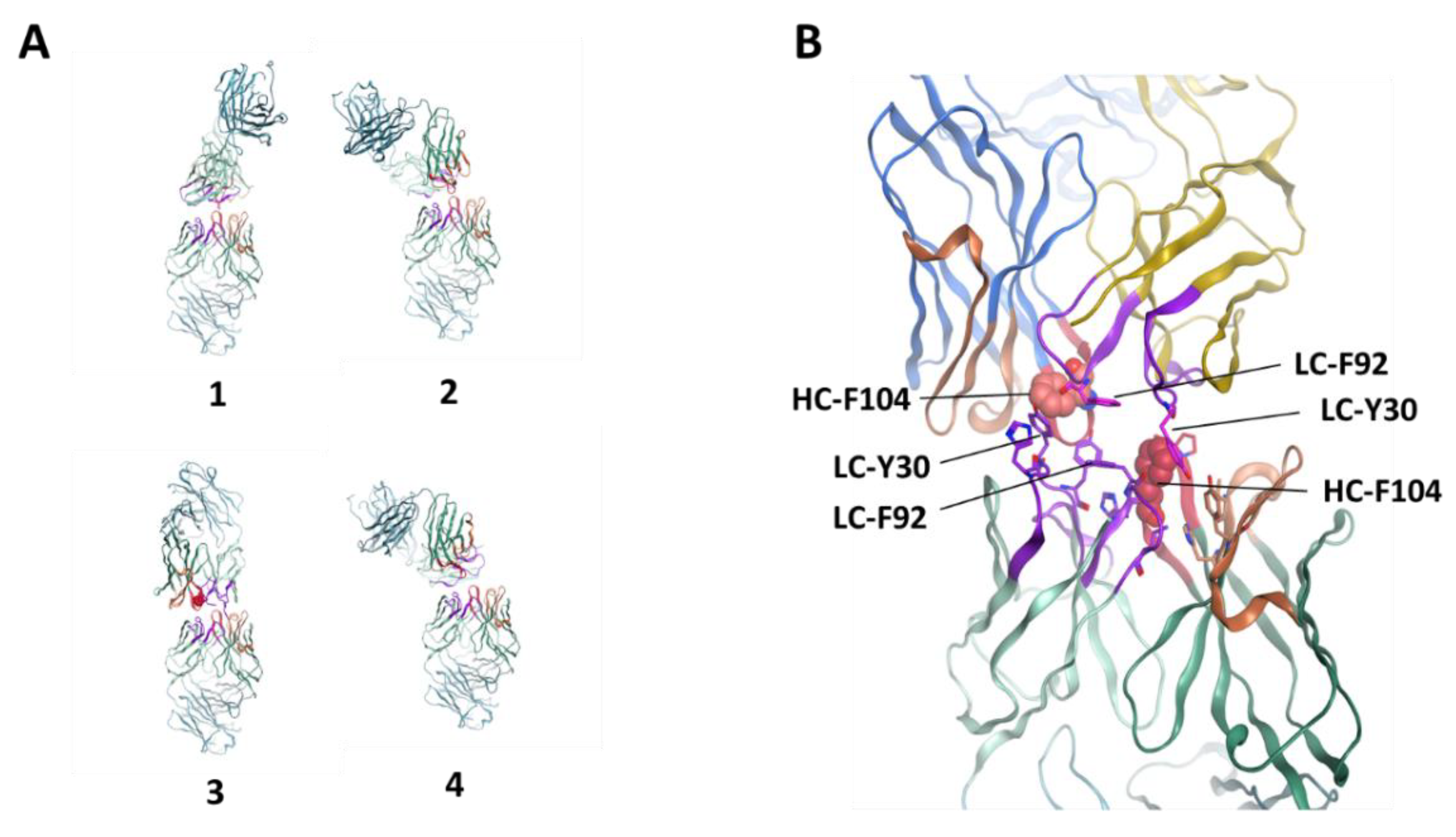
3.4. Homology Modeling by Patch Analysis and Self-Docking
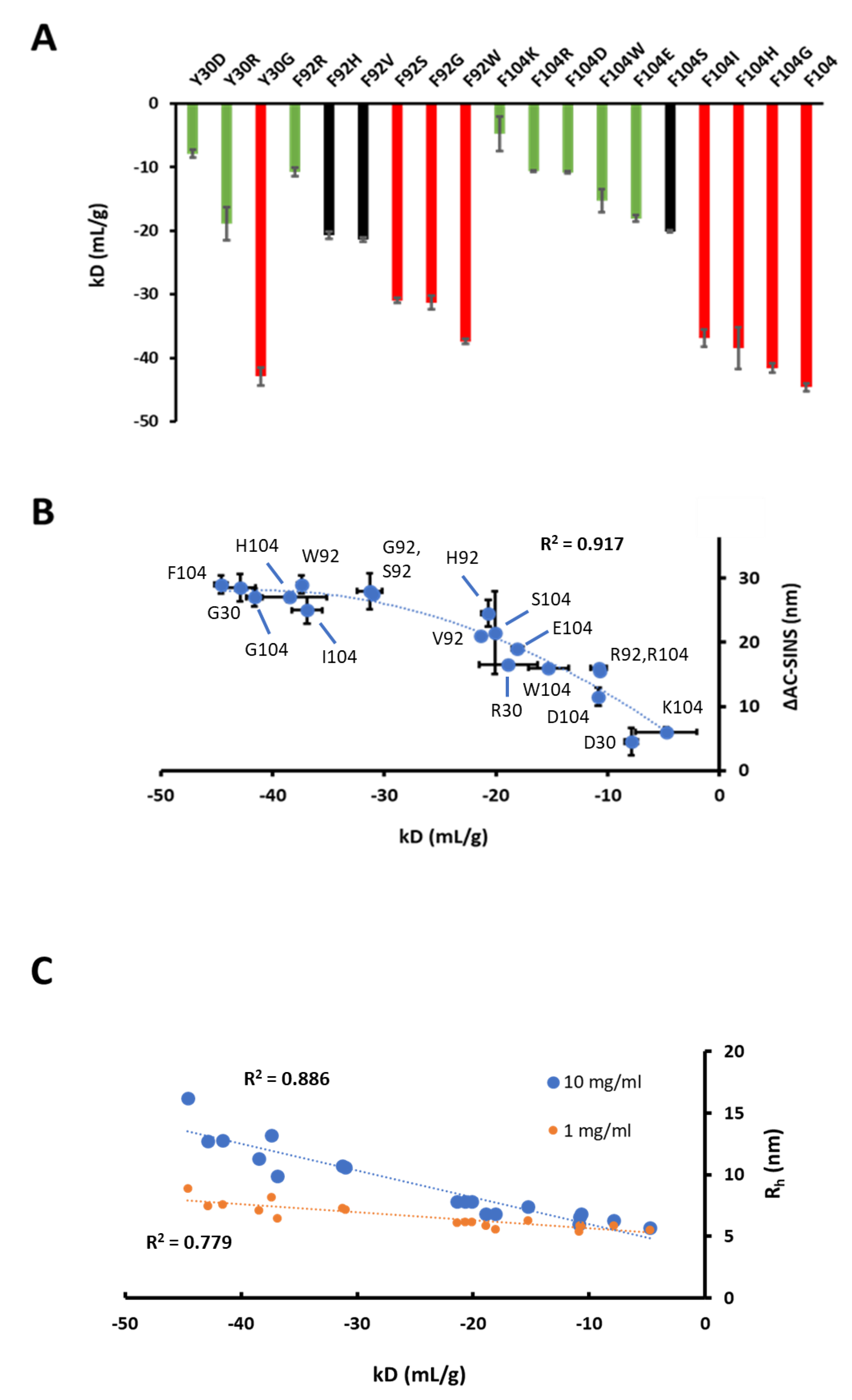
3.5. Mutagenesis of Residues Revealed by Homology Model Patch Analysis and Self-Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, R.; Hwang, Y.; Liu, I.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2019, 11, 219–238. [Google Scholar] [CrossRef]
- Shire, S.J. Formulation and manufacturability of biologics. Curr. Opin. Biotechnol. 2009, 20, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Le Basle, Y.; Chennell, P.; Tokhadze, N.; Astier, A.; Sautou, V. Physicochemical stability of monoclonal antibodies: A review. J. Pharm. Sci. 2020, 109, 169–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowitcz, B.; Caffry, I.; Yu, Y.; et al. Biophysical features of the clinical mAb landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Bailly, M.; Mieczkowski, C.; Juan, V.; Metwally, E.; Tomazela, D.; Baker, J.; Uchida, M.; Kofman, E.; Raoufi, F.; Motlagh, S.; et al. Predicting antibody developability profiles through early stage discovery screening. mAbs 2020, 12, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Singh, S.; Zeng, D.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef]
- Jarasch, A.; Koll, H.; Regula, J.T.; Bader, M.; Papadimitriou, A.; Kettenberger, H. Developability assessment during the selection of novel therapeutic antibodies. J. Pharm. Sci. 2015, 104, 1885–1898. [Google Scholar] [CrossRef]
- Daugherty, A.L.; Mrsny, R.J. Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef]
- Avery, L.B.; Wade, J.; Wang, M.; Tam, A.; King, A.; Piche-Nicholas, N.; Kavosi, M.S.; Penn, S.; Cirelli, D.; Kurz, J.C.; et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. mAbs 2018, 10, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Hotzel, I.; Theil, F.P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Bumbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.; Sun, T.; Jain, T.; Caffry, I.; Yu, Y.; Cao, Y.; Lynaugh, H.; Brown, M.; Vásquez, M.; Wittrup, K.D.; et al. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. mAbs 2015, 7, 770–777. [Google Scholar] [CrossRef]
- Geng, S.B.; Cheung, J.K.; Narasimhan, C.; Shameen, M.; Tessier, P.M. Improving monoclonal antibody selection and engineering using measurements of colloidal protein interactions. J. Pharm. Sci. 2014, 103, 3356–3363. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Nguyen, M.D.; Andya, J.D.; Shire, S.J. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J. Pharm. Sci. 2005, 94, 1928–1940. [Google Scholar] [CrossRef]
- Laue, T.M.; Shire, S.J. The molecular interaction process. J. Pharm. Sci. 2020, 109, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethea, D.; Wu, S.J.; Luo, J.; Hyun, L.; Lacy, E.R.; Teplyakov, A.; Jacobs, S.A.; O’Neil, K.T.; Gilliland, G.L.; Feng, Y. Mechanisms of self association of a human monoclonal antibody CNTO607. Protein Eng. Des. Sel. 2012, 25, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saluja, A.; Kalonia, D.S. Nature and consequences of protein–protein interactions in high protein concentration solutions. Int. J. Pharm. 2008, 358, 1–15. [Google Scholar] [CrossRef]
- Tomar, D.S.; Kumar, S.; Singh, S.K.; Goswami, S.; Li, L. Molecular basis of high viscosity in concentrated antibody solutions: Strategies for high concentration drug product development. mAbs 2016, 8, 216–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromwell, M.; Felten, C.; Flores, H.; Liu, J.; Shire, S.J. Self-association of therapeutic proteins: Implications for product development. In Misbehaving Proteins; Murphy, R.M., Tsai, A.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 313–330. [Google Scholar]
- Warne, N.W. Development of high concentration protein biopharmaceuticals: The use of platform approaches in formulation development. Eur. J. Pharm. Biopharm. 2009, 78, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Schrag, J.D.; Picard, M.E.; Gaudreault, F.; Gagnon, L.P.; Baardsnes, J. Binding symmetry and surface flexibility mediate antibody self-association. mAbs 2019, 11, 1300–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef]
- Lehermayr, C.; Mahler, H.C.; Mader, K.; Fischer, S. Assessment of net charge and protein-protein interactions of different monoclonal antibodies. J. Pharm. Sci. 2011, 100, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Laue, T.M.; Kalonia, D.S.; Singh, S.N.; Shire, S.J. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol. Pharm. 2012, 9, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, R.; Parupudi, A.; Casas-Finet, J.; Gadre, D.; Sathish, H. Mechanism of reversible self-association of a monoclonal antibody: Role of electrostatic and hydrophobic interactions. J. Pharm. Sci. 2014, 104, 577–586. [Google Scholar] [CrossRef]
- Salinas, B.A.; Sathish, H.A.; Bishop, S.M.; Harn, N.; Carpenter, J.F.; Randolph, T.W. Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation. J. Pharm. Sci. 2010, 99, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casaz, P.; Boucher, E.; Wollacott, R.; Pierce, B.G.; Rivera, R.; Sedic, M.; Ozturk, S.; Thomas, W.D., Jr.; Wang, Y. Resolving self-association of a therapeutic antibody by formulation optimization and molecular approaches. mAbs 2014, 6, 1533–1539. [Google Scholar] [CrossRef] [Green Version]
- Kanai, S.; Liu, J.; Patapoff, T.W.; Shire, S.J. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J. Pharm. Sci. 2008, 97, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Helk, B.; Kumar, S.; Mody, N.; Sathish, H.A.; Samra, H.S.; Buck, P.M.; Li, L.; Trout, B.L. Computational tool for the early screening of monoclonal antibodies for their viscosities. mAbs 2016, 8, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apgar, J.R.; Tam, A.S.P.; Sorm, R.; Moesta, S.; King, A.C.; Yang, H.; Kelleher, K.; Murphy, D.; D’Antona, A.M.; Yan, G.; et al. Modeling and mitigation of high concentration antibody viscosity through structure-based computer-aided protein design. PLoS ONE 2020, 5, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kumar, S.; Buck, P.M.; Burns, C.; Lavoie, J.; Singh, S.K.; Warne, N.W.; Nichols, P.; Luksha, N.; Boardman, D. Concentration dependent viscosity of monoclonal antibody solutions: Explaining experimental behavior in terms of molecular properties. Pharm. Res. 2014, 31, 3161–3178. [Google Scholar] [CrossRef]
- Shan, L.; Mody, N.; Sormani, P.; Rosenthal, K.L.; Damschroder, M.M.; Esfandiary, R. Developability assessment of engineered monoclonal antibody variants with a complex self-association behavior using complementary analytical and in silico tools. Mol. Pharm. 2018, 15, 5697–5710. [Google Scholar] [CrossRef]
- Raybould, M.I.J.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.K.; Patapoff, T.W.; Kabakoff, B.; Pai, S.; Hilario, E.; Zhang, B.; Li, C.; Borisov, O.; Kelley, R.F.; Chorny, I.; et al. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 2014, 111, 18601–18606. [Google Scholar] [CrossRef] [Green Version]
- Cromwell, M.E.; Hilario, E.; Jacobson, F. Protein aggregation and bioprocessing. AAPS J. 2006, 8, E572–E579. [Google Scholar] [CrossRef] [Green Version]
- Monkos, K. Concentration and temperature dependence of viscosity in lysozyme aqueous solutions. Biochim. Biophys. Acta 1997, 1339, 304–310. [Google Scholar] [CrossRef]
- Yadav, S.; Shire, S.J.; Kalonia, D.S. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. J. Pharm. Sci. 2010, 99, 4812–4826. [Google Scholar] [CrossRef]
- Scherer, T.M.; Liu, J.; Shire, S.J.; Minton, A.P. Intermolecular interactions of IgG1 monoclonal antibodies at high concentrations characterized by light scattering. J. Phys. Chem. B 2010, 114, 12948–12957. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Hasegawa, J.; Kobayashi, N.; Kishi, N.; Uchiyama, S.; Fukui, K. Behavior of monoclonal antibodies: Relation between the second virial coefficient (B(2)) at low concentrations and aggregation propensity and viscosity at high concentrations. Pharm. Res. 2012, 29, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Borwankar, A.U.; Dear, B.J.; Twu, A.; Hung, J.J.; Dinin, A.K.; Wilson, B.K.; Yue, J.; Maynard, J.A.; Truskett, T.M.; Johnston, K.P. Viscosity reduction of a concentrated monoclonal antibody with arginine·HCl and arginine·glutamate. Ind. Eng. Chem. Res. 2016, 55, 11225–11234. [Google Scholar] [CrossRef]
- Inoue, N.; Takai, E.; Arakawa, T.; Shiraki, K. Specific decrease in solution viscosity of antibodies by arginine for therapeutic formulations. Mol. Pharm. 2014, 11, 1889–1896. [Google Scholar] [CrossRef]
- Connolly, B.D.; Petry, C.; Yadav, S.; Demeule, B.; Ciaccio, N.; Moore, J.M.; Shire, S.J.; Gokarn, Y.R. Weak interactions govern the viscosity of concentrated antibody solutions: High-throughput analysis using the diffusion interaction parameter. Biophys. J. 2012, 103, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saluja, A.; Fesinmeyer, R.M.; Hogan, S.; Brems, D.N.; Gokarn, Y.R. Diffusion and sedimentation interaction parameters for measuring the second virial coefficient and their utility as predictors of protein aggregation. Biophys. J. 2010, 99, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, P.M.; Wu, J.; Dickinson, C.D. Emerging methods for identifying monoclonal antibodies with low propensity to self-associate during the early discovery process. Expert Opin. Drug Deliv. 2014, 11, 461–465. [Google Scholar] [CrossRef]
- Geng, S.B.; Wittekind, M.; Vigil, A.; Tessier, P.M. Measurements of monoclonal antibody self-association are correlated with complex biophysical properties. Mol. Pharm. 2016, 13, 1636–1645. [Google Scholar] [CrossRef]
- Sule, S.V.; Dickinson, C.D.; Lu, J.; Chow, C.K.; Tessier, P.M. Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol. Pharm. 2013, 10, 1322–1331. [Google Scholar] [CrossRef]
- Jayaraman, J.; Wu, J.; Brunelle, M.C.; Cruz, A.M.M.; Goldberg, D.S.; Lobo, B.; Shah, A.; Tessier, P.M. Plasmonic measurements of monoclonal antibody self-association using self-interaction nanoparticle spectroscopy. Biotechnol. Bioeng. 2014, 111, 1513–1520. [Google Scholar] [CrossRef]
- Howlett, G.J.; Minton, A.P.; Rivas, G. Analytical ultracentrifugation for the study of protein association and assembly. Curr. Opin. Chem. Biol. 2006, 10, 430–436. [Google Scholar] [CrossRef]
- Jimenez, M.; Rivas, G.; Minton, A.P. Quantitative characterization of weak self-association in concentrated solutions of immunoglobulin G via the measurement of sedimentation equilibrium and osmotic pressure. Biochemistry 2007, 46, 8373–8378. [Google Scholar] [CrossRef] [Green Version]
- Esfandiary, R.; Hayes, D.B.; Parupudi, A.; Casas-Finet, J.; Bai, S.; Samra, H.S.; Shah, A.U.; Sathishi, H.A. A Systematic multitechnique approach for detection and characterization of reversible self-association during formulation development of therapeutic antibodies. J. Pharm. Sci. 2012, 102, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Caffry, I.; Wu, J.; Geng, S.B.; Jain, T.; Sun, T.; Reid, F.; Cao, Y.; Estep, P. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. mAbs 2014, 6, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.; Nicolai, T. Dynamic Light Scattering: The Method and Some Applications; Oxford University Press: New York, NY, USA, 1993. [Google Scholar]
- Berteau, C.; Filipe-Santos, O.; Wang, T.; Rojas, H.E.; Granger, C.; Schwarzenbach, F. Evaluation of the impact of viscosity, injection volume, and injection flow rate on subcutaneous injection tolerance. Med. Devices Evid. Res. 2015, 8, 473–484. [Google Scholar]
- Tilegenova, C.; Izadi, S.; Yin, J.; Huang, C.S.; Wu, J.; Ellerman, D.; Hymowitz, S.G.; Walters, B.; Salisbury, C.; Carter, P.J. Dissecting the molecular basis of high viscosity of monospecific and bispecific IgG antibodies. mAbs 2020, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Miyajima, M.; Wakiyama, N.; Kubota, K.; Hasegawa, J.; Uchiyama, S.; Fukui, K. Fc domain mediated self-association of an IgG1 monoclonal antibody under a low ionic strength condition. J. Biosci. Bioeng. 2011, 112, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kuriyan, J.; Eisenberg, D. The origin of protein interactions and allostery in colocalization. Nature 2007, 450, 983–990. [Google Scholar] [CrossRef]
- Kastritis, P.L.; Bonvin, A.M. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Roberts, V.A.; Rittenberg, M.B. Random point mutations in an antibody CDR2 sequence: Many mutated antibodies lose their ability to bind antigen. J. Exp. Med. 1992, 176, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Ambrosetti, F.; Jimenez-Garcia, B.; Roel-Touris, J.; Bonvin, A.M. Modeling antibody-antigen complexes by information-driven docking. Structure 2020, 28, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pedotti, M.; Simonelli, L.; Livoti, E.; Varani, L. Computational docking of antibody-antigen complexes, opportunities and pitfalls illustrated by influenza hemagglutinin. Int. J. Mol. Sci. 2011, 12, 226–251. [Google Scholar] [CrossRef] [Green Version]
- Teplyakov, A.; Obmolova, G.; Wu, S.J.; Luo, J.; Kang, J.; O’Neil, K.; Gilliland, G.L. Epitope mapping of anti-interleukin-13 neutralizing antibody CNTO607. J. Mol. Biol. 2009, 389, 115–123. [Google Scholar] [CrossRef]
- Shetata, L.; Maurer, D.P.; Wec, A.Z.; Lilov, A.; Champney, A.; Sun, T.; Archambault, K.; Burnina, I.; Lynaugh, H.; Zhi, X.; et al. Affinity maturation enhances antibody specificity but compromises conformational stability. Cell Rep. 2019, 28, 3300–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | KD (nM) | KD/KDref | kD (mL/g) |
|---|---|---|---|
| F104 | 0.35 | 1 | −44.6 |
| F104W | 0.16 | 0.44 | −15.3 |
| F104I | 108 | 305 | −36.9 |
| F104H | 1372 | 3879 | −38.5 |
| F104D | NB | NB | −10.9 |
| F104K | NB | NB | −4.8 |
| F104E | NB | NB | −18.1 |
| F104S | NB | NB | −20.1 |
| F104G | NB | NB | −41.6 |
| F104R | NB | NB | −10.7 |
| Y30R | 4.36 | 12.3 | −18.9 |
| Y30H | 5.5 | 15.6 | NA |
| Y30N | 45.5 | 129 | NA |
| Y30D | 60.9 | 172 | −7.9 |
| Y30Q | 82.6 | 234 | NA |
| Y30G | 170 | 481 | −42.9 |
| F92W | 5.51 | 15.6 | −37.4 |
| F92H | 35.1 | 99.2 | −20.7 |
| F92V | 38.7 | 109 | −21.4 |
| F92R | 50.2 | 142 | −10.8 |
| F92S | 62.7 | 177 | −31 |
| F92G | 142 | 402 | −31.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mieczkowski, C.; Cheng, A.; Fischmann, T.; Hsieh, M.; Baker, J.; Uchida, M.; Raghunathan, G.; Strickland, C.; Fayadat-Dilman, L. Characterization and Modeling of Reversible Antibody Self-Association Provide Insights into Behavior, Prediction, and Correction. Antibodies 2021, 10, 8. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010008
Mieczkowski C, Cheng A, Fischmann T, Hsieh M, Baker J, Uchida M, Raghunathan G, Strickland C, Fayadat-Dilman L. Characterization and Modeling of Reversible Antibody Self-Association Provide Insights into Behavior, Prediction, and Correction. Antibodies. 2021; 10(1):8. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010008
Chicago/Turabian StyleMieczkowski, Carl, Alan Cheng, Thierry Fischmann, Mark Hsieh, Jeanne Baker, Makiko Uchida, Gopalan Raghunathan, Corey Strickland, and Laurence Fayadat-Dilman. 2021. "Characterization and Modeling of Reversible Antibody Self-Association Provide Insights into Behavior, Prediction, and Correction" Antibodies 10, no. 1: 8. https://0-doi-org.brum.beds.ac.uk/10.3390/antib10010008